
Multicomponent Vaccines against Group A Streptococcus Can Effectively Target Broad Disease Presentations

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Growth Conditions
2.2. Preparation of Recombinant GAS Antigens
2.3. Animals and Immunisations
2.4. Purification of IgG from Mouse Sera
2.5. ELISA to Measure IgG Antibody Titre
2.6. Fractionation of GAS Cultures for Protein Analysis
2.7. Visualisation of Protein Antigens
2.8. Immunofluorescence Staining and Flow Cytometry Analysis
2.9. Opsonophagocytosis Assay (OPA)
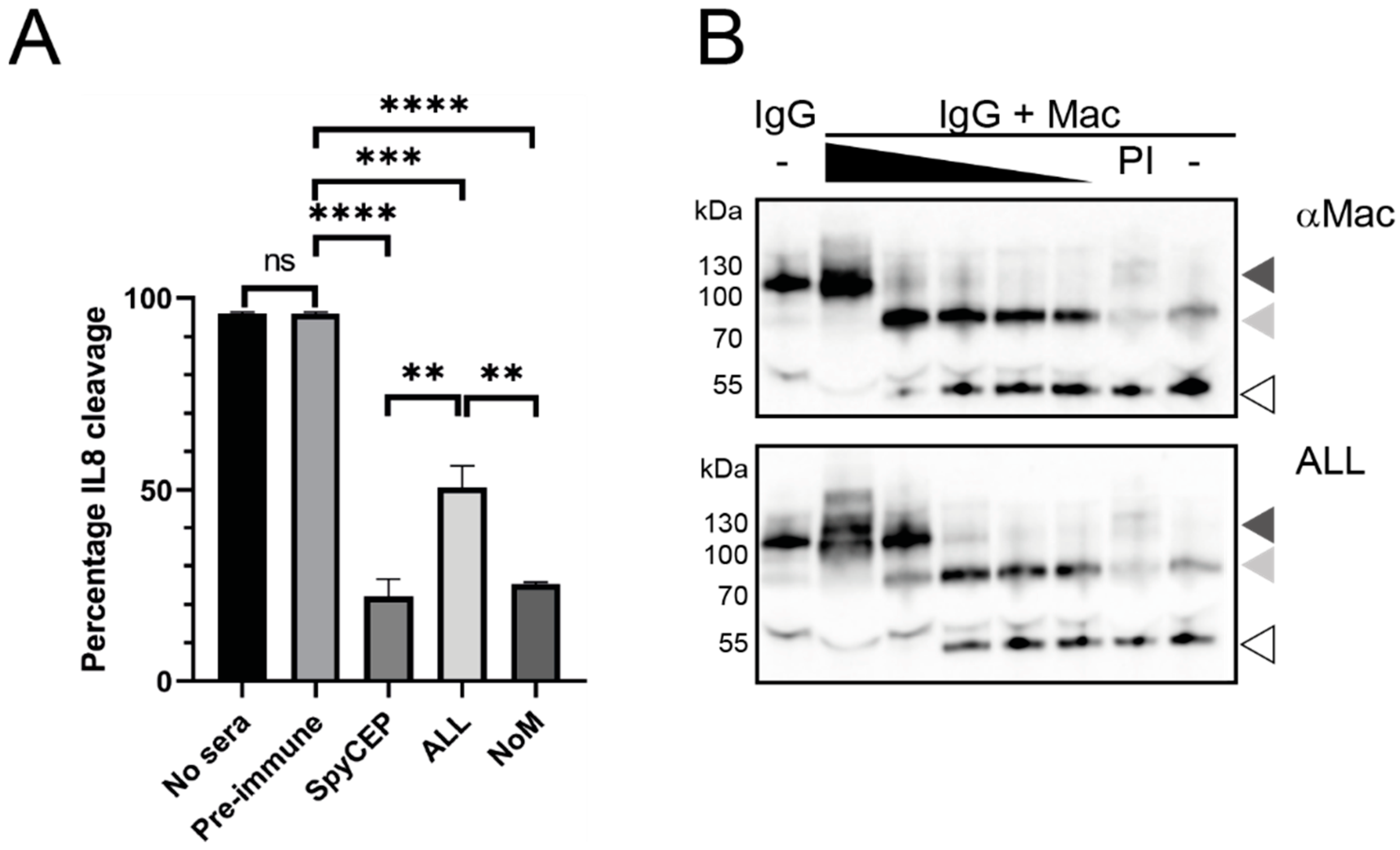
2.10. IL-8 Cleavage Assay
2.11. IgG Cleavage Assay
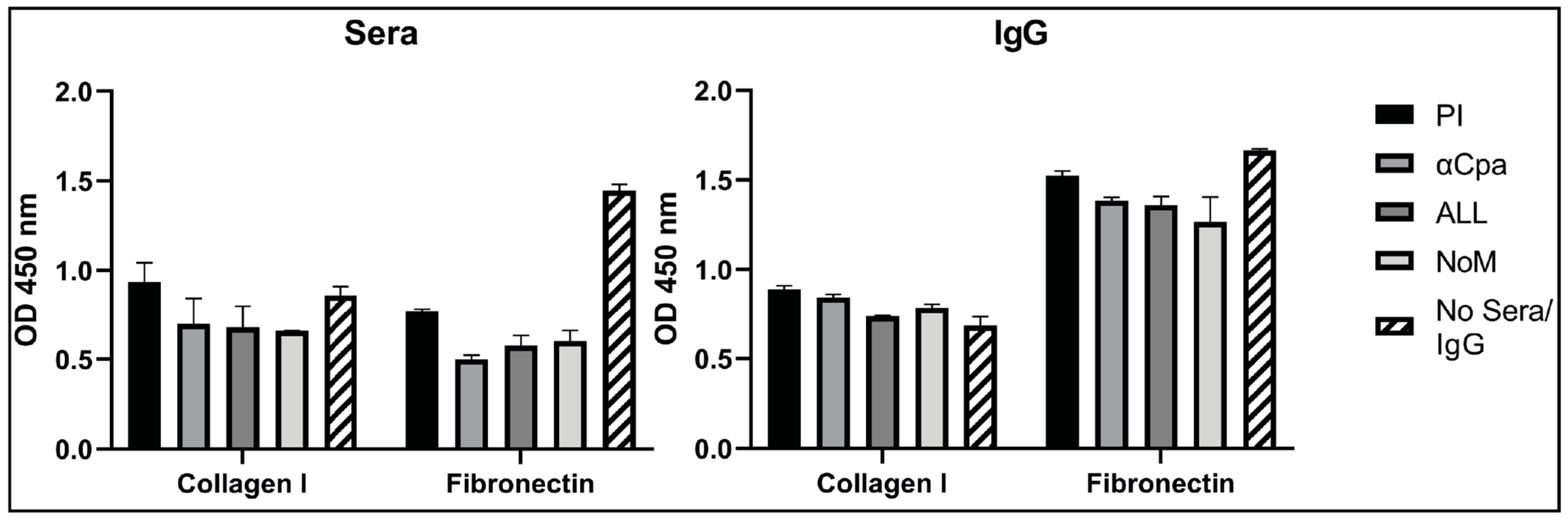
2.12. Binding Assays
3. Results
3.1. Protein Production and Characterisation
3.2. Immunisation with Recombinant Protein Antigens Induces a High IgG Antibody Response for Most Candidates
3.3. Immunisation in Combination Can Alter the Immune Response to Protein Antigens
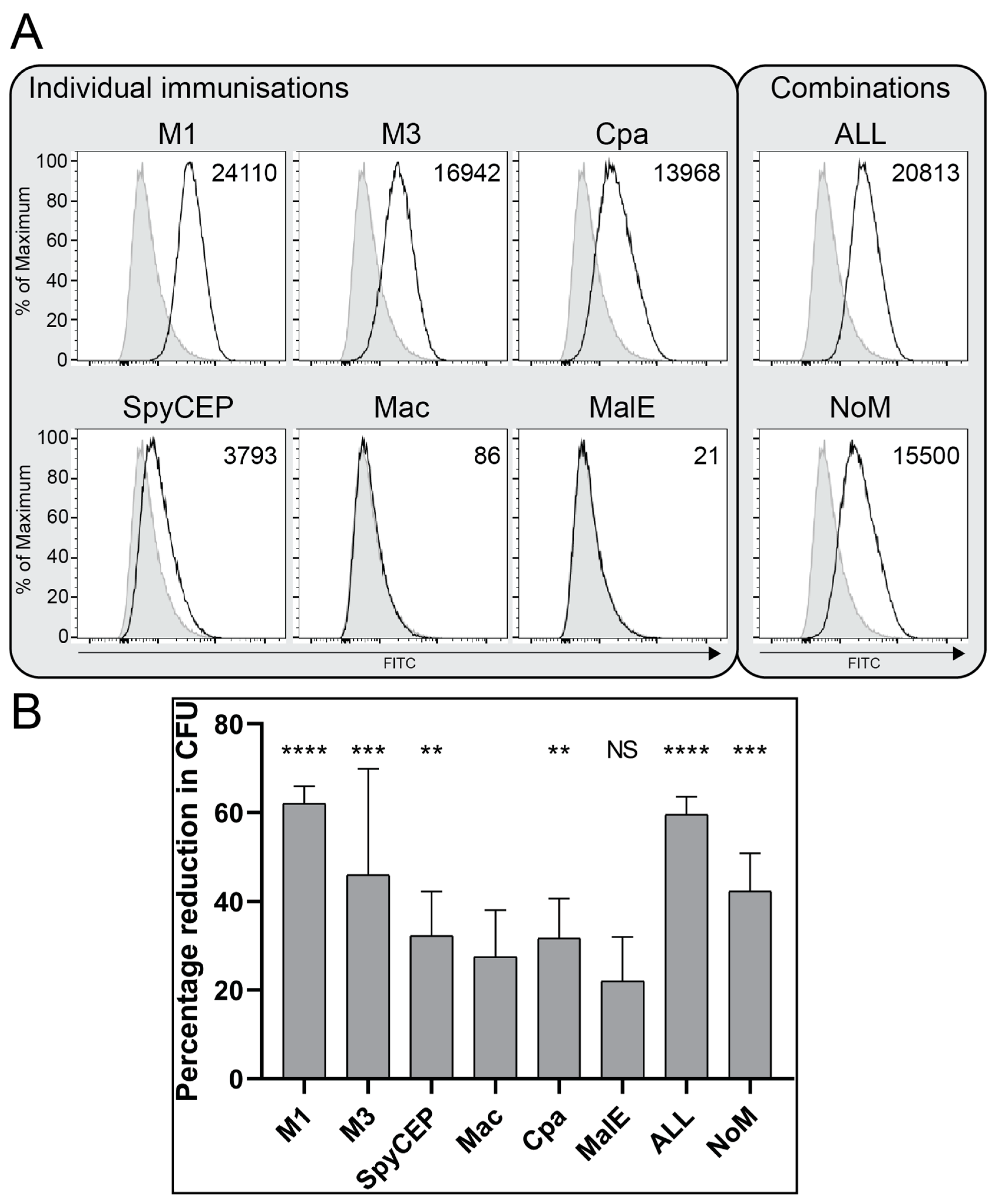
3.4. Antibodies Bind the Surface of GAS and Demonstrate Functional Killing Activity
3.5. Immune Sera Can Block the Functional Activity of Secreted GAS Virulence Factors
3.6. IgGs Are Not Effective at Blocking the Binding Activity of Cpa to Fibronectin and Collagen
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watkins, D.A.; Johnson, C.O.; Colquhoun, S.; Karthikeyan, G.; Beaton, A.; Bukhman, G.; Forouzanfar, M.H.; Longenecker, C.T.; Mayosi, B.M.; Mensah, G.A.; et al. Global, Regional, and National Burden of Rheumatic Heart Disease, 1990–2015. N. Engl. J. Med. 2017, 377, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Vekemans, J.; Gouvea-Reis, F.; Kim, J.H.; Excler, J.-L.; Smeesters, P.; O’Brien, K.L.; A Van Beneden, C.; Steer, A.C.; Carapetis, J.; Kaslow, D.C. The Path to Group A Streptococcus Vaccines: World Health Organization Research and Development Technology Roadmap and Preferred Product Characteristics. Clin. Infect. Dis. 2019, 69, 877–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastural, É.; McNeil, S.A.; MacKinnon-Cameron, D.; Ye, L.; Langley, J.M.; Stewart, R.; Martin, L.H.; Hurley, G.J.; Salehi, S.; Penfound, T.A.; et al. Safety and immunogenicity of a 30-valent M protein-based group a streptococcal vaccine in healthy adult volunteers: A