1. Introduction
Influenza is a major cause of respiratory infections around the world. The global influenza disease burden has been estimated to approach up to 1 billion infections, 3–5 million cases of severe disease and 300,000–500,000 deaths annually [
1]. In addition, influenza pandemics occur at various times and cause varying levels of severity and death [
2]. Seasonal influenza virus vaccine is the most utilized method to reduce the impact of influenza infection. The two major glycoproteins on the surface of influenza are hemagglutinin (HA) and neuraminidase (NA). Antibodies to HA are generally considered the key mediators of influenza immunity, and inhibition of HA activity has been the primary measure of vaccine effectiveness. Point mutations causing HA antigenic drift is the primary reason for variable and limited effectiveness of the seasonal influenza vaccine. While point mutations in the NA can occur, antigenic drift is lower than for HA among influenza A viruses [
3,
4]. In fact, a recent study showed that humans infected with influenza develop NA-reactive antibodies that are broadly reactive and protective [
5].
New influenza vaccine strategies that focus on inducing antibodies to multiple key proteins could promote broad influenza immunity across seasonal and pandemic strains. In humans, HA-reactive antibodies constitute a subset of neutralizing antibodies that may be important in protection [
6]. Neuraminidase drifts independently of HA, and immunity to NA has been correlated with protection [
7]. The extracellular domain of Matrix 2 (M2e) protein is a highly conserved region in influenza A viruses [
8]. Immunity to influenza induced by M2e may include both innate and adaptive immune responses and therefore has been considered an important target for developing a universal influenza vaccine [
8,
9]. The HA and NA peptides are highly conserved and located near or at the protein area where the receptor binding sites reside [
10,
11,
12]. These epitopes classically are impacted by induction of antibodies through the adaptive immune system and a major target for influenza vaccines. To promote broad immunity across Groups 1 and 2 influenza viruses, structural mapping of the HA and NA proteins was performed to identify highly conserved epitope sequences. These peptide sequences were then combined and arranged as continuous sequences to form an HA and NA composite peptide antigen. The extracellular domain of the M2 matrix protein (M2e) is highly conserved and constitutes a set of epitopes that may function through different arms of the innate immune system [
8,
13]. Antibodies to M2e may not directly neutralize influenza viruses but utilize other immunologic mechanisms such as NK-mediated ADCC (antibody-dependent cellular cytotoxicity). The M2e epitope has also been considered an important target for developing a universal influenza A vaccine [
8]. A composite M2e peptide was constructed by utilizing a highly conserved region of the M2e and then repeating a portion of the sequence [
8,
13]. A tetanus toxoid T-cell epitope was also added to the C-terminal end. These composite peptide vaccines containing highly conserved HA, NA and M2e epitopes were designed to induce broad immunity across seasonal and pandemic influenza strains. Since these peptide vaccines are also synthetically produced, they avoid propagation of the virus in eggs and therefore can be rapidly and efficiently manufactured.
It has been proposed that carrier proteins and adjuvants are necessary to elicit a strong immune response to peptide-based influenza vaccines [
8,
14]. Initial preliminary studies demonstrated that immunizing mice with these influenza composite peptides conjugated to a carrier protein, and with an adjuvant, induced broadly reactive, neutralizing antibodies across both Groups 1 and 2 influenza A virus strains (unpublished data). In addition, these composite influenza peptide/conjugate vaccines, administered with the saponin adjuvant Quil-A
®, produced higher influenza IgG titers in mice against HA, NA and M2e, to include IgG1, IgG2a and IgG2b (unpublished data).
Previous studies demonstrated that liposomes could serve as vehicles both for adjuvants and for inducing antibodies and cellular immunity to immunogenic peptides [
15]. Liposomes containing monophosphoryl lipid A (now referred to as Army Liposome Formulation, or ALF) were potent and effective for inducing antibodies and cell-mediated immunity both to conjugated and unconjugated HIV-1 peptides [
15,
16], and even for inducing antibodies to haptens for a candidate heroin vaccine [
17,
18] or for a combination heroin-HIV vaccine [
19]. ALF-type adjuvants were also combined with different types of antigens in numerous phase 1 and phase 2 human clinical trials [
20].
In 2015, liposomes with 55 mol % cholesterol and containing both monophosphoryl lipid A and QS21 saponin (ALFQ) were introduced as a novel and highly potent adjuvant formulation [
21]. ALFQ is being used in three ongoing human phase 1 clinical trials for two different candidate malaria vaccines and a COVID vaccine and is projected for use in other types of candidate vaccines [
22]. Because of the effectiveness of ALF-type adjuvants for induction of antibodies both to conjugated and unconjugated peptides, and because ALFQ has now been introduced as an adjuvant for candidate human vaccines, ALFQ was employed in the present study to explore its effectiveness as an adjuvant for inducing broad binding and neutralizing antibodies to composite influenza peptide antigens. In this report, we demonstrate that ALFQ adjuvant improved the immunogenicity of both conjugated and unconjugated composite influenza peptides and promoted the induction of a balanced Th1/Th2 response while increasing its neutralization breadth against both Group 1 and Group 2 influenza viruses.
2. Materials and Methods
2.1. Cell Culture
Madin-Darby canine kidney (MDCK) cells (ATCC, Manassas, VA, USA) were maintained in Eagle’s Minimal Essential Medium (EMEM) (Lonza, Basel, Switzerland) containing 5% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA), 2 mM L-glutamine (Sigma-Aldrich, St. Louis, MO, USA), 1% sodium pyruvate and 1% non-essential amino acids (NEAA) (Thermo Fisher Scientific, Waltham, MA, USA), 1% amphotericin B (Sigma-Aldrich, St. Louis, MO, USA) and 0.1% gentamycin (Thomas Scientific, Swedesboro, NJ, USA).
