
Impact of Transcriptome and Gut Microbiome on the Response of HIV-1 Infected Individuals to a Dendritic Cell-Based HIV Therapeutic Vaccine

 , , and
, , and 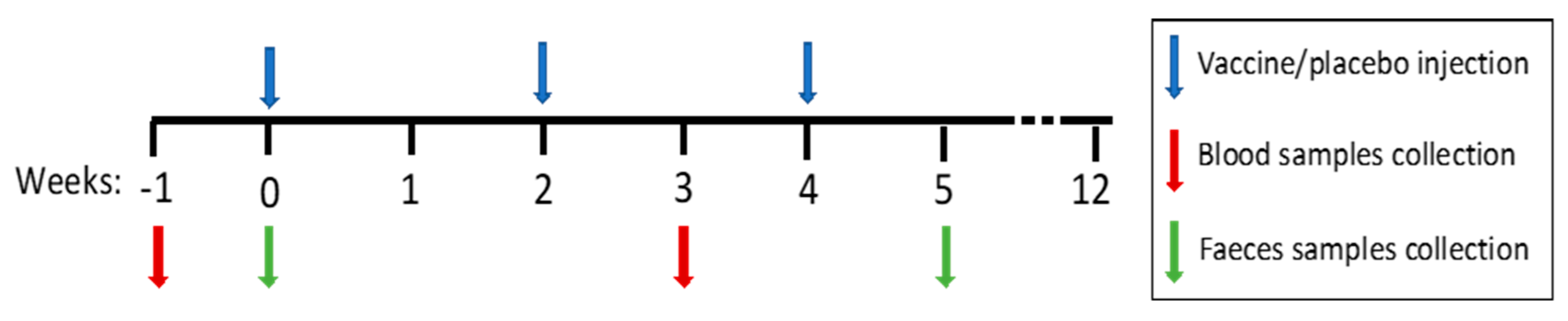{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
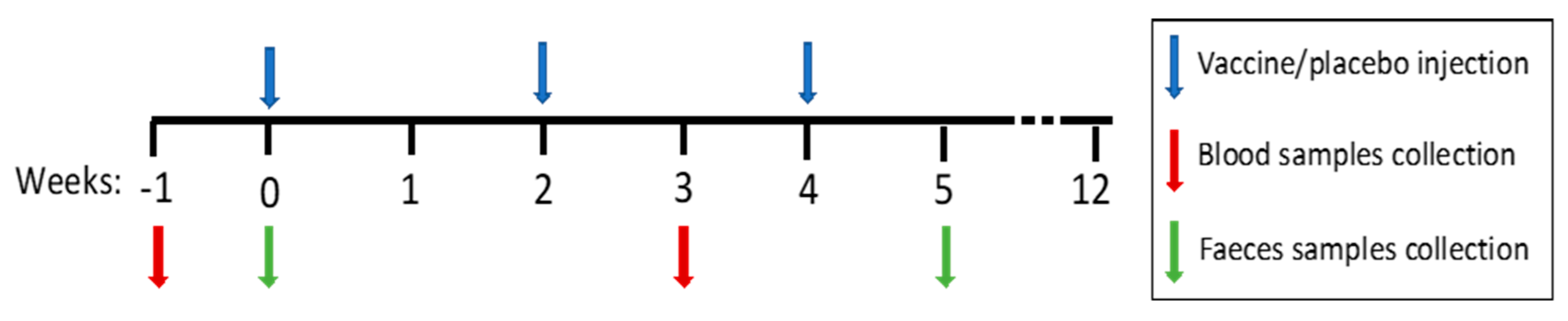
2.1. Patients and Study Design
- ARM I (n = 8): VACCINE. Autologous differentiated adult dendritic cells from monocytes of peripheral blood nonexpanded pulsed with autologous inactivated HIV virus.
- ARM II (n = 6): VACCINE + IFN. Autologous differentiated adult dendritic cells from monocytes of peripheral blood nonexpanded pulsed with autologous inactivated HIV virus with INF.