randomized, controlled phase I study. Vaccine 2020, 38, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Sekuloski, S.; Batzloff, M.R.; Griffin, P.; Parsonage, W.; Elliott, S.; Hartas, J.; O’Rourke, P.; Marquart, L.; Pandey, M.; Rubin, F.A.; et al. Evaluation of safety and immunogenicity of a group A streptococcus vaccine candidate (MJ8VAX) in a randomized clinical trial. PLoS ONE 2018, 13, e0198658. [Google Scholar] [CrossRef]
- Dale, J.B.; Fischetti, V.A.; Carapetis, J.; Steer, A.C.; Sow, S.; Kumar, R.; Mayosi, B.; Rubin, F.A.; Mulholland, K.; Hombach, J.M.; et al. Group A streptococcal vaccines: Paving a path for accelerated development. Vaccine 2013, 31, B216–B222. [Google Scholar] [CrossRef]
- Rivera-Hernandez, T.; Pandey, M.; Henningham, A.; Cole, J.; Choudhury, B.; Cork, A.J.; Gillen, C.M.; Ghaffar, K.A.; West, N.P.; Silvestri, G.; et al. Differing Efficacies of Lead Group A Streptococcal Vaccine Candidates and Full-Length M Protein in Cutaneous and Invasive Disease Models. mBio 2016, 7, e00618-16. [Google Scholar] [CrossRef] [Green Version]
- Bensi, G.; Mora, M.; Tuscano, G.; Biagini, M.; Chiarot, E.; Bombaci, M.; Capo, S.; Falugi, F.; Manetti, A.G.O.; Donato, P.; et al. Multi High-Throughput Approach for Highly Selective Identification of Vaccine Candidates: The Group A Streptococcus Case. Mol. Cell. Proteom. 2012, 11, 111.015693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reglinski, M.; Lynskey, N.N.; Choi, Y.J.; Edwards, R.J.; Sriskandan, S. Development of a multicomponent vaccine for Streptococcus pyogenes based on the antigenic targets of IVIG. J. Infect. 2016, 72, 450–459. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Hernandez, T.; Carnathan, D.G.; Jones, S.; Cork, A.J.; Davies, M.R.; Moyle, P.M.; Toth, I.; Batzloff, M.R.; McCarthy, J.; Nizet, V.; et al. An Experimental Group A Streptococcus Vaccine That Reduces Pharyngitis and Tonsillitis in a Nonhuman Primate Model. mBio 2019, 10, e00693-19. [Google Scholar] [CrossRef] [Green Version]
- Carapetis, J.; Steer, A.C.; Mulholland, E.K.; Weber, M. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 2005, 5, 685–694. [Google Scholar] [CrossRef]