2.2. Influenza Viruses
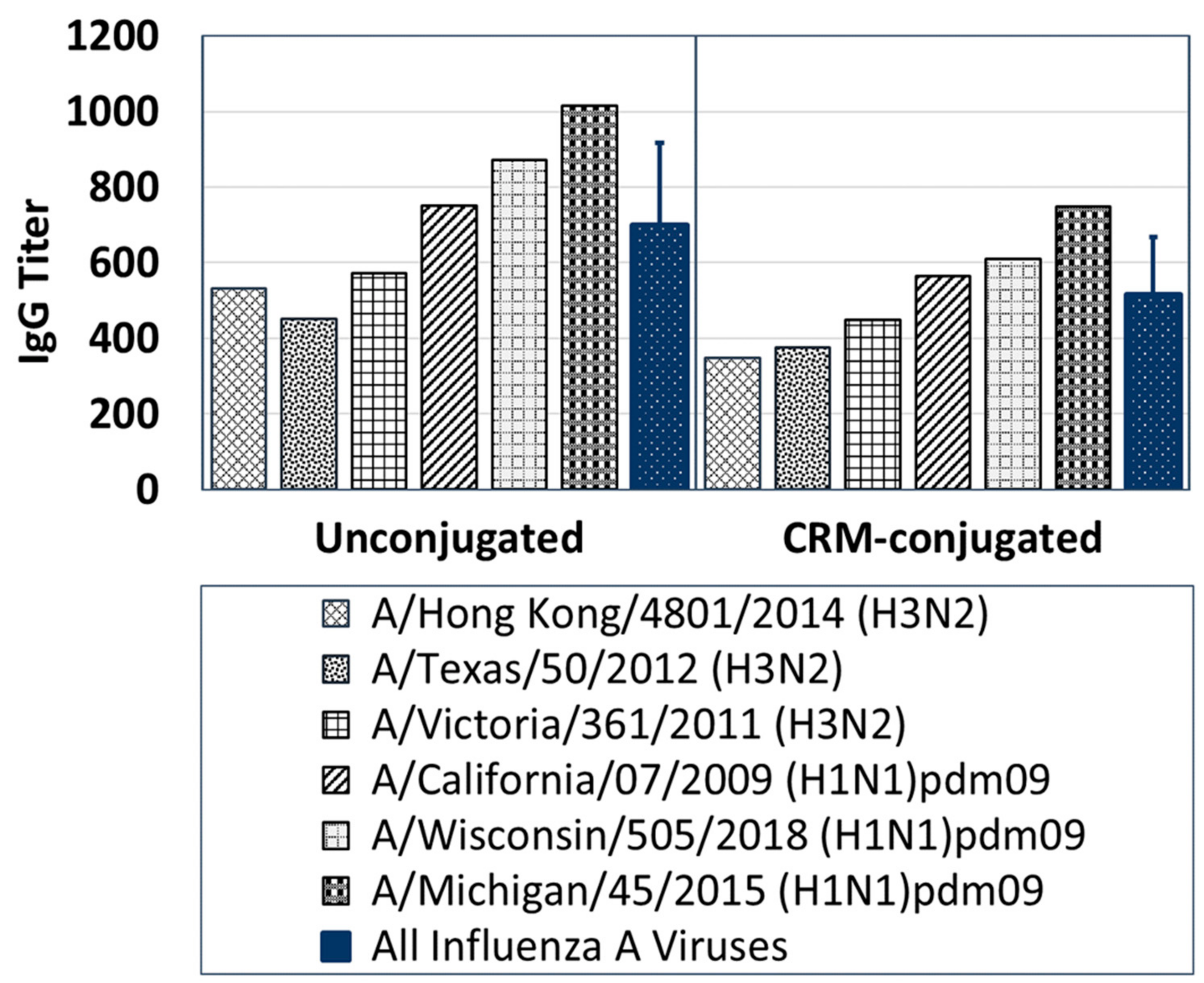
Several influenza viruses were obtained from the Influenza Reagents Resource (IRR) (Manassas, VA, USA), established by the Centers for Disease Control and Prevention (CDC), USA. Group 1 virus strains used include A/Michigan/45/2015 (H1N1) pdm09, A/California/07/2009 (H1N1) pdm09, and A/Wisconsin/505/2018 (H1N1) pdm09. Group 2 influenza virus strains used include A/Hong Kong/4801/2014 (H3N2), A/Victoria/361/2011 (H3N2) and A/Texas/50/2012 (H3N2). All viruses were propagated by infection of MDCK monolayers for 3 days at 37 °C and 5% CO2. Virus concentrations were determined by the standard tissue culture infectious dose (TCID50) assay on MDCK cells and aliquots were stored at −80 °C.
2.3. Influenza Peptides
Mice immunization studies were performed using a vaccine formulation of two composite influenza peptides, Flu Pep11 and Flu Pep5906, adjuvanted with ALFQ to evaluate humoral and cytokine immune responses. The individual and composite flu peptides were synthesized under a contract with Atlantic Peptides, Lewisburg, PA, USA. Flu Pep11, comprising HA epitopes (Flu Pep3 and Flu Pep6) and an NA epitope (Flu Pep10), was used in conjunction with Flu Pep5906, comprising M2e epitope and a tetanus T-cell epitope. The individual and composite peptides were used in immunoassays to evaluate binding and functional activities of anti-influenza antibodies.
2.4. Selection of Conserved HA and NA Epitopes
A primary amino acid multiple sequence alignment was performed using sequences derived from more than 2000 H1 and H3 influenza A field strains and representative H5 strains available from the literature and via public domain sequences deposited in NCBI at:
www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi?go=database (accessed on 13 March 2010) and through a personal database (L.T. Daum, unpublished). H3 and H1 vaccine and reference strains and H5 peptides representing clades 1, 1′, 2, and 3 were also included. Highly conserved peptide regions of influenza A hemagglutinin and neuraminidase were identified through multiple sequence alignments of several thousand strains. Sequences were obtained using the Influenza Virus Resource website (available at:
http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html (accessed on 13 March 2010)), and represent strains obtained from human, avian, and/or mammalian sources. Peptides were selected based on conserved HA and NA regions deduced from multiple sequence alignments (LaserGene, DNAStar Inc., Madison, WI, USA). The protein epitope 5′-GNLIAP-3′, corresponding to H3 amino acid positions 249–254, is a highly conserved region in H3 and H1 subtypes, while GNFIAP is highly conserved in H5. The GNLIAP region is partially exposed on the distal HA1 surface and is not located within previously proposed antigenic/immunodominant sites (A–E). Furthermore, WGVHHP, corresponding to H3 amino acid positions 180–185, is highly conserved in H3 and H1 subtypes. The corresponding region on H5, WGIHHP, differs by one residue-isoleucine. To demonstrate the utility of small, conserved influenza epitopes to induce immunity, several HA and NA conserved regions were conjugated to a protein carrier or unconjugated. These small, conserved influenza epitopes included sequence GNLFIAP, which is a novel combination of GN with both the L and F and then IAP. This contains all residues for H1, H3 and H5 subtypes.