- ARM III (n = 7): PLACEBO. Autologous differentiated adult dendritic cells from monocytes of peripheral blood nonexpanded.
- ARM IV (n = 8): PLACEBO + IFN. Autologous differentiated adult dendritic cells from monocytes of peripheral blood nonexpanded with INF.
2.2. RNA Extraction, mRNA Library Preparation, and Sequencing
2.3. Stool Samples Collection, DNA Extraction, and Sequence
2.4. Bioinformatic and Statistical Analysis
3. Results
3.1. Patients
3.2. Vaccine Effect on the PBMC Transcriptome
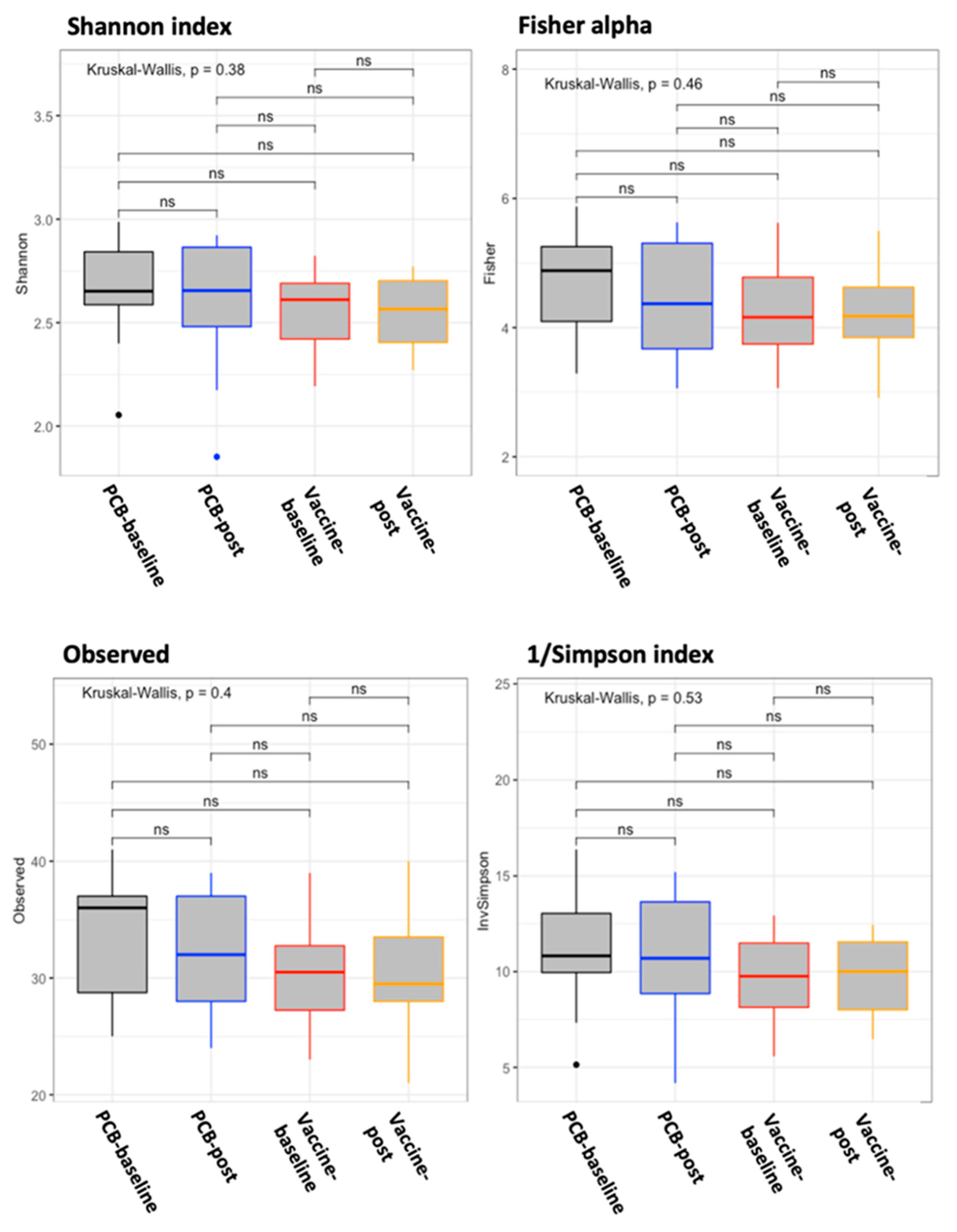
