
Free Energy Analyses of Cell-Penetrating Peptides Using the Weighted Ensemble Method


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Equilibrium Simulations
2.2. Weighted Ensemble (WE) Simulations
3. Results
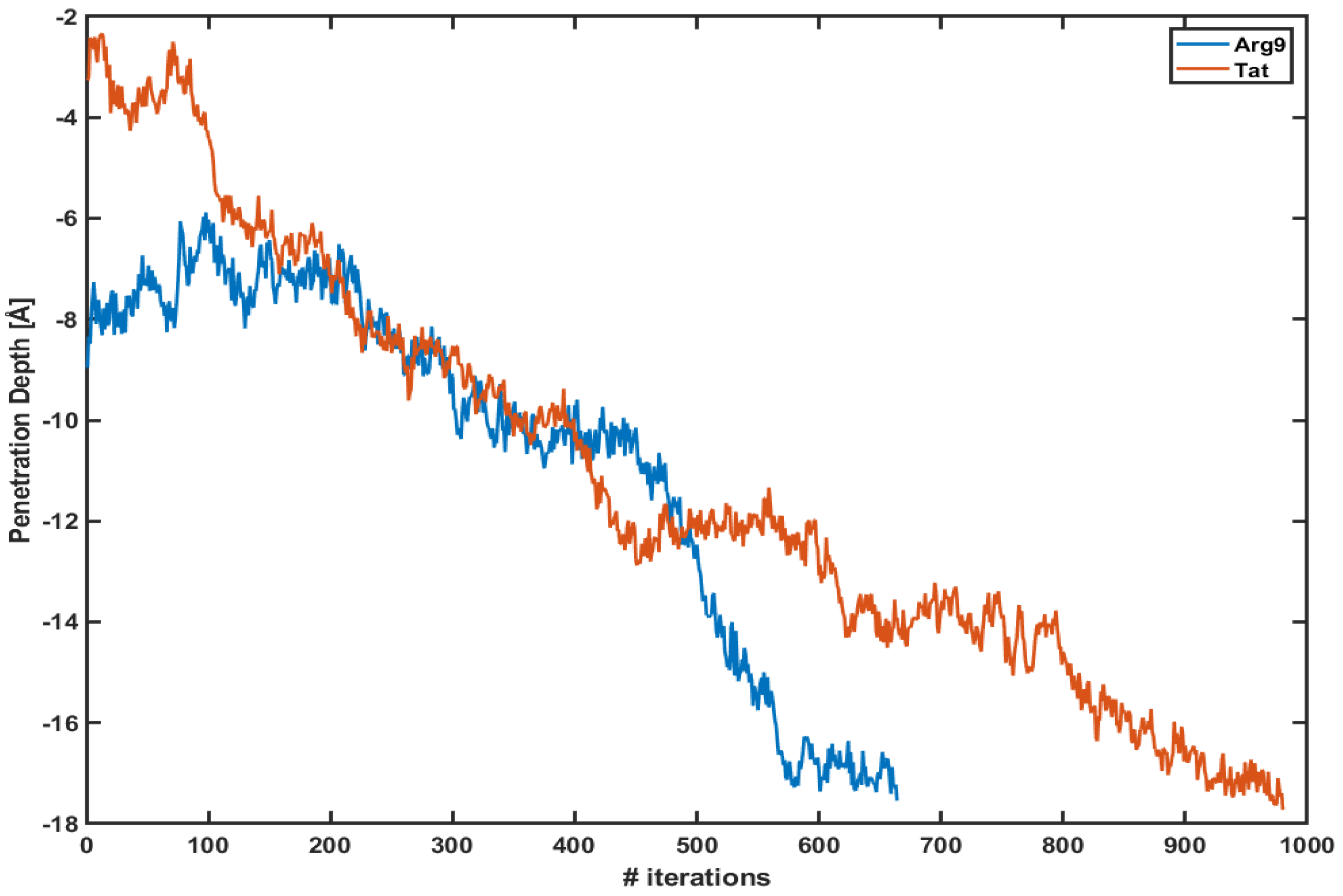
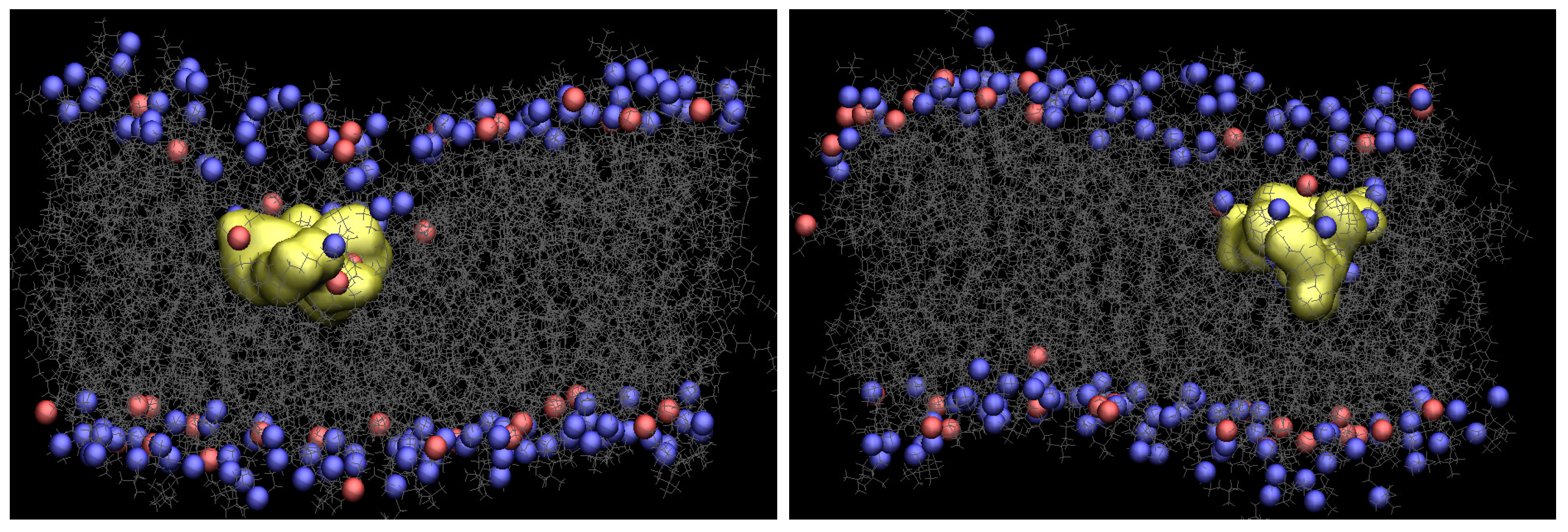
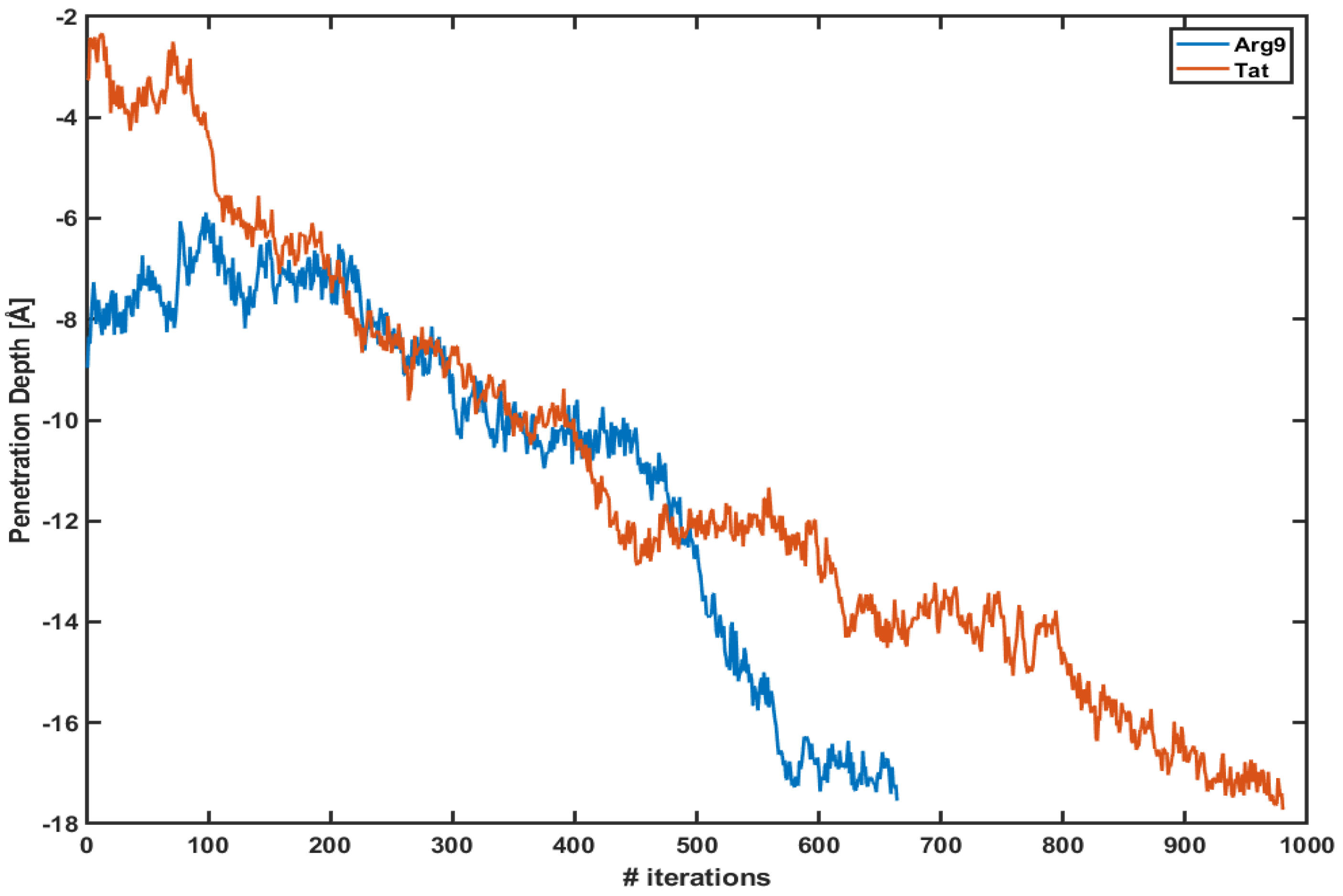
3.1. WE Simulations Show Much-Enhanced Penetration within a Short Amount of Time
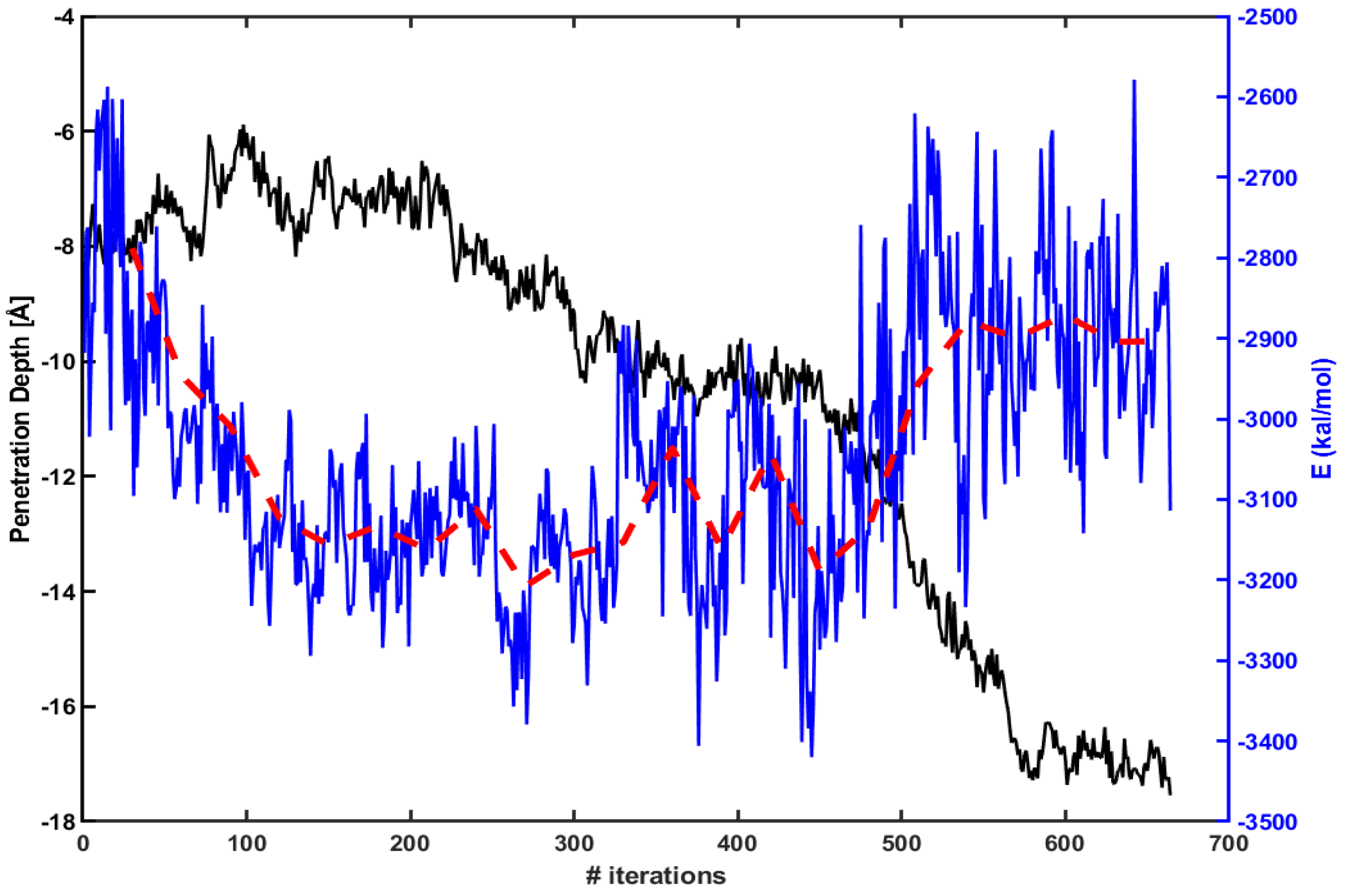
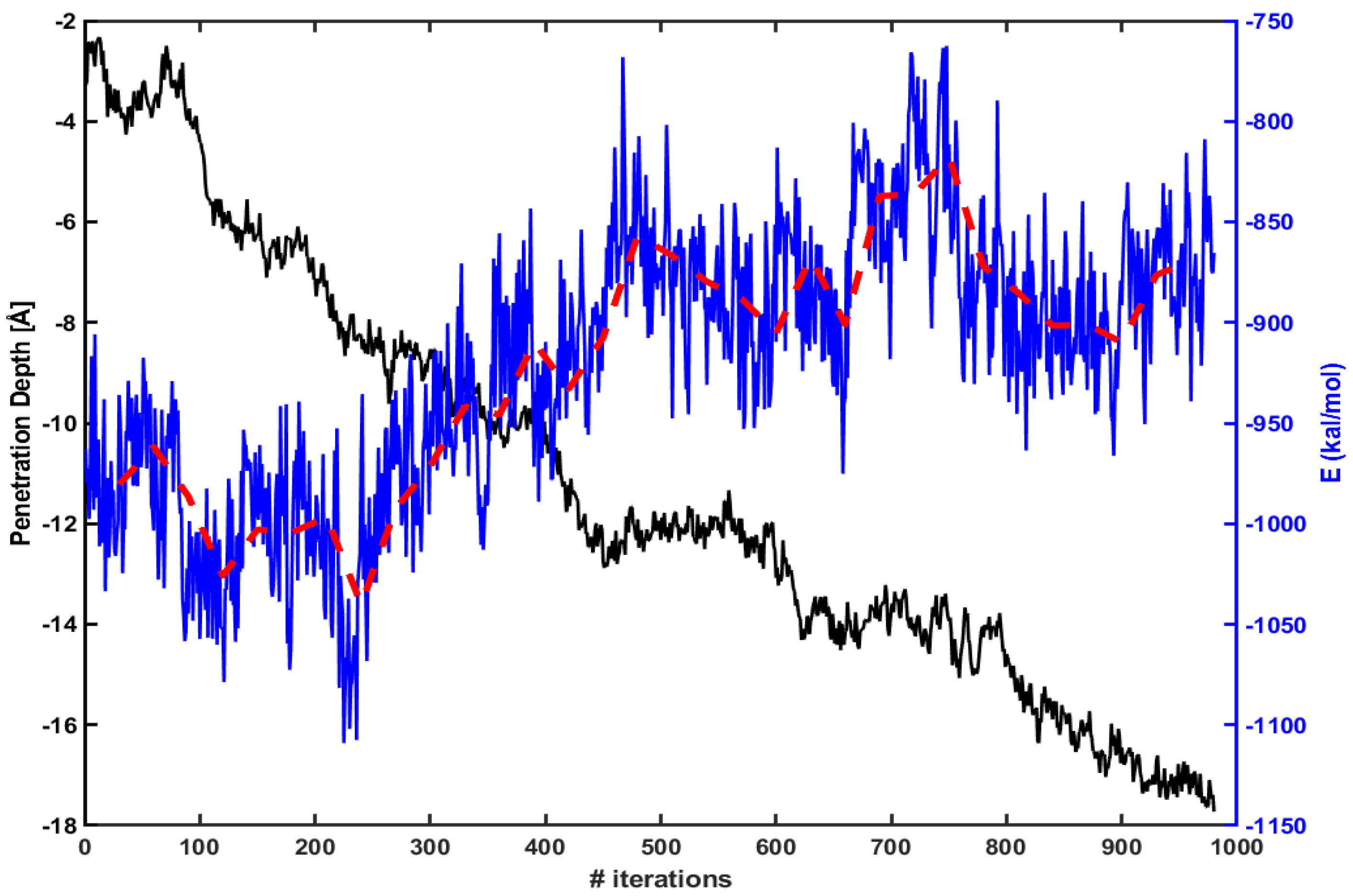
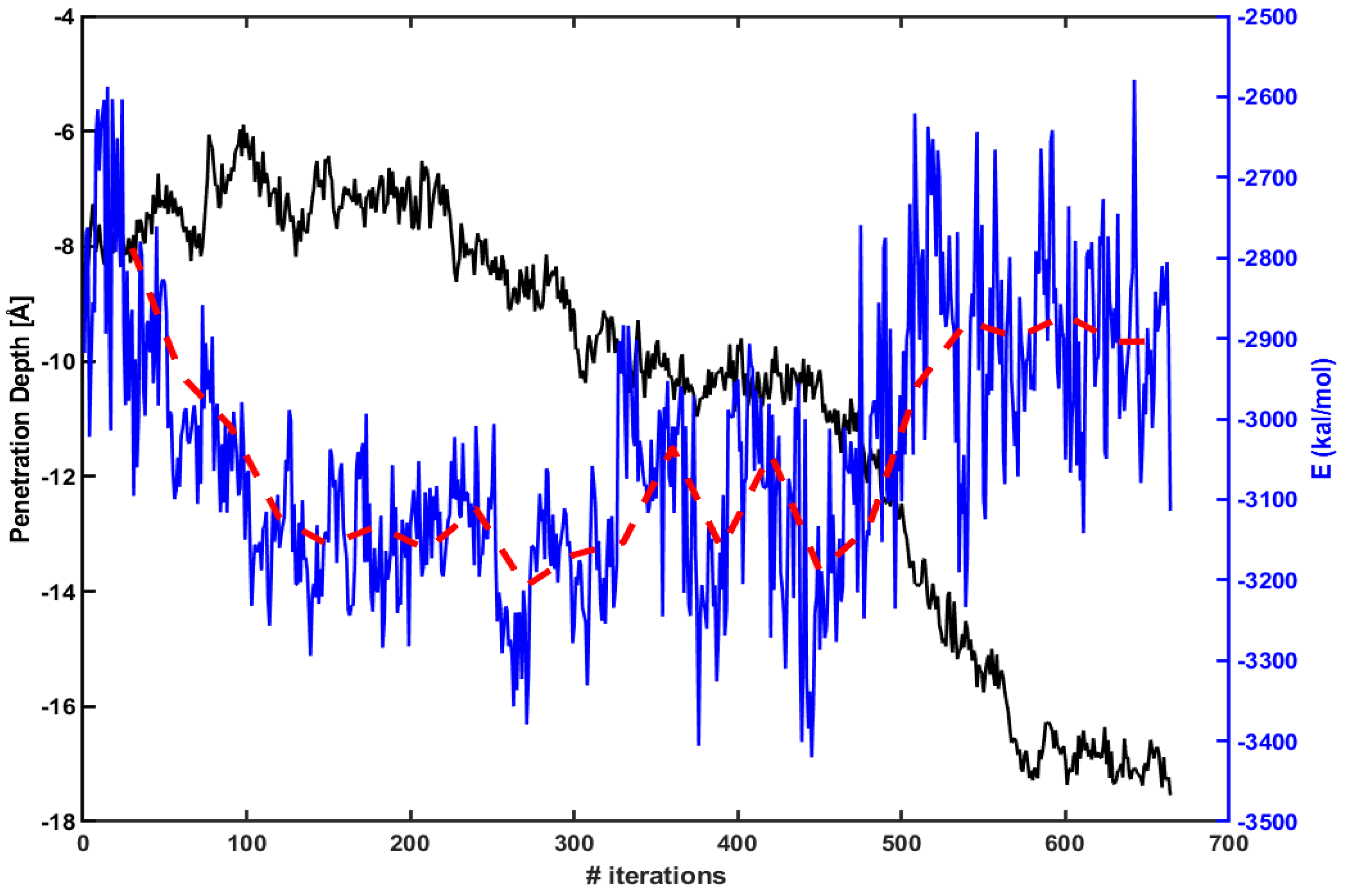
3.2. Electrostatic Energy between a CPP and a Model Membrane Plays a Role during the Translocation
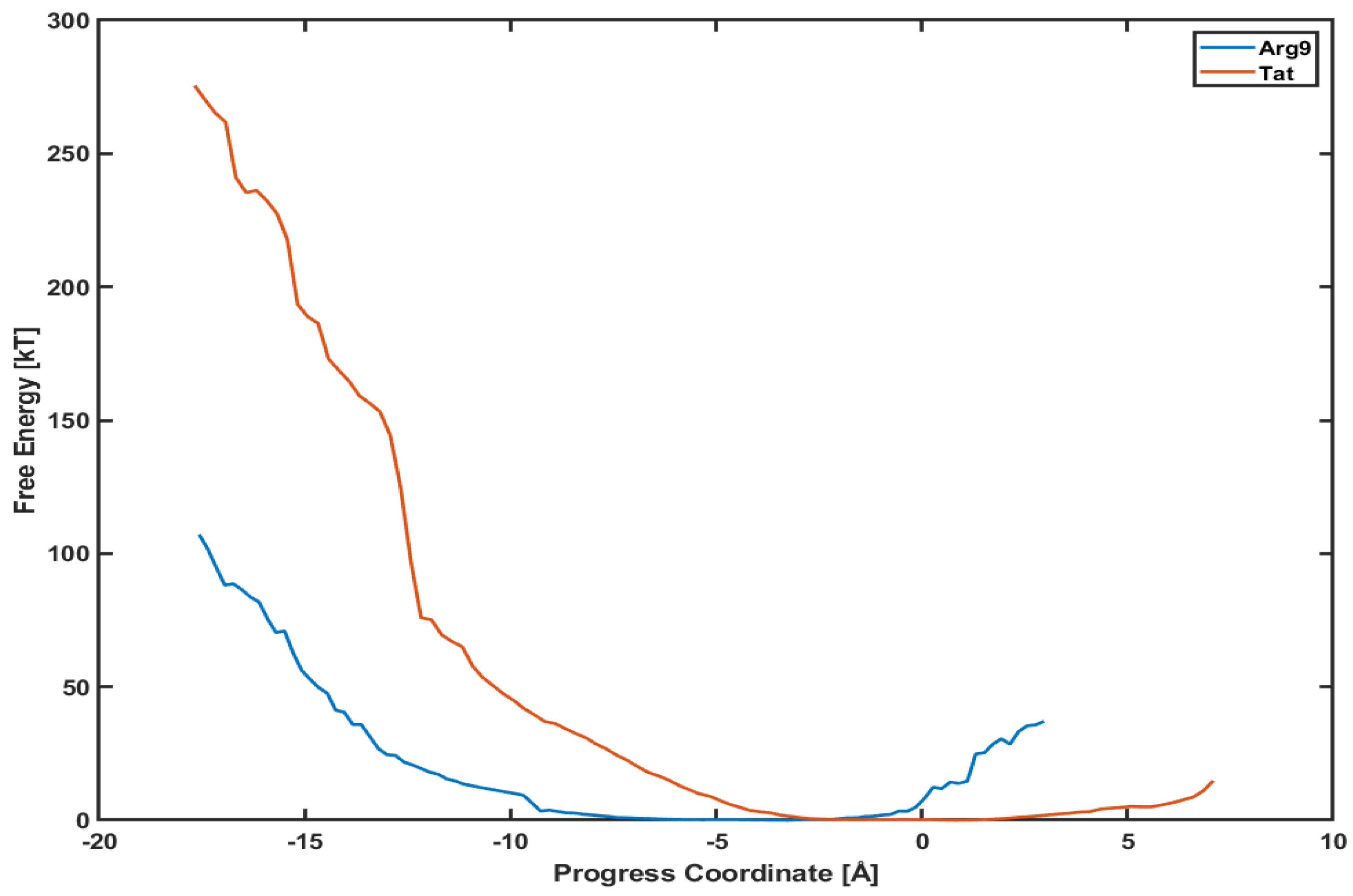
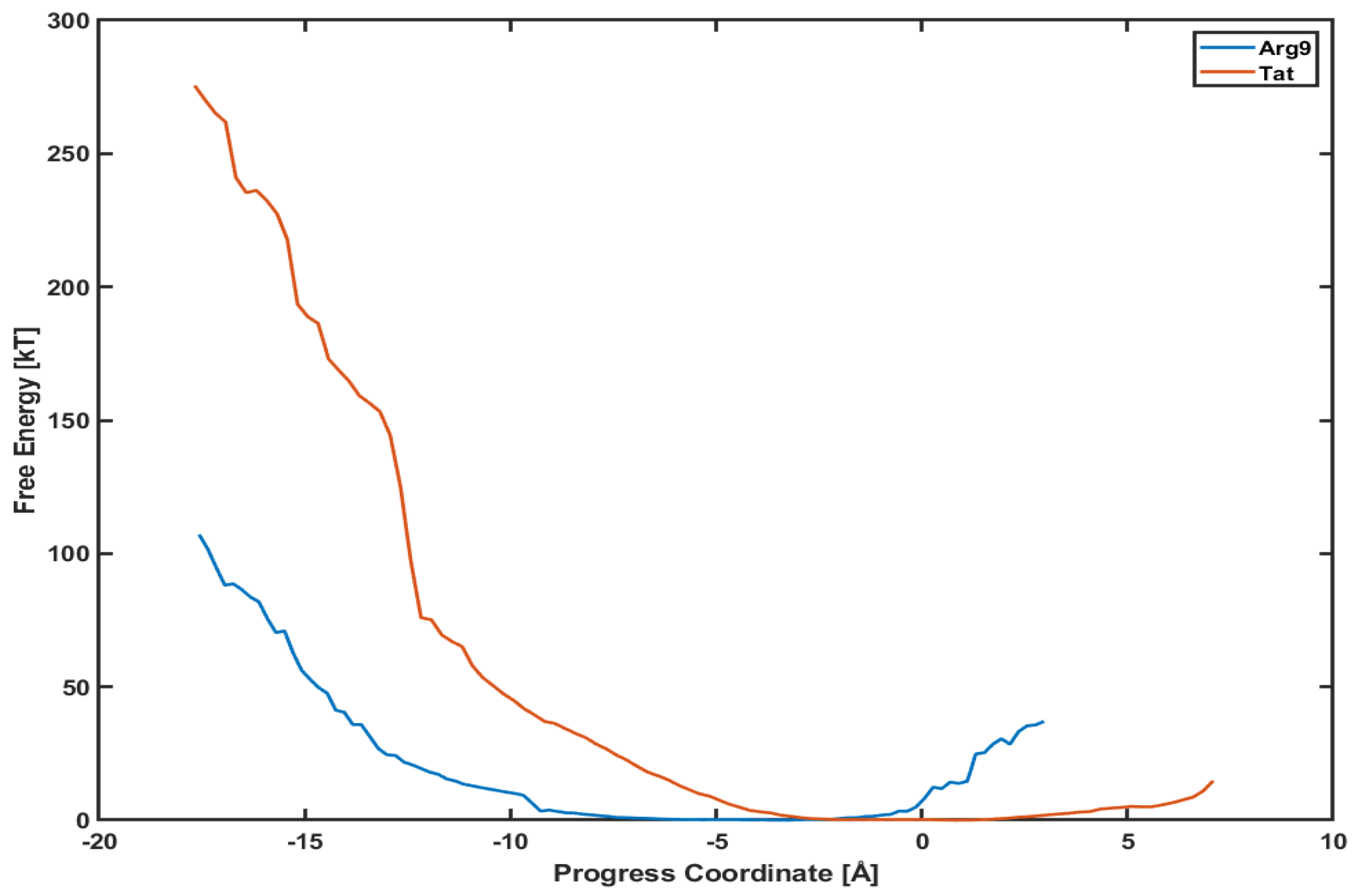
3.3. The Free Energy Profiles Based on the WE Simulation Data Show That Arg Penetrates through a DOPC/DOPG(4:1) Model Membrane Much Easier Than Tat Does
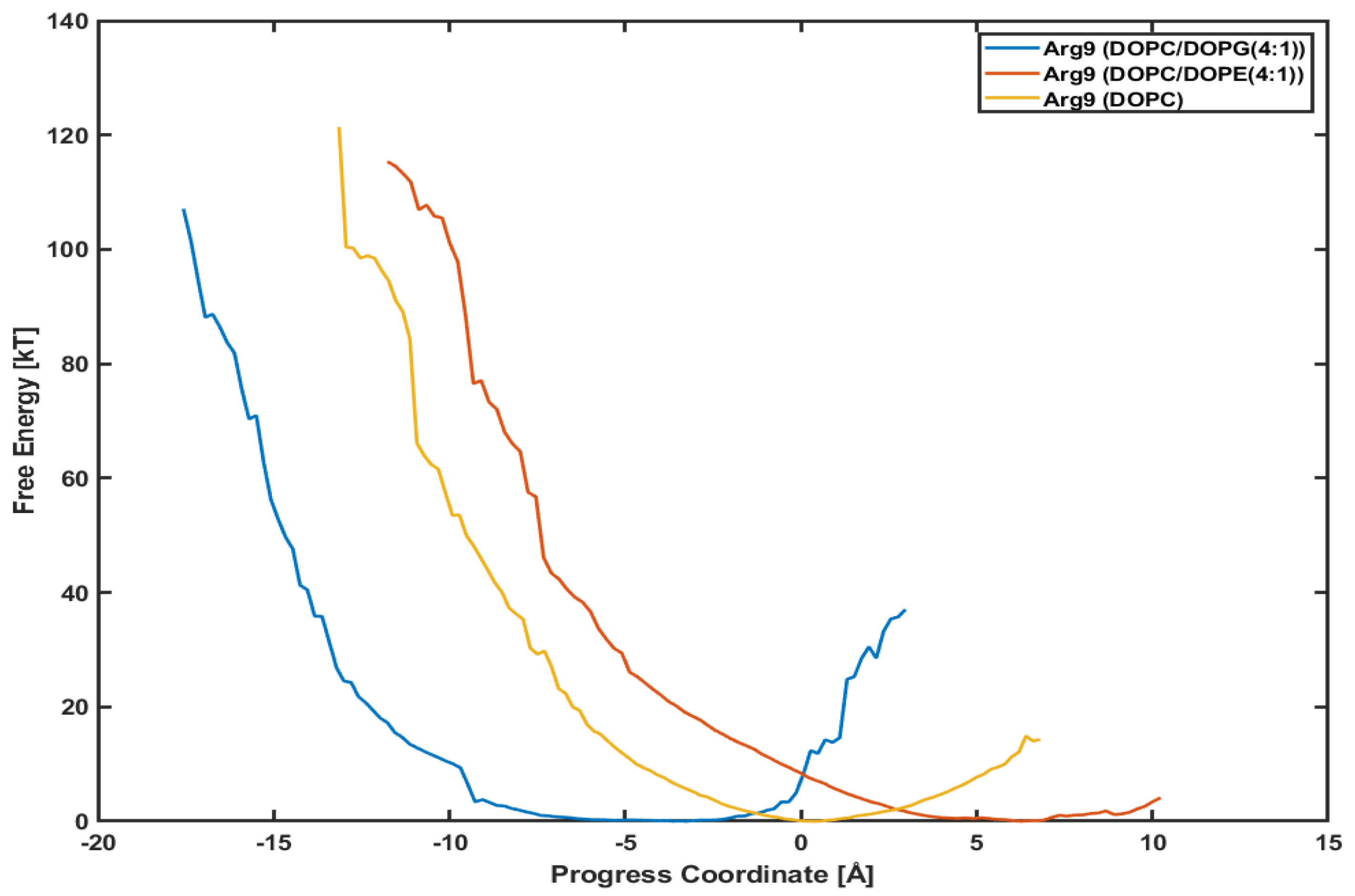
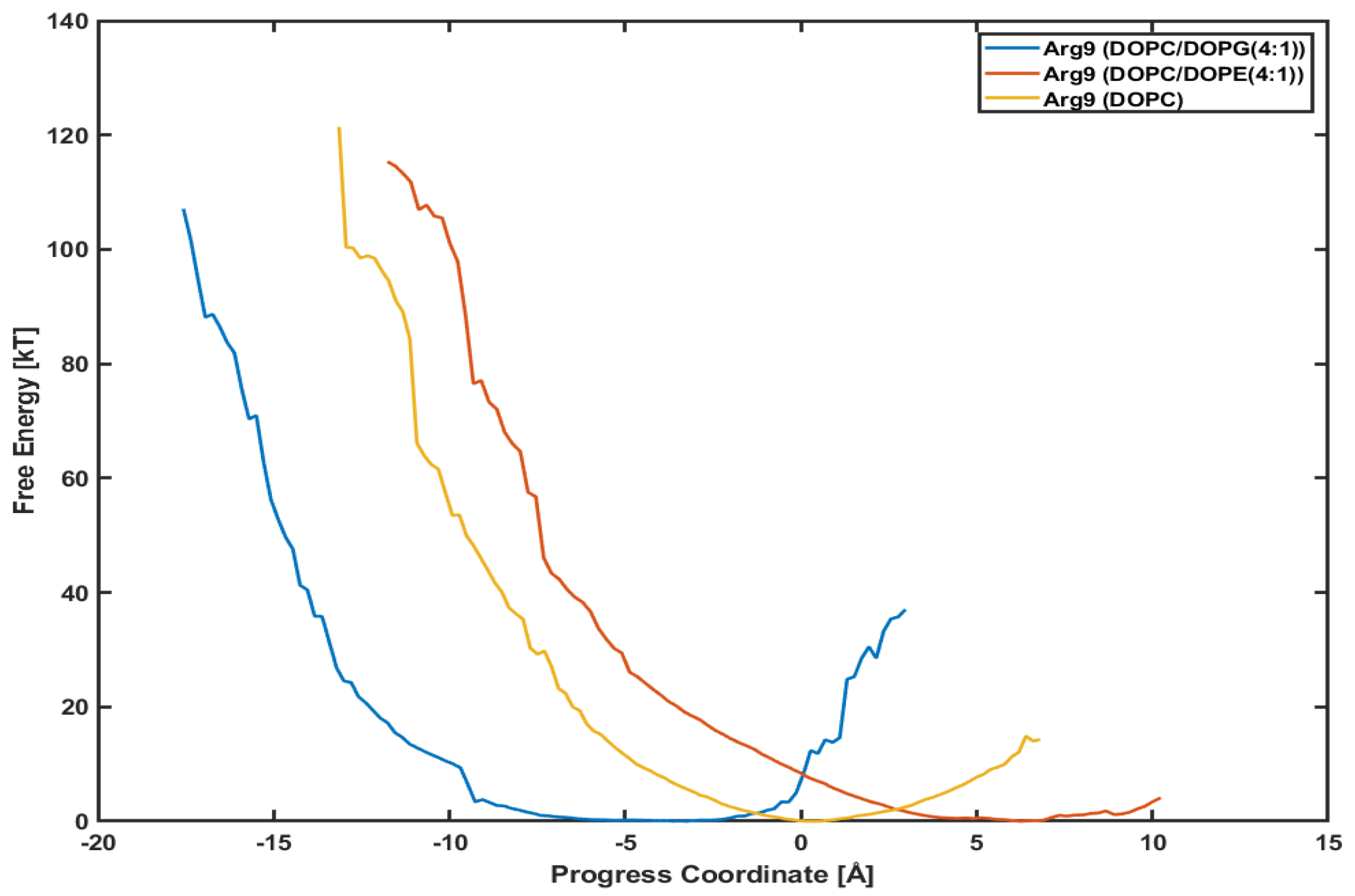
3.4. Different Lipid Compositions Slightly Affect the PMF Profiles of Arg
4. Discussion
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CPP | Cell-Penetrating Peptide |
| WE | Weighted Ensemble |
| DNA | DeoxyriboNucleic Acid |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPG | 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] |