Three-dimensional (3-D) folded HA and NA proteins were constructed based on the nearest homolog available in the Protein Sequencing Database with either Swiss PDF Viewer (Version 3.7) or Mol* viewer (PDB Protein Data Bank;
www.rcsb.org (accessed on 13, March, 2010)) (
Figure 1). Conserved residues observed in sequence alignments are depicted in the mature HA or NA proteins (
Figure 1). For HA monomer, 3-D HA structure was derived from A/Wisconsin/67/2005 (H3N2) vaccine strain and constructed using Swiss PDF Viewer, while for the NA monomer, the 3-D NA structure was derived from A/Perth/16/2009 (H3N2) (GenBank 6BR6_A) and constructed using Mol* viewer (PDB Protein Data Bank;
www.rcsb.org (accessed on 13, March, 2010)) (
Figure 1). GNLFIAP, referred to as Flu Pep3, is a composite of GNLIAP (H3/H1 subtypes) and GNFIAP (H5 subtypes). WGVIHHP, referred to as Flu Pep6, is a composite of WGVHHP (H3/H1 subtypes) and WFIHHP (H5 subtype). GNLFIAPWGVIHHPHYEECSCY, referred to as Flu Pep11, is a composite that contains these three epitopes: GNLFIAP, WGVIHHP and HYEECSCY (
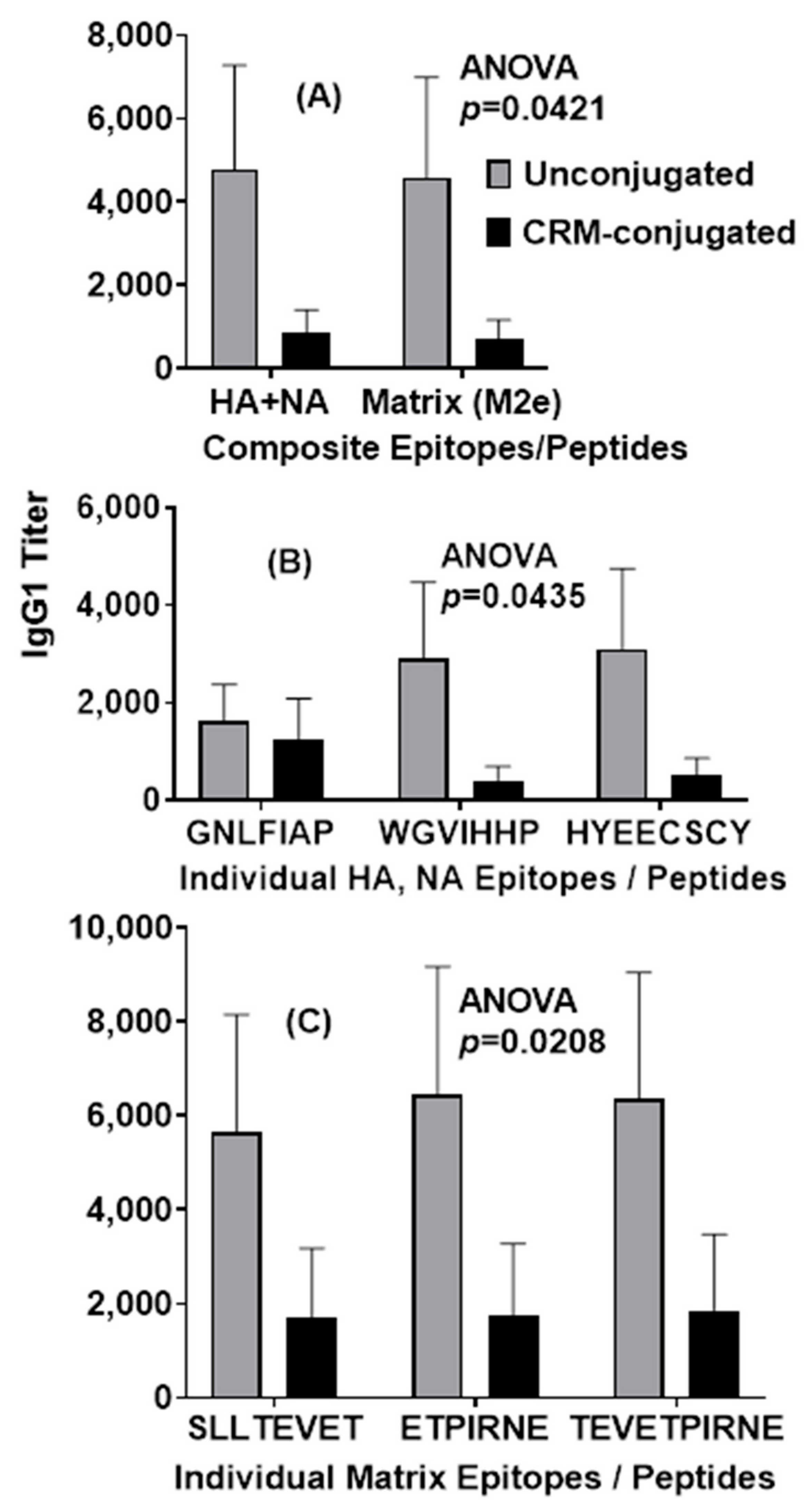
Table 1). The NA monomer consists of a conserved epitope sequence with residues HYEECSCY (N1 subtype), referred to as Flu Pep10. This conserved epitope varies by a single amino acid between N1 and N2 neuraminidase with a Y (N1) and V (N2) at residue 276. Monoclonal antibodies to the HYEECSCY epitope were shown to neutralize both H1N1 and H3N2 influenza viruses (unpublished data).
2.5. Selection of Conserved M2e Epitopes
Because there is high M2e sequence conservation among human influenza viruses, M2e has been long proposed as an ideal universal influenza A vaccine candidate [
9]. The highly conserved region of M1 and M2 (M2e sequence: MSLLTEVETPIRNEWGCRCNDSSD) was utilized to construct an M2e composite peptide that provides epitope repetition and a C-terminal T-cell epitope [
8,
13]. Selected epitopes were synthesized as a composite M2e vaccine with the C-terminal T-cell epitope and were utilized as a peptide vaccine with and without CRM conjugation. M2e Composite Peptide Vaccine—SLLTEVETPIRNEWGLLTEVETPIRQYIKANSKFIGITE, is referred to as Flu Pep5906 (
Table 1).