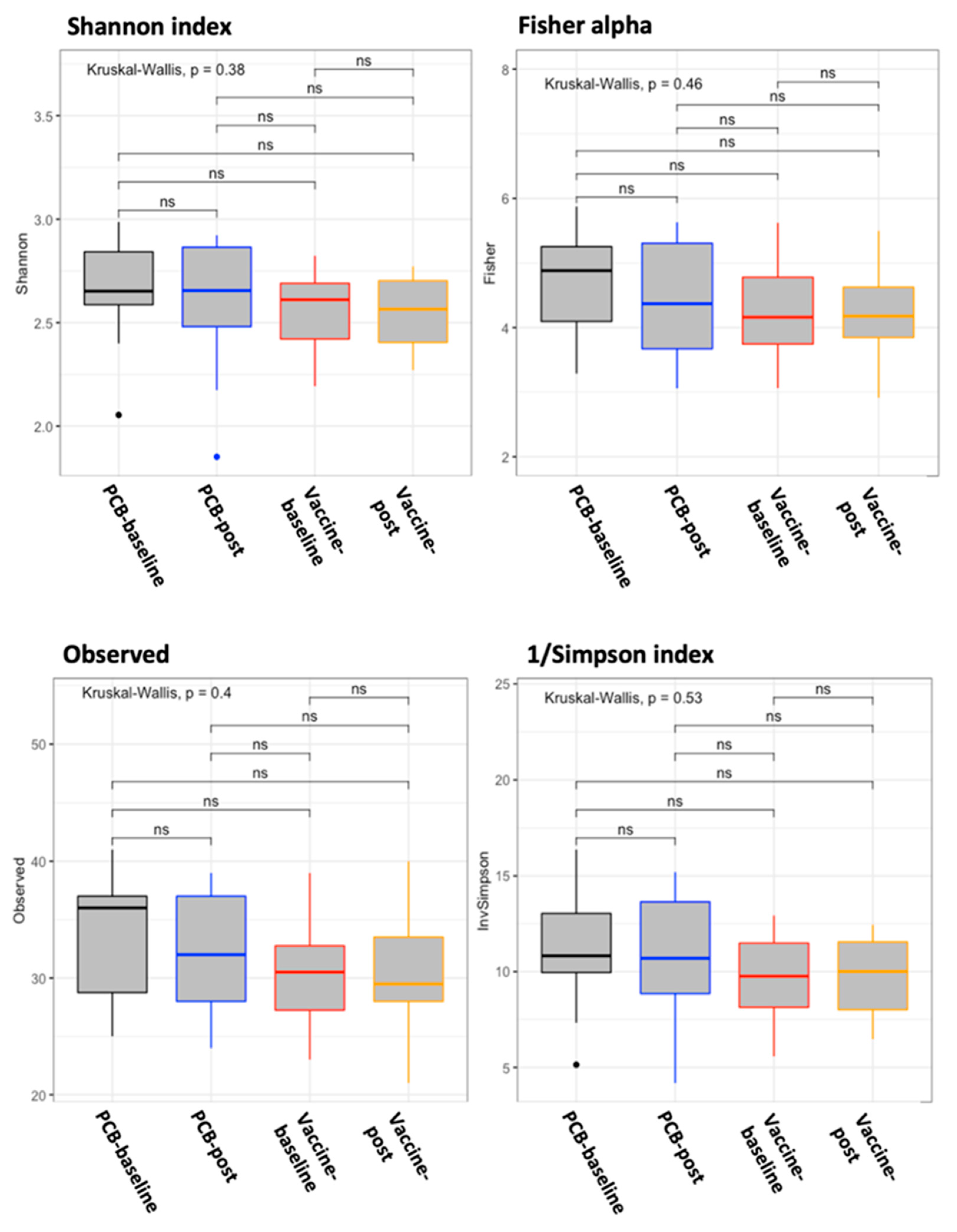
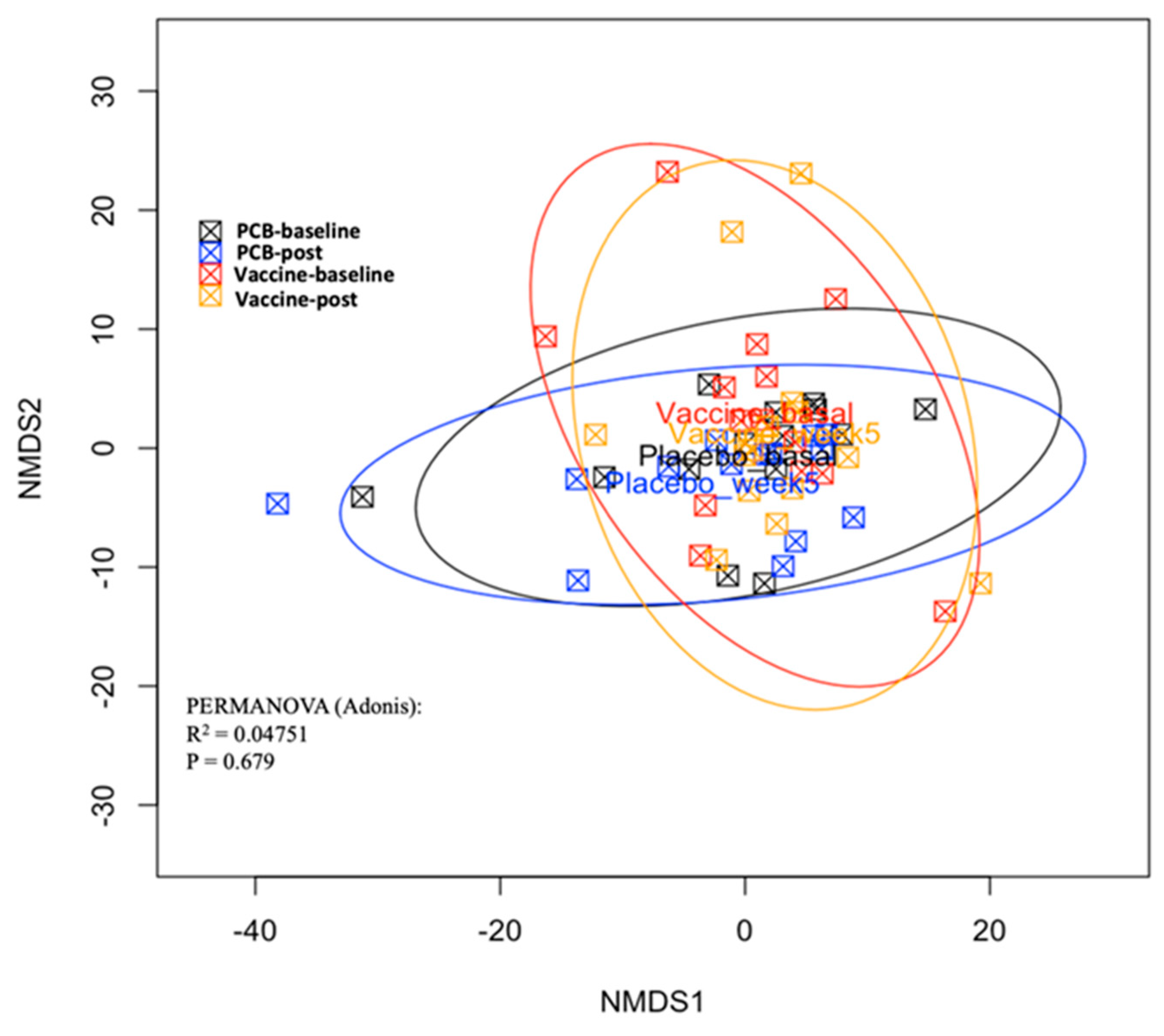
3.3. Vaccine Effect on the Gut Microbiome