- Davies, M.R.; McIntyre, L.; Mutreja, A.; Lacey, J.; Lees, J.A.; Towers, R.J.; Duchene, S.; Smeesters, P.; Frost, H.R.; Price, D.J.; et al. Atlas of group A streptococcal vaccine candidates compiled using large-scale comparative genomics. Nat. Genet. 2019, 51, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steer, A.C.; Carapetis, J.; Dale, J.; Fraser, J.D.; Good, M.F.; Guilherme, L.; Moreland, N.; Mulholland, E.K.; Schodel, F.; Smeesters, P. Status of research and development of vaccines for Streptococcus pyogenes. Vaccine 2016, 34, 2953–2958. [Google Scholar] [CrossRef]
- Jones, S.; Moreland, N.; Zancolli, M.; Raynes, J.; Loh, J.M.; Smeesters, P.; Sriskandan, S.; Carapetis, J.; Fraser, J.D.; Goldblatt, D. Development of an opsonophagocytic killing assay for group a streptococcus. Vaccine 2018, 36, 3756–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGregor, R.; Jones, S.; Jeremy, R.M.; Goldblatt, D.; Moreland, N.J. An Opsonophagocytic Killing Assay for the Evaluation of Group A Streptococcus Vaccine Antisera. Methods Mol. Biol. 2020, 2136, 323–335. [Google Scholar] [CrossRef]
- Rivera-Hernandez, T.; Rhyme, M.S.; Cork, A.J.; Jones, S.; Segui-Perez, C.; Brunner, L.; Richter, J.; Petrovsky, N.; Lawrenz, M.; Goldblatt, D.; et al. Vaccine-Induced Th1-Type Response Protects against Invasive Group A Streptococcus Infection in the Absence of Opsonizing Antibodies. mBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Kreikemeyer, B.; Nakata, M.; Oehmcke, S.; Gschwendtner, C.; Normann, J.; Podbielski, A. Streptococcus pyogenes Collagen Type I-binding Cpa Surface Protein. J. Biol. Chem. 2005, 280, 33228–33239. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.D.; Pointon, J.A.; Abbot, E.; Kang, H.J.; Baker, E.; Hirst, B.H.; Wilson, J.; Banfield, M.; Kehoe, M.A. Roles of Minor Pilin Subunits Spy0125 and Spy0130 in the Serotype M1 Streptococcus pyogenes Strain SF370. J. Bacteriol. 2010, 192, 4651–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelburne, S.A.; Fang, H.; Okorafor, N.; Sumby, P.; Sitkiewicz, I.; Keith, D.; Patel, P.; Austin, C.; Graviss, E.A.; Musser, J.M.; et al. MalE of Group A Streptococcus Participates in the Rapid Transport of Maltotriose and Longer Maltodextrins. J. Bacteriol. 