| DOPE | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine |
| Arg(R9) | a peptide with RRRRRRRRR |
| Tat | a peptide with GRKKRRQRRRPPQ |
References
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Pisa, M.D.; Chassaing, G.; Swiecicki, J. Translocation Mechanism(s) of Cell-Penetrating Peptides: Biophysical Studies Using Artificial Membrane Bilayers. Biochemistry 2015, 54, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, U.; Futaki, S.; Gräslund, A. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Fretz, M.M.; Penning, N.A.; Al-Taei, S.; Futaki, S.; Takeuchi, T.; Nakase, I.; Storm, G.; Jones, A.T. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem. J. 2007, 403, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [Green Version]
- Herce, H.D.; Garcia, A.E.; Litt, J.; Kane, R.S.; Martin, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yesylevskyy, S.; Marrink, S.J.; Mark, A. Alternative mechanisms for the interaction of the cell-penetrating peptides penetratin and the TAT peptide with lipid bilayers. Biophys. J. 2009, 97, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Zorko, M.; Langel, U. Cell-penetrating peptides: Mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 2005, 57, 529–545. [Google Scholar] [CrossRef]
- Choe, S. Molecular dynamics studies of interactions between Arg9(nona-arginine) and a DOPC/DOPG(4:1) membrane. AIP Adv. 2020, 10, 105103. [Google Scholar] [CrossRef]
- Pourmousa, M.; Wong-ekkabut, J.; Patra, M.; Karttunen, M. Molecular Dynamic Studies of Transportan Interacting with a DPPC Lipid Bilayer. J. Phys. Chem. B 2013, 117, 230–241. [Google Scholar] [CrossRef]
- Teseia, G.; Vazdarb, M.; Jensenc, M.R.; Cragnella, C.; Masond, P.E.; Heydae, J.; Skepo, M.; Jungwirthd, P.; Lunda, M. Self-association of a highly charged arginine-rich cell-penetrating peptide. Proc. Nalt. Acad. Sci. USA 2017, 114, 11428–11433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choong, F.H.; Yap, B.K. Cell-Penetrating Peptides: Correlation between peptide-lipid interaction and penetration efficiency. ChemPhysChem 2021, 22, 493–498. [Google Scholar] [CrossRef]
- Akhunzada, M.J.; Chandramouli, B.; Bhattacharjee, N.; Macchi, S.; Cardarelli, F.; Brancato, G. The role of Tat peptide self-aggregation in membrane pore stabilization: Insights from a computational study. Phys. Chem. Chem. Phys. 2017, 19, 27603–27610. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, D.M.; Chong, L.T. Weighted Ensemble Simulation: Review of Methodology, Applications, and Software. Annu. Rev. Biophys. 2017, 46, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Bogetti, A.T.; Mosto1an, B.; Dickson, A.; Pratt, A.; Saglam, A.S.; Harrison, P.O.; Adelman, J.L.; Dudek, M.; Torrillo, P.A.; DeGrave, A.J.; et al. A Suite of Tutorials for the WESTPA Rare-Events Sampling Software. Living J. Comp. Mol. Sci. 2019, 1, 10607. [Google Scholar]
- Zwier, M.C.; Adelman, J.L.; Kaus, J.W.; Pratt, A.J.; Wong, K.F.; Rego, N.B.; Suarez, E.; Lettieri, S.; Wang, D.W.; Grabe, M.; et al. WESTPA: An interoperable, highly scalable software package for weighted ensemble simulation and analysis. J. Chem. Theory Comput. 2015, 11, 800–809. [Google Scholar] [CrossRef] [Green Version]
- Crosio, M.A.; Via, M.A.; Cámara, C.I.; Mangiarotti, A.; Pópolo, M.G.D.; Wilke, N. Interaction of a polyarginine peptide with membranes of different mechanical properties. Biomolecules 2019, 9, 625. [Google Scholar] [CrossRef] [Green Version]
- Sharmin, S.; Islam, M.Z.; Karal, M.A.S.; Shibly, S.U.A.; Dohra, H.; Yamazaki, M. Effects of Lipid Composition on the Entry of Cell-Penetrating Peptide Oligoarginine into Single Vesicles. Biochemistry 2016, 55, 4154–4165. [Google Scholar] [CrossRef]