2.6. Peptide Conjugation to CRM197
For Flu Pep11, thiol-ether chemistry was used to link cysteine residue to CRM197 (hereafter referred to as CRM) pre-labeled with 15 maleimides: 4 peptides (2.5 K) per CRM (Fina Biosolutions, Rockville, MD, USA). The conjugate was purified by dialysis (Hermanson G.T, Bioconjugate Techniques 3rd Edition) [
23].
For Flu Pep5906, the peptide was labeled with a thiol linker N-Succinimidyl 3- (2-pyridildithio) propionate (SPDP) (CovaChem, Illinois, IL, USA) and then conjugated to CRM pre-labeled with 15 maleimides: 8 peptides (4.5 K) per CRM. The conjugate was purified by dialysis (Hermanson G.T., Bioconjugate Techniques, 3rd Edition) [
23].
2.7. ALFQ Formulation with Composite Influenza Peptides
Dimyristoyl phosphatidylcholine (DMPC), dimyristoyl phosphatidylglycerol (DMPG) cholesterol (Chol), and synthetic monophosphoryl lipid A (MPLA, 3D-PHAD
®) were purchased from Avanti Polar Lipids. DMPC and Chol were dissolved in chloroform, and DMPG and 3D-PHAD
® were dissolved in chloroform:methanol (9:1). QS21 was purchased from Desert King International and was dissolved in Sorensen PBS, pH 6.2 and filtered. For vaccine preparations adjuvanted with ALF containing QS21 (ALFQ), lipids were mixed in a molar ratio of 9:1:12.2:0.114 (DMPC:DMPG:Chol: 3D-PHAD
®), dried by rotary evaporation followed by overnight desiccation, rehydrated by adding Sorensen PBS, pH 6.2, processed by microfluidization to form small unilamellar vesicles (SUV), and filtered [
21]. QS21 was added to the SUVs to form ALFQ (DMPC:DMPG:Chol:MPLA:QS21; 9:1:12.2:0.114:0.044) [
22].
Vaccines were prepared to contain either unconjugated Flu Pep11 and Flu Pep5906 or CRM-conjugated Flu Pep11 and Flu Pep5906. Vaccines containing CRM-conjugated peptides were prepared by adding 35, 17.5, 7, or 3.5 µg of each peptide to 186.4 µL of ALFQ (containing 751 µg/mL of 3D-PHAD) and then diluting to a 350 µL final volume with DPBS. Unconjugated flu peptide 11 and peptide 5906 were dissolved in 50% DPBS and then combined. Vaccines containing unconjugated peptides were prepared by adding 17.5, 7, or 3.5 µg of each peptide to 186.4 µL of ALFQ (containing 751 µg/mL of 3D-PHAD) and then diluting to a 350 µL final volume with DPBS. The vials were vortexed at slow speed for 1 min and then put on a roller for 15 min. The vials were stored at 4 °C prior to immunization. Each vaccine dose contained 20 µg 3D-PHAD, 10 µg QS21 (ALFQ), and 2.5, 1, or 0.5 µg of each peptide in 50 µL volume.