3.4. Post-Hoc Analysis: Response Effect
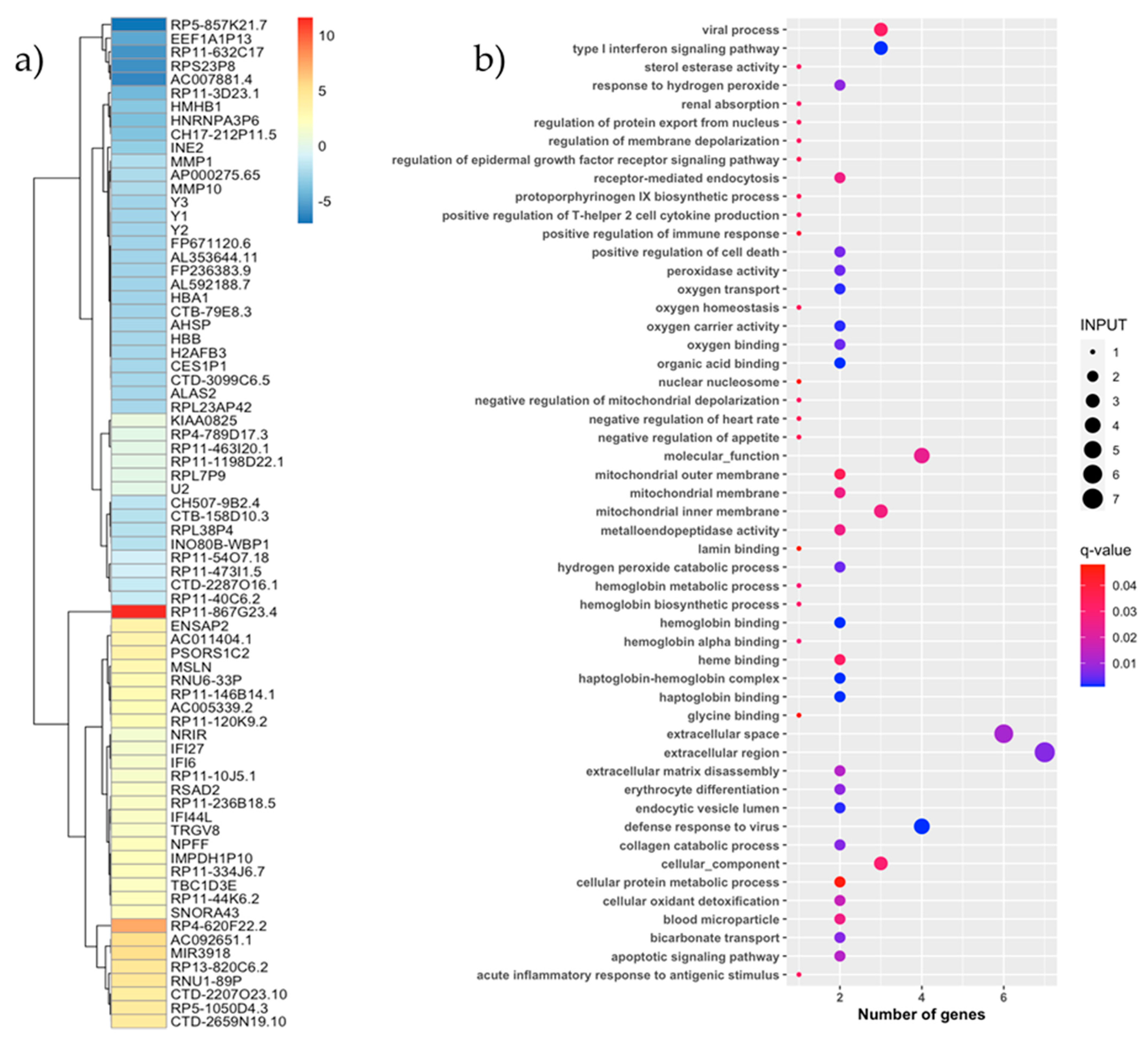
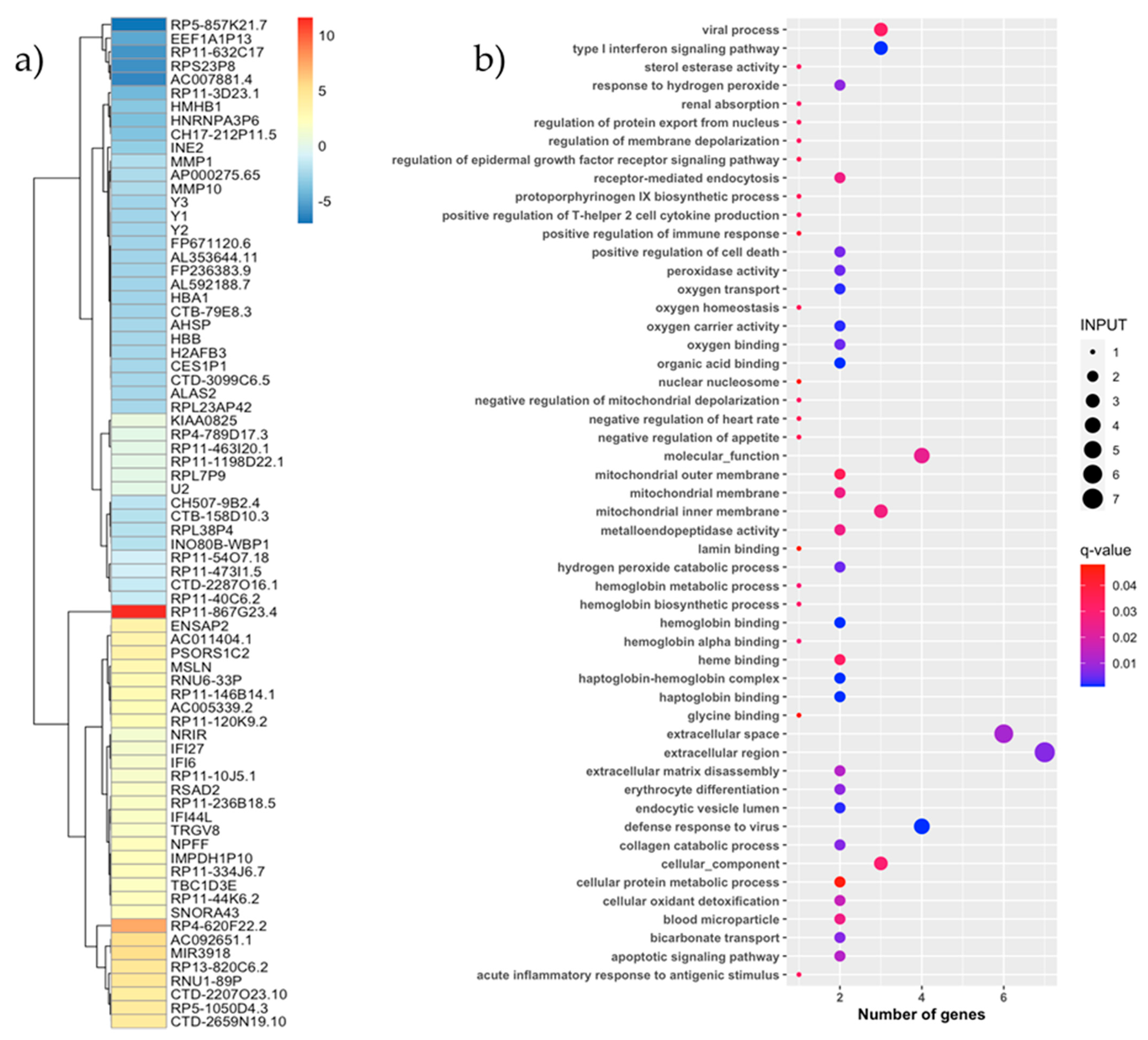