- Phillips, J.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Vogel, A.; Correia, I.; Lequin, O.; Olausson, B.E.S.; Desbat, B.; Sagan, S.; Alves, I.D. Membrane interactions of two arginine-rich peptides with different cell internalization capacities. Biochim. Biophys. Acta 2012, 1818, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Roux, B. The membrane potential and its representation by a constant electric field in computer simulations. Biophys. J. 2008, 95, 4205–4216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Galassi, V.; Wilke, N. On the Coupling between Mechanical Properties and Electrostatics in Biological Membranes. Membranes 2021, 11, 478. [Google Scholar] [CrossRef]
- Huang, K.; Garcia, A.E. Free energy of translocating an arginine-rich cell-penetrating peptide across a lipid bilayer suggests pore formation. Biophys. J. 2013, 104, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Guterstam, P.; Madani, F.; Hirose, H.; Takeuchi, T.; Futaki, S.; Andaloussi, S.E.; Gräslund, A.; Langel, Ü. Elucidating cell-penetrating peptide mechanisms of action for membrane interaction, cellular uptake, and translocation utilizing the hydrophobic counter-anion pyrenebutyrate. Biochim. Biophys. Acta 2009, 1788, 2509–2517. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.Z.; Sharmin, S.; Moniruzzaman, M.; Yamazaki, M. Elementary processes for the entry of cell-penetrating peptides into lipid bilayer vesicles and bacterial cells. Appl. Microbiol. Biotech. 2018, 102, 3879–3892. [Google Scholar] [CrossRef]
- Freites, J.A.; Tobias, D.J.; von Heijne, G.; White, S.H. Interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. USA 2005, 102, 15059–15064. [Google Scholar] [CrossRef] [Green Version]
- Dorairaj, S.; Allen, T.W. On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc. Natl. Acad. Sci. USA 2007, 104, 4943–4948. [Google Scholar] [CrossRef] [Green Version]
- MacCallum, J.L.; Bennett, W.F.D.; Tieleman, D.P. Partitioning of amino acid side chains into lipid bilayers: Results from computer simulations and comparison to experiment. J. Gen. Physiol. 2007, 129, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Hecht, K.A.; Grabe, M. A continuum method for determining protein insertion energies and the problem of charged residues. J. Gen. Physiol. 