2007, 189, 2610–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelburne, S.A.; Sumby, P.; Sitkiewicz, I.; Okorafor, N.; Granville, C.; Patel, P.; Voyich, J.; Hull, R.; DeLeo, F.; Musser, J.M. Maltodextrin Utilization Plays a Key Role in the Ability of Group A Streptococcus to Colonize the Oropharynx. Infect. Immun. 2006, 74, 4605–4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, B.; DeLeo, F.; Hoe, N.P.; Graham, M.R.; Mackie, S.M.; Cole, R.L.; Liu, M.; Hill, H.R.; Low, D.E.; Federle, M.J.; et al. Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhibits opsonophagocytosis. Nat. Med. 2001, 7, 1298–1305. [Google Scholar] [CrossRef]
- Lei, B.; DeLeo, F.; Reid, S.D.; Voyich, J.M.; Magoun, L.; Liu, M.; Braughton, K.R.; Ricklefs, S.; Hoe, N.P.; Cole, R.L.; et al. Opsonophagocytosis-Inhibiting Mac Protein of Group A Streptococcus: Identification and Characteristics of Two Genetic Complexes. Infect. Immun. 2002, 70, 6880–6890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Pawel-Rammingen, U. Streptococcal IdeS and Its Impact on Immune Response and Inflammation. J. Innate Immun. 2012, 4, 132–140. [Google Scholar] [CrossRef]
- Edwards, R.J.; Taylor, G.W.; Ferguson, M.; Murray, S.; Rendell, N.; Wrigley, A.; Bai, Z.; Boyle, J.; Finney, S.J.; Jones, A.; et al. Specific C-Terminal Cleavage and Inactivation of Interleukin-8 by Invasive Disease Isolates ofStreptococcus pyogenes. J. Infect. Dis. 2005, 192, 783–790. [Google Scholar] [CrossRef] [Green Version]
- McKenna, S.; Malito, E.; Rouse, S.L.; Abate, F.; Bensi, G.; Chiarot, E.; Micoli, F.; Mancini, F.; Moriel, D.G.; Grandi, G.; et al. Structure, dynamics and immunogenicity of a catalytically inactive CXC chemokine-degrading protease SpyCEP from Streptococcus pyogenes. Comput. Struct. Biotechnol. J. 2020, 18, 650–660. [Google Scholar] [CrossRef]
- Kurupati, P.; Turner, C.; Tziona, I.; Lawrenson, R.A.; Alam, F.M.; Nohadani, M.; Stamp, G.W.; Zinkernagel, A.; Nizet, V.; Edwards, R.J.; et al. Chemokine-cleaving Streptococcus pyogenes protease SpyCEP is necessary and sufficient for bacterial dissemination within soft tissues and the respiratory tract. Mol. Microbiol. 2010, 76, 1387–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingaretti, C.; Falugi, F.; Nardi-Dei, V.; Pietrocola, G.; Mariani, M.; Liberatori, S.; Gallotta, M.; Tontini, M.; Tani, C.; Speziale, P.; et al. Streptococcus pyogenes SpyCEP: A chemokine-inactivating protease with unique structural and biochemical features. FASEB J. 2010, 24, 2839–2848. [Google Scholar] [CrossRef]