2.8. Mice Immunizations Using ALFQ Formulated Composite Peptides
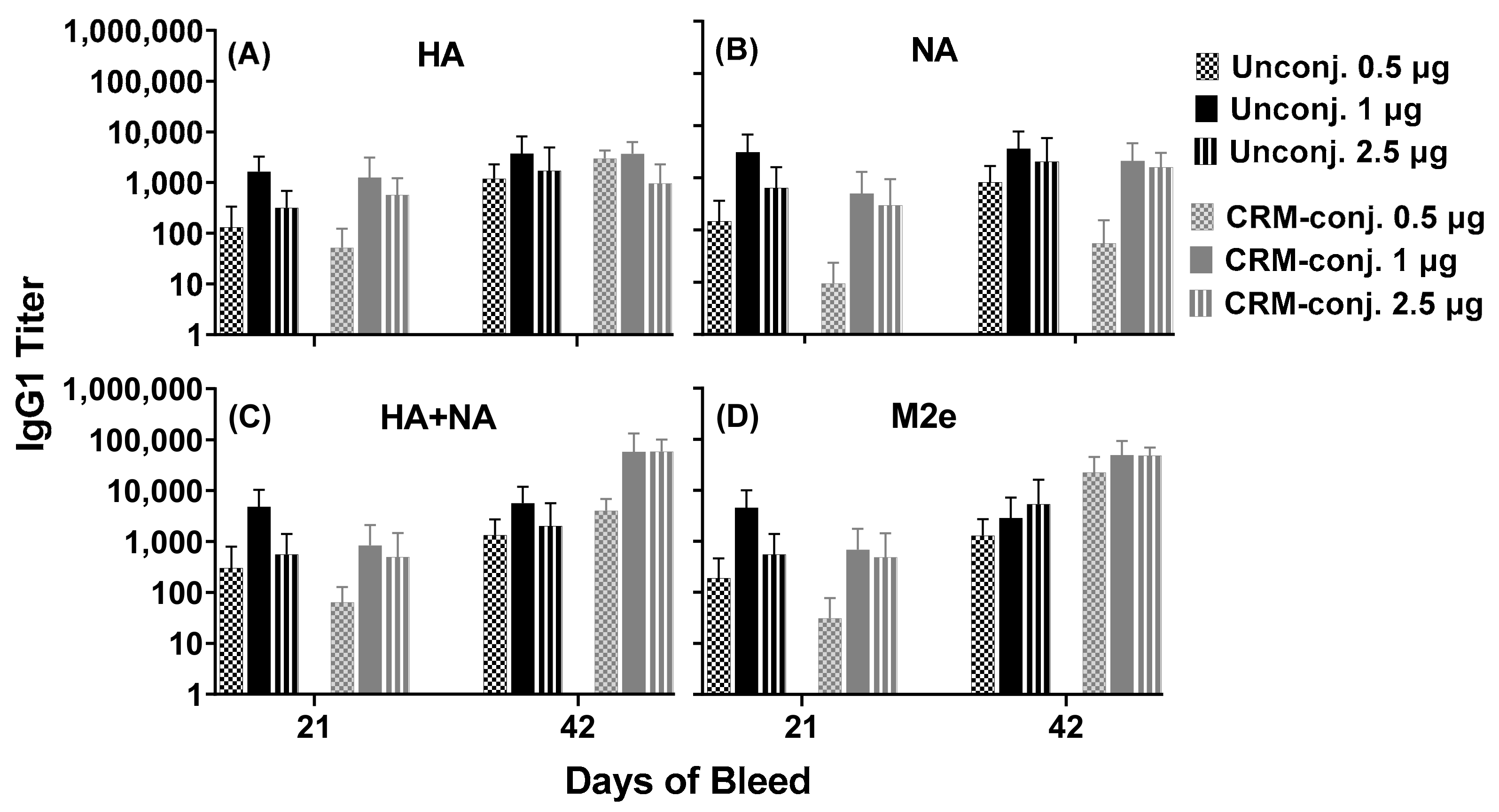
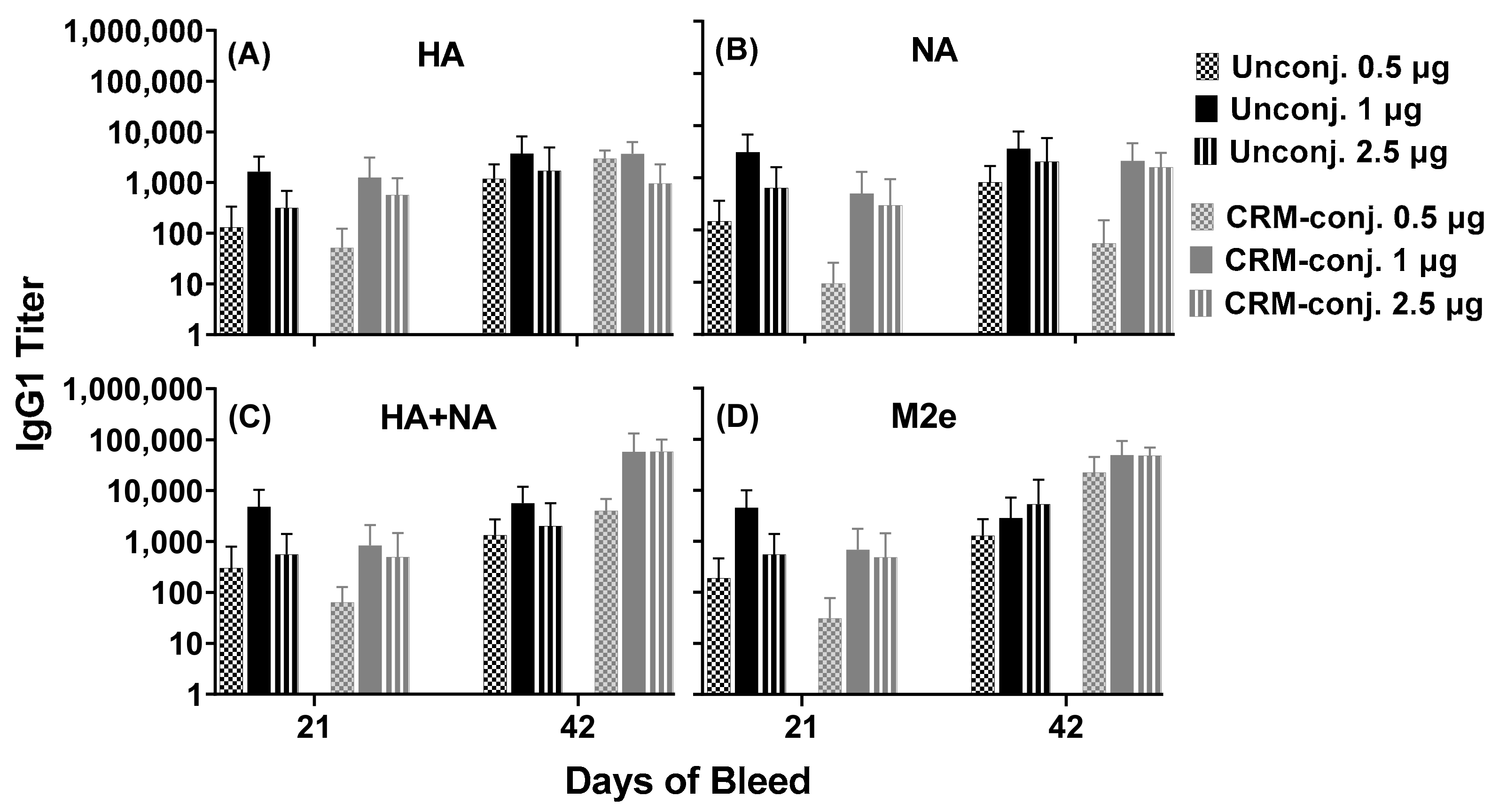
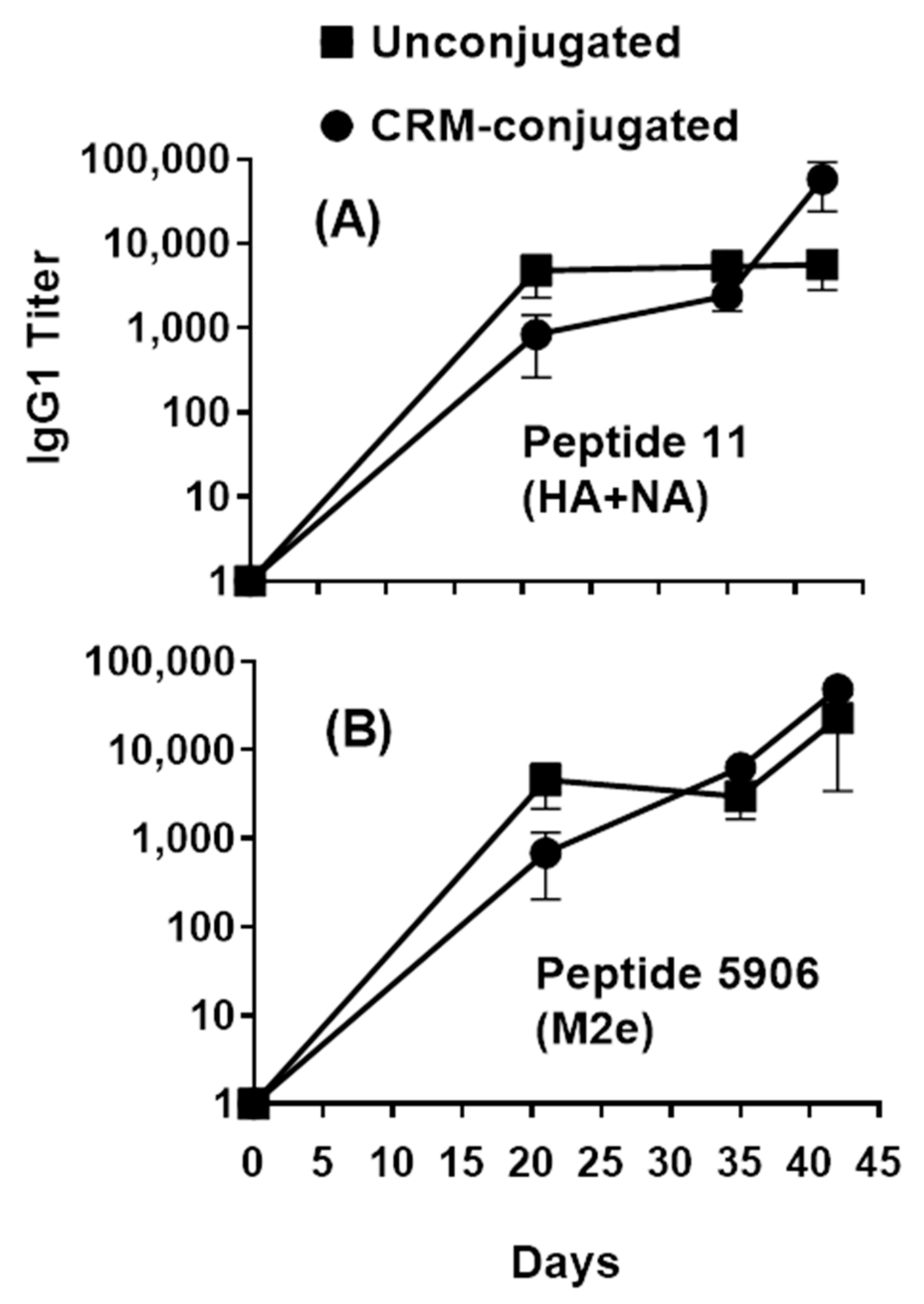
All animal procedures were approved by the Institutional Animal Care and Use Committee (Sigmovir Biosystems Inc., Rockville, MD, USA, IACUC Protocol #71). Female Institute of Cancer Research (ICR-CD-1®) mice, 3–4 weeks of age, were obtained from Envigo (Indianapolis, IN, USA) for use in this study. Mice were housed in groups of 5 and maintained in a 12 h light–dark cycle with ad libitum access to feed and water. All mice were given a 2-week acclimatization period. Mice were randomly assigned to one of the 6 treatments (n = 5). The composite unconjugated or CRM-conjugated influenza peptides (Flu Pep11 + Flu Pep5906, in combination), formulated with ALFQ, were injected intramuscularly at doses of 0.5, 1, and 2.5 µg/mouse (n = 5 mice/dose/group). Mice were immunized on day 0 and booster immunizations were given on days 21 and 35. Submandibular bleeds were obtained on day 7 (pre-immunization), and days 7, 21, 28, 35, 42 and 56. Approximately 150–200 µL of blood were collected at each bleed and processed for serological testing.
2.9. Antisera ELISA: Detection of Antibodies That Bind to Influenza Peptides/Viruses
Serum anti-influenza levels were evaluated using peptides/antigens and live influenza viruses (H3N2 and H1N1). Next, 96-well NUNC MaxiSorp ELISA plates (Fisher Scientific, Pittsburg, PA, USA) were coated with peptides (1 µg/mL) and live influenza viruses (105 TCID50 per well) in PBS at 100 µL per well for 18–24 h at room temperature (RT) and 2–8 °C, respectively. Antigen-coated plates were blocked with 3% normal goat serum (NGS, Southern Biotech, Birmingham, AL, USA) in PBS and subsequently washed with PBS-0.05% Tween 20 (PBS-T, Fisher Scientific, Pittsburg, PA, USA) using an ELx405 Automated Plate Washer (BioTek, Winooski, VT, USA). Serial dilutions of individual serum samples were added in duplicate. Anti-influenza antibodies were detected with Horse Radish Peroxidase (HRP)-conjugated, gamma-specific, goat anti-mouse IgG (Southern Biotech, Birmingham, AL, USA) or HRP-conjugated, IgG isotype-specific goat anti-mouse IgG1, IgG2a, IgG2b, IgG3 (Southern Biotech, Birmingham, AL, USA). TMB Substrate Solution (Fisher Scientific, Pittsburg, PA, USA) was added prior to being quenched with TMB STOP solution (Fisher Scientific, Pittsburg, PA, USA). The absorbance values (450 nM) of each well were