3.5. PBMC Transcriptome Analysis by Response Effect
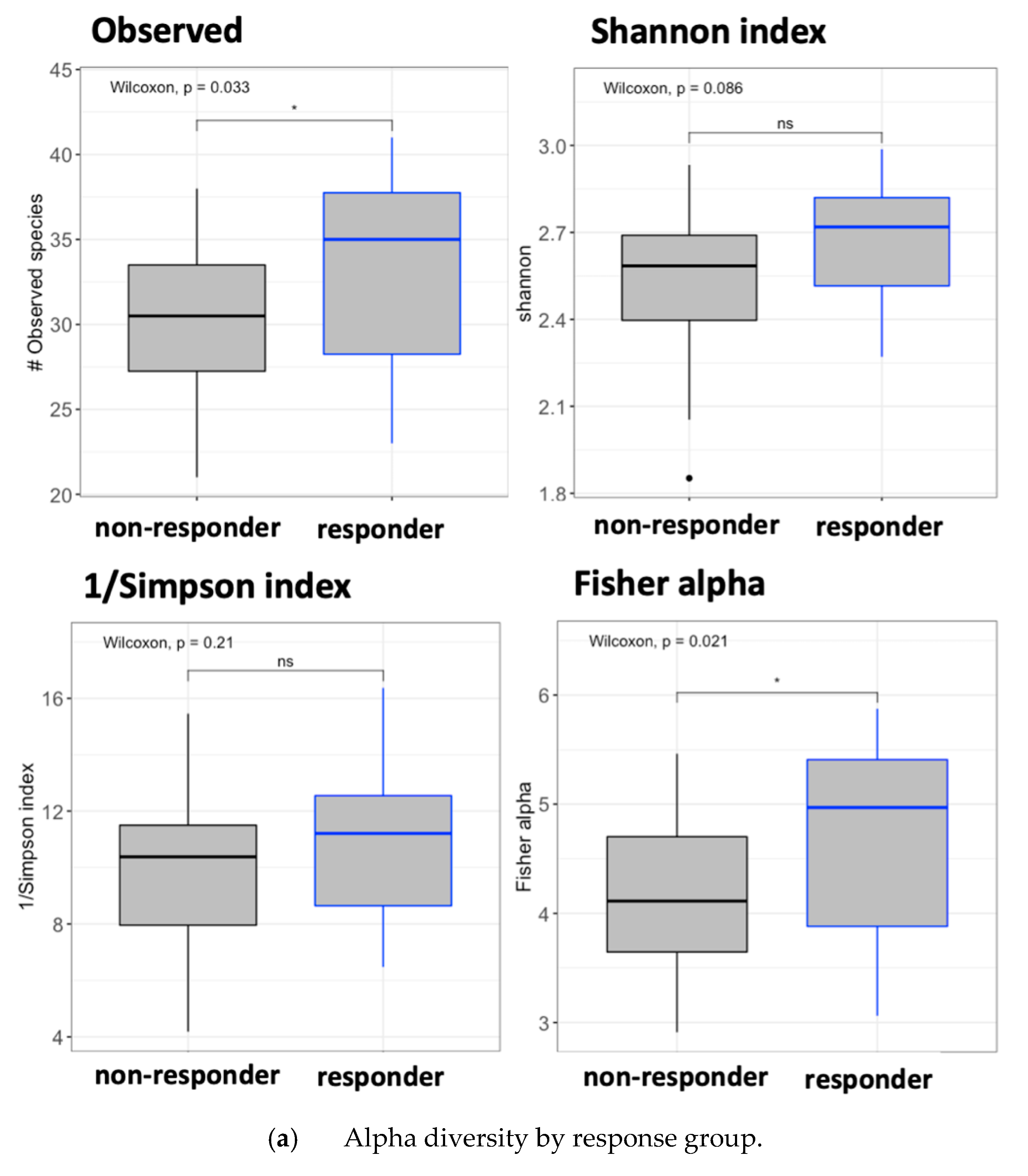
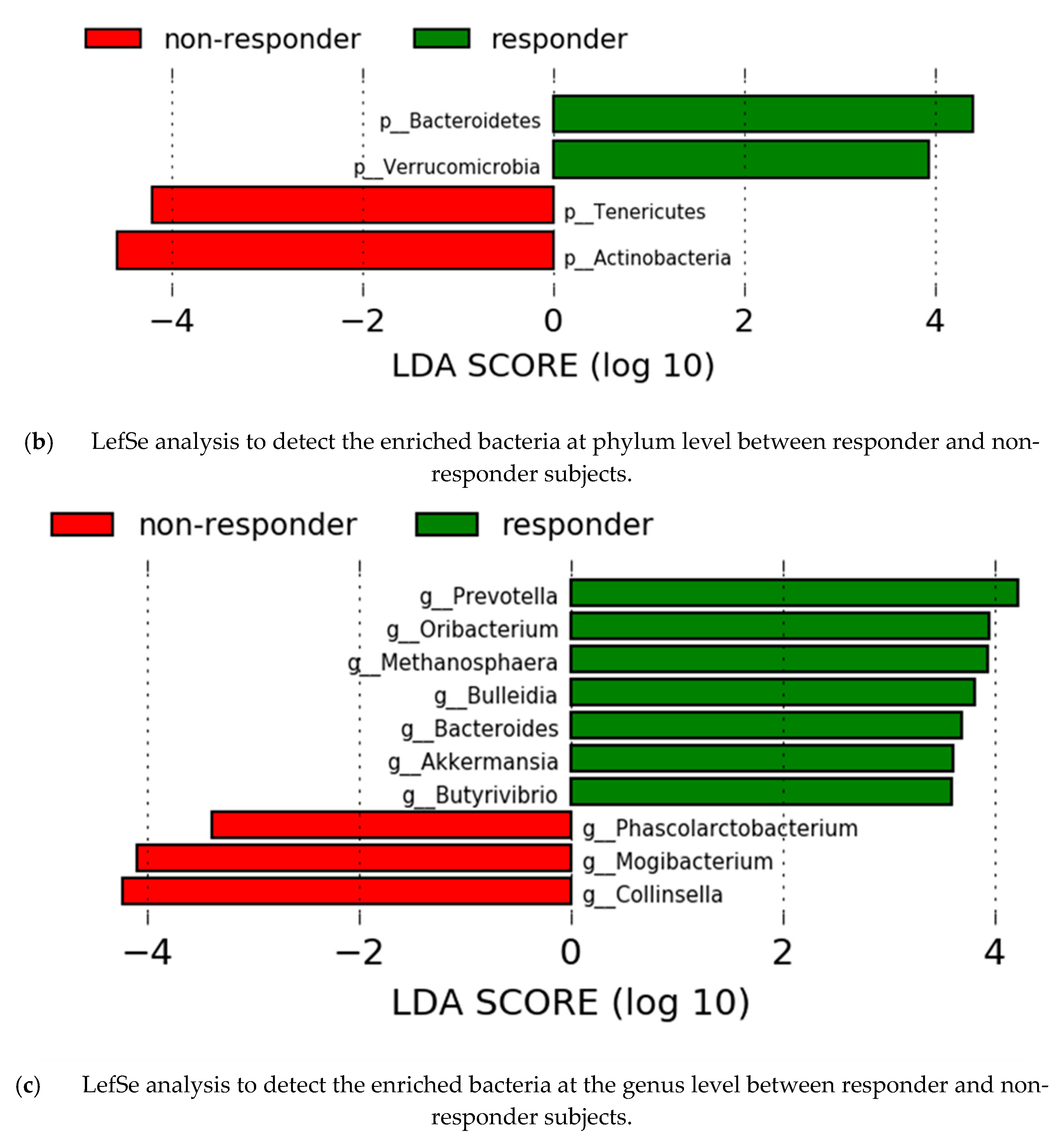