- Turner, C.; Kurupati, P.; Wiles, S.; Edwards, R.J.; Sriskandan, S. Impact of immunization against SpyCEP during invasive disease with two streptococcal species: Streptococcus pyogenes and Streptococcus equi. Vaccine 2009, 27, 4923–4929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, J.B.; Penfound, T.A.; Chiang, E.Y.; Walton, W.J. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of group A streptococci. Vaccine 2011, 29, 8175–8178. [Google Scholar] [CrossRef] [Green Version]
- Agniswamy, J.; Lei, B.; Musser, J.M.; Sun, P.D. Insight of Host Immune Evasion Mediated by Two Variants of Group A Streptococcus Mac Protein. J. Biol. Chem. 2004, 279, 52789–52796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, J.M.S.; Lorenz, N.; Tsai, C.J.-Y.; Khemlani, A.H.J.; Proft, T. Mucosal vaccination with pili from Group A Streptococcus expressed on Lactococcus lactis generates protective immune responses. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lei, B.; Liu, M.; Chesney, G.L.; Musser, J.M. Identification of New Candidate Vaccine Antigens Made byStreptococcus pyogenes:Purification and Characterization of 16 Putative Extracellular Lipoproteins. J. Infect. Dis. 2004, 189, 79–89. [Google Scholar] [CrossRef] [Green Version]
- A Hayman, W.; Brandt, E.R.; A Relf, W.; Cooper, J.; Saul, A.; Good, M.F. Mapping the minimal murine T cell and B cell epitopes within a peptide vaccine candidate from the conserved region of the M protein of group A streptococcus. Int. Immunol. 1997, 9, 1723–1733. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, C.L.; Groves, N.; Andrews, N.; Litt, D.J.; Fry, N.K.; Southern, J.; Miller, E. The Genomics of Streptococcus Pneumoniae Carriage Isolates from UK Children and Their Household Contacts, Pre-PCV7 to Post-PCV13. Genes 2019, 10, 687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, C.Y.M.; Anderson, E.L.; Döhrmann, S.; Tran, D.N.; Olson, J.; von Pawel-Rammingen, U.; Nizet, V. IgG Protease Mac/IdeS Is Not Essential for Phagocyte Resistance or Mouse Virulence of M1T1 Group A Streptococcus. mBio 2013, 4, e00499-13. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- Tiouajni, M.; Durand, D.; Blondeau, K.; Graille, M.; Urvoas, A.; Valerio-Lepiniec, M.; Guellouz, A.; Aumont-Nicaise, M.; Minard, P.; Van Tilbeurgh, H. Structural and functional analysis of the fibronectin-binding protein FNE fromStreptococcus equispp.equi. FEBS J. 2014, 281, 5513–5531. [Google Scholar] [CrossRef]