obtained using a SpectraMax Plus Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). Pre-immunization sera were used as negative controls. Antisera titers were calculated using the formula: EXP (((LN (b) − LN (a))/(d − c)) * (E − c) + LN (a)), where A = dilution giving absorbance above the desired titer point, B = dilution giving absorbance below the desired titer point, C = absorbance value at dilution A, D = absorbance value at dilution B, E = Titer Point selected (absorbance value of 0.5). Absorbance values consisted of OD readings at 450 nM.
2.10. Microneutralization Assay for Determination of Serum Neutralizing Antibodies
MDCK cells were seeded for 3–4 days in 96-well plates (Corning, Tewksbury, MA, USA). Pooled anti-influenza sera samples in quadruplets were heat-inactivated at 56 °C for 30 min and serially diluted twofold from a starting dilution of 1:40 in TPCK-trypsin medium in a 96-well plate. Then, 120 µL per well of influenza A viruses (H3N2 or H1N1) were added to each well containing 120 µL of diluted antisera and incubated for 2 h at 37 °C/5% CO2. This addition of an equal volume of virus into the anti-influenza sera samples further increased the antisera dilution twofold to a starting dilution of 1:80 and a final dilution of 1:5120. Confluent MDCK cell monolayers in 96-well plates were washed with PBS (Fisher Scientific, Pittsburg, PA, USA) using an ELx405 Automated Plate Washer (BioTek, Winooski, VT, USA) 10 min before the next step. After the 2 h incubation, 100 µL/well of each dilution of antisera-virus mixture were transferred to quadruplicate wells of pre-washed MDCK cell monolayers. Wells containing virus dilutions only and MDCK cells only were included in each plate as positive and negative controls for infectivity, respectively. The assay plates were incubated at 37 °C/5% CO2 for 4 days. After incubation, the plates were stained with Crystal Violet and scored by recording cytopathic effect (CPE) of virus on MDCK cell monolayer. Clear wells representing absence of MDCK cells were scored as positive CPE and indicated an absence of neutralization of viral replication by the antisera tested. Wells with a blue coloration representing presence of MDCK cells were scored as negative CPE and indicated neutralization of viral replication by the antisera tested. Neutralizing titers were reported as reciprocal of the highest dilution that showed complete protection of the MDCK cell monolayer, e.g., 1:2560 antisera dilution = 2560 neutralizing titer.