2008, 131, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Yang, X.; Lin, Z.; He, B.; Mei, D.; Wang, D.; Zhang, H.; Zhang, H.; Dai, W.; Wang, X.; et al. The use of electronic-neutral penetrating peptides cyclosporin A to deliver pro-apoptotic peptide: A possibly better choice than positively charged TAT. J. Control. Release 2017, 261, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, M.; Takeuchi, T.; Nakase, I.; Jones, A.T.; Futaki, S. Cellular Internalization and Distribution of Arginine-Rich Peptides as a Function of Extracellular Peptide Concentration, Serum, and Plasma Membrane Associated Proteoglycans. Bioconjugate Chem. 2008, 19, 656–664. [Google Scholar] [CrossRef]
- Sun, C.; Shen, W.C.; Tu, J.; Zaro, J.L. Interaction between Cell-Penetrating Peptides and Acid-Sensitive Anionic Oligopeptides as a Model for the Design of Targeted Drug Carriers. Mol. Pharm. 2014, 11, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Ouahab, A.; Cheraga, N.; Onoja, V.; Shen, Y.; Tu, J. Novel pH-sensitive charge-reversal cell penetrating peptide conjugated PEG-PLA micelles for docetaxel delivery: In vitro study. Int. J. Pharm. 2014, 466, 233–245. [Google Scholar] [CrossRef]