- Pointon, J.A.; Smith, W.D.; Saalbach, G.; Crow, A.; Kehoe, M.A.; Banfield, M.J. A Highly Unusual Thioester Bond in a Pilus Adhesin Is Required for Efficient Host Cell Interaction*. J. Biol. Chem. 2010, 285, 33858–33866. [Google Scholar] [CrossRef] [Green Version]
- Linke-Winnebeck, C.; Paterson, N.; Young, P.G.; Middleditch, M.J.; Greenwood, D.; Witte, G.; Baker, E.N. Structural Model for Covalent Adhesion of the Streptococcus pyogenes Pilus through a Thioester Bond. J. Biol. Chem. 2014, 289, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluckiger, U.; Jones, K.F.; Fischetti, V.A. Immunoglobulins to Group A Streptococcal Surface Molecules Decrease Adherence to and Invasion of Human Pharyngeal Cells. Infect. Immun. 1998, 66, 974–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reglinski, M.; Gierula, M.; Lynskey, N.N.; Edwards, R.J.; Sriskandan, S. Identification of the Streptococcus pyogenes surface antigens recognised by pooled human immunoglobulin. Sci. Rep. 2015, 5, 15825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cáceres, C.; Cardenas-Garcia, S.; Jain, A.; Gay, L.; Carnaccini, S.; Seibert, B.; Ferreri, L.; Geiger, G.; Jasinskas, A.; Nakajima, R.; et al. Development of a Novel Live Attenuated Influenza A Virus Vaccine Encoding the IgA-Inducing Protein. Vaccines 2021, 9, 703. [Google Scholar] [CrossRef] [PubMed]
- Mawas, F.; Newman, G.; Burns, S.; Corbel, M.J. Suppression and Modulation of Cellular and Humoral Immune Responses toHaemophilus influenzaeType B (Hib) Conjugate Vaccine in Hib–Diphtheria-Tetanus Toxoids–Acellular Pertussis Combination Vaccines: A Study in a Rat Model. J. Infect. Dis. 2005, 191, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Radcliff, F.J.; Fraser, J.D.; Proft, T. Vaccination with Streptococcus pyogenes nuclease A stimulates a high antibody response but no protective immunity in a mouse model of infection. Med. Microbiol. Immunol. 2014, 204, 185–191. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen (SF370) | Function | Location | Colonisation | Invasion | Immune Evasion | Vaccine | Titre in IVIG | Ref. |