2.11. Preparation of Supernatants for Cytokine Determination
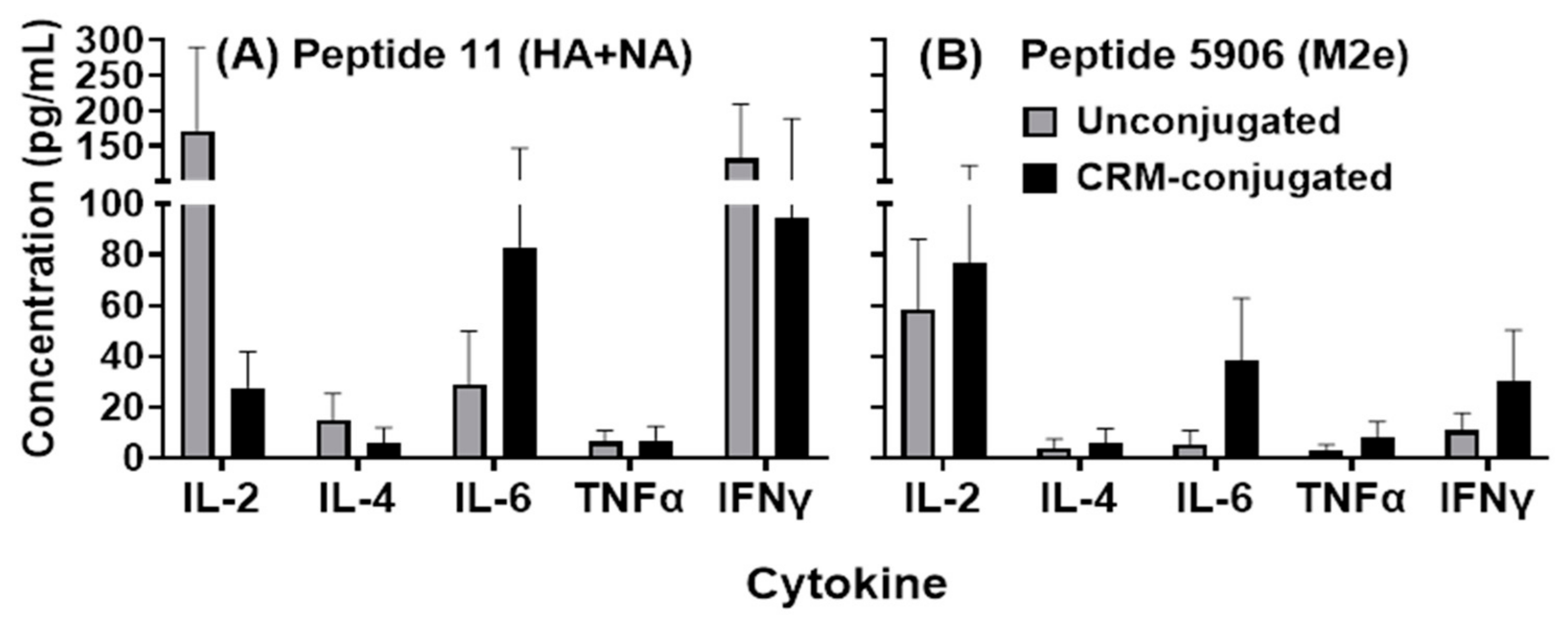
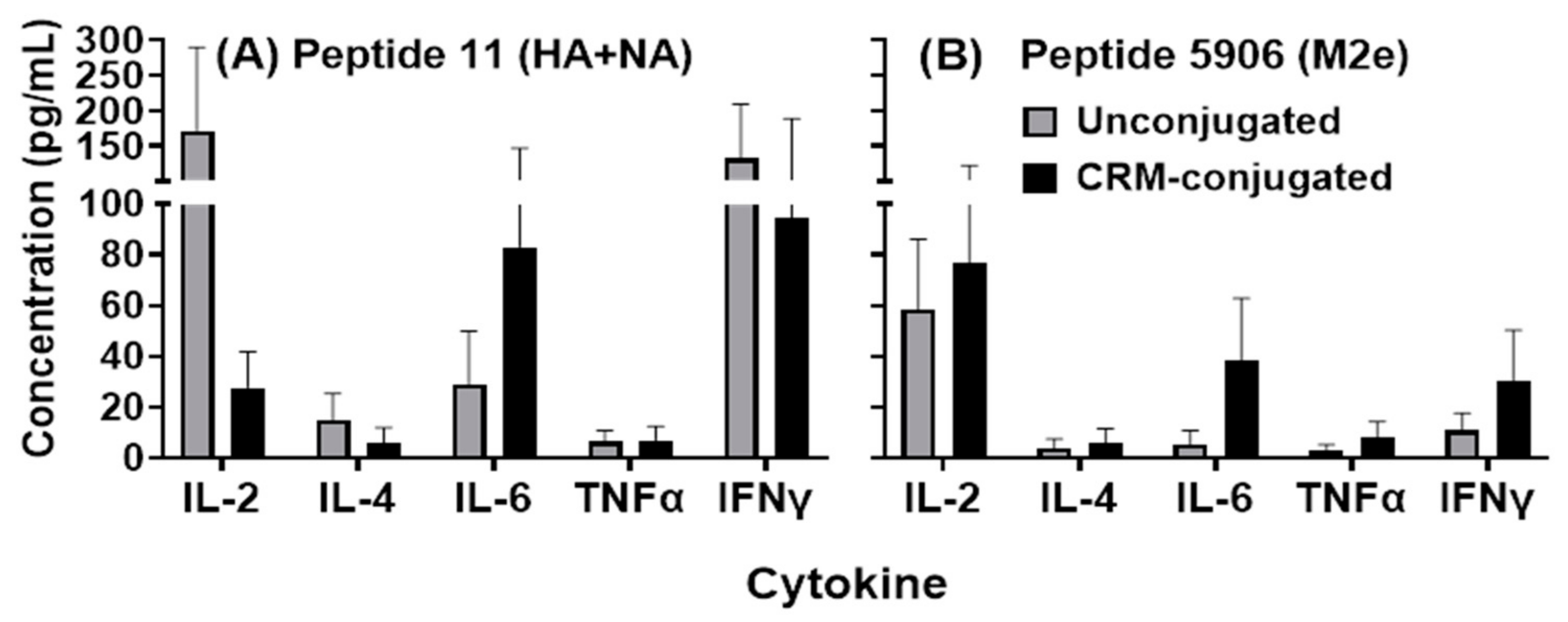
Spleens were collected from immunized mice on day 100 ± 7. Each spleen was removed and placed into a sterile 50 mL conical tube containing 25 mL of serum-free, high-glucose DMEM (Quality Biological Inc, Gaithersburg, MD, USA) supplemented with 2 mM L-glutamine (Corning Inc., Corning, NY, USA) and 50 µg/mL of gentamicin sulfate (Corning Inc., Corning, NY, USA) (SF-DMEM). Spleens were perfused with 10 mL of SF-DMEM. A single-cell suspension of splenocytes was prepared by gently pressing the spleen through a 60-mesh cell sieve and passed through a 70 µm Falcon cell strainer (Fisher Scientific, Hampton, NH, USA) into a 50 mL conical tube. The resulting cell suspension was pelleted by centrifugation and the supernatant discarded. Red blood cell lysis buffer (Sigma-Aldrich, St. Louis, MO, USA) was added to the pellet for 1 min at room temperature with gentle shaking and the cells were resuspended into 25 mL of SF-DMEM. The cells were again pelleted by centrifugation and, after removal of the supernatant, the cells were resuspended at a concentration of 4 × 106 cells/mL in cDMEM (DMEM supplemented with 10% heat-inactivated FBS) (Cytiva, Marlborough, MA, USA), 2 mM L-glutamine, and 50 µg/mL of gentamicin sulfate. One mL of cell suspension was seeded into each well of a 24-well culture dish and 1 mL of Flu Pep11 or Flu Pep5906 in cDMEM were added to individual wells at final concentration of 30 µg/mL. Control wells received cDMEM alone. The cells were incubated in a humidified atmosphere at 37 °C/5% CO2 for 68–72 h. Cultures were harvested and centrifuged. Cell-free supernatants were aliquoted and stored at −20 °C until evaluated for cytokine concentrations. Cytokines and chemokines were assayed using a Quansys Biosystems multiplex system Q-PlexTM Array (Logan, UT, USA), according to the instructions of the manufacturer.
2.12. Statistical Analysis
Data was analyzed using GraphPad Prism 9 (San Diego, CA, USA). Antisera titer results were expressed as mean (±standard deviation and/or standard error of mean) from n = 5 mice per dose/group with significance threshold set at p < 0.05 using the two-way ANOVA with a Šidák correction for multiple comparisons. Statistical significance in neutralization titer results was evaluated using a Student t-test. Cytokine results were evaluated using the two-way ANOVA with a Šidák correction for multiple comparisons and significance threshold also set at p < 0.05.
4. Discussion
One strategy for the development of peptide vaccines entails the incorporation of multiple copies of the same epitope to improve the humoral immune response to the target antigen. However, a more desirable approach that combines multiple epitopes from different key antigens could provide better coverage of pathogen antigen diversity and reduce the risk of pathogen escape due to immune pressure [