- Chen, W.; Morrow, B.H.; Shi, C.; Shen, J.K. Recent development and application of constant pH molecular dynamics. Mol. Simul. 2014, 40, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Radak, B.K.; Chipot, C.; Suh, D.; Jo, S.; Jiang, W.; Phillips, J.C.; Schulten, K.; Roux, B. Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput. 2017, 13, 5933–5944. [Google Scholar] [CrossRef]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Hung, W.C.; Chen, F.Y.; Huang, H.W. Manybody effect of antimicrobial peptides: On the correlation between lipid’s spontaneous curvature and pore formation. Biophys. J. 2005, 89, 4006–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choe, S. Free Energy Analyses of Cell-Penetrating Peptides Using the Weighted Ensemble Method. Membranes 2021, 11, 974. https://doi.org/10.3390/membranes11120974
Choe S. Free Energy Analyses of Cell-Penetrating Peptides Using the Weighted Ensemble Method. Membranes. 2021; 11(12):974. https://doi.org/10.3390/membranes11120974
Chicago/Turabian StyleChoe, Seungho. 2021. "Free Energy Analyses of Cell-Penetrating Peptides Using the Weighted Ensemble Method" Membranes 11, no. 12: 974. https://doi.org/10.3390/membranes11120974
APA StyleChoe, S. (2021). Free Energy Analyses of Cell-Penetrating Peptides Using the Weighted Ensemble Method. Membranes, 11(12), 974. https://doi.org/10.3390/membranes11120974