|---|---|---|---|---|---|---|---|---|
| M protein (emm1, SPy_2018) | Main virulence factor | Cell wall | 30 valent J8 StreptinCor | 24,628 (M1) 17,392 (M3) | [28] | |||
| SpyCEP (SPy_0416) | IL-8 protease | Cell wall/secreted | GSK combo, Combo5 | 6038 | [28] | |||
| Mac (IdeS) (SPy_0861) | IgG protease | Secreted | - | 12,203 | [22,29] | |||
| Cpa (AP1) (SPy_0125) | Pilin tip | Cell wall | L. lactis | 3329 | [17,30] | |||
| MalE (SPy_1294) | Maltodextrin binding, saliva survival | Membrane | - | 828 | [19,31] |
| Group | Geometric Mean Titre of IgG Response against Individual Antigens (95% CI) | |||||
|---|---|---|---|---|---|---|
| M1 | M3 | SpyCEP | Mac | Cpa | MalE | |
| Pre-immune | 1 | 1 | 11 | 1 | 1 | 88 |
| Individual | 34,542 (16,943–70,422) | 97,715 (38,142–250,333) | 125,934 (57,608–275,299) | 4856 (1945–12,127) | 2870 (188–43,844) | 93 (34–258) |
| Combination “ALL” | 29,503 (20,367–42,736) | 60,579 (33,142–110,729) | 31,881 ** (16,146–62,950) | 10,549 * (7333–15,175) | 51,741 *** (39,617–67,575) | 268 * (174–411) |
| “NoM” | 155 (94–257) | 11 (1–101) | 115,946 ++ (67,399–199,462) | 12,208 (7287–20,453) | 11,800 + (3529–39,458) | NT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaw, H.A.; Ozanne, J.; Burns, K.; Mawas, F. Multicomponent Vaccines against Group A Streptococcus Can Effectively Target Broad Disease Presentations. Vaccines 2021, 9, 1025. https://doi.org/10.3390/vaccines9091025
Shaw HA, Ozanne J, Burns K, Mawas F. Multicomponent Vaccines against Group A Streptococcus Can Effectively Target Broad Disease Presentations. Vaccines. 2021; 9(9):1025. https://doi.org/10.3390/vaccines9091025
Chicago/Turabian StyleShaw, Helen A., James Ozanne, Keira Burns, and Fatme Mawas. 2021. "Multicomponent Vaccines against Group A Streptococcus Can Effectively Target Broad Disease Presentations" Vaccines 9, no. 9: 1025. https://doi.org/10.3390/vaccines9091025
APA StyleShaw, H. A., Ozanne, J., Burns, K., & Mawas, F. (2021). Multicomponent Vaccines against Group A Streptococcus Can Effectively Target Broad Disease Presentations. Vaccines, 9(9), 1025. https://doi.org/10.3390/vaccines9091025