24]. The HA, NA and matrix proteins provide a diverse set of highly conserved influenza virus epitopes that we incorporated into our composite influenza peptide immunogens. While HA has been a focus for many seasonal vaccines, other influenza proteins such as NA and M2e have been proposed as important vaccine components to induce broad neutralizing antibodies [
5,
6,
7,
8,
9,
10]. Monoclonal antibodies to each of the peptide epitopes in the composite antigens used in this study were shown to neutralize influenza viruses across Groups 1 and 2 contemporary strains (unpublished data). Additionally, the inclusion of a T-cell epitope may further promote broad immunity. Synthetic peptide vaccines eliminate the need for the cumbersome and time-consuming process of egg-based production, removing the possibility of antigenic changes during vaccine development [
24,
25].
Small peptide antigens are often poorly immunogenic unless they are conjugated to a carrier such as detoxified diphtheria or tetanus toxins. However, in this study we have shown that composite peptides comprising of small HA, NA and M2e highly conserved epitopes (conjugated and unconjugated) induced a robust antibody response when formulated with ALFQ adjuvant. As little as 1 µg of unconjugated composite peptide vaccine formulated with the ALFQ adjuvant induced broad influenza humoral immunity. Being able to induce high levels of peptide-specific antibodies without the need for conjugation simplifies the manufacturing process of peptide vaccines. Rapid induction of antibodies was observed after the initial immunization and both Th1 and Th2 immune responses were detected by antibody and cytokine analysis. These preliminary studies strongly suggest that both the targeted epitopes and the adjuvant formulation are important for induction of broadly neutralizing antibodies to influenza across Groups 1 and 2 influenza strains.
The ALF family of adjuvants has been used with a variety of antigens [
22], and liposomes containing both MPLA and QS21 (AS01 adjuvant, GSK) are present in the first FDA-approved liposome-based vaccine, Shingrix, (GSK), a vaccine for the prevention of herpes zoster caused by a previous infection with varicella virus (chicken pox) [
22]. ALFQ was shown to be safe with no adverse events in two different types of candidate malaria vaccines (unpublished data), and a phase 1 clinical trial with QS21 as the adjuvant component together with a spike ferritin nanoparticle construct for a COVID-19 vaccine is currently in progress. The unique formulation of influenza composite peptides and ALFQ provides stimulation of the immune system at many points. Previous studies have also shown that ALFQ provides a more balanced Th1 and Th2 response when compared to other liposomal adjuvants [
22]. Moreover, the ability to promote broad immunologic stimulation with little or no apparent toxicity may allow enhanced immunity with very low levels of antigen.
Further immunogenicity studies are being conducted to analyze low-dose immunizations using various routes of administration. In addition, influenza challenge studies are being designed to evaluate protection against Group 1 and Group 2 influenza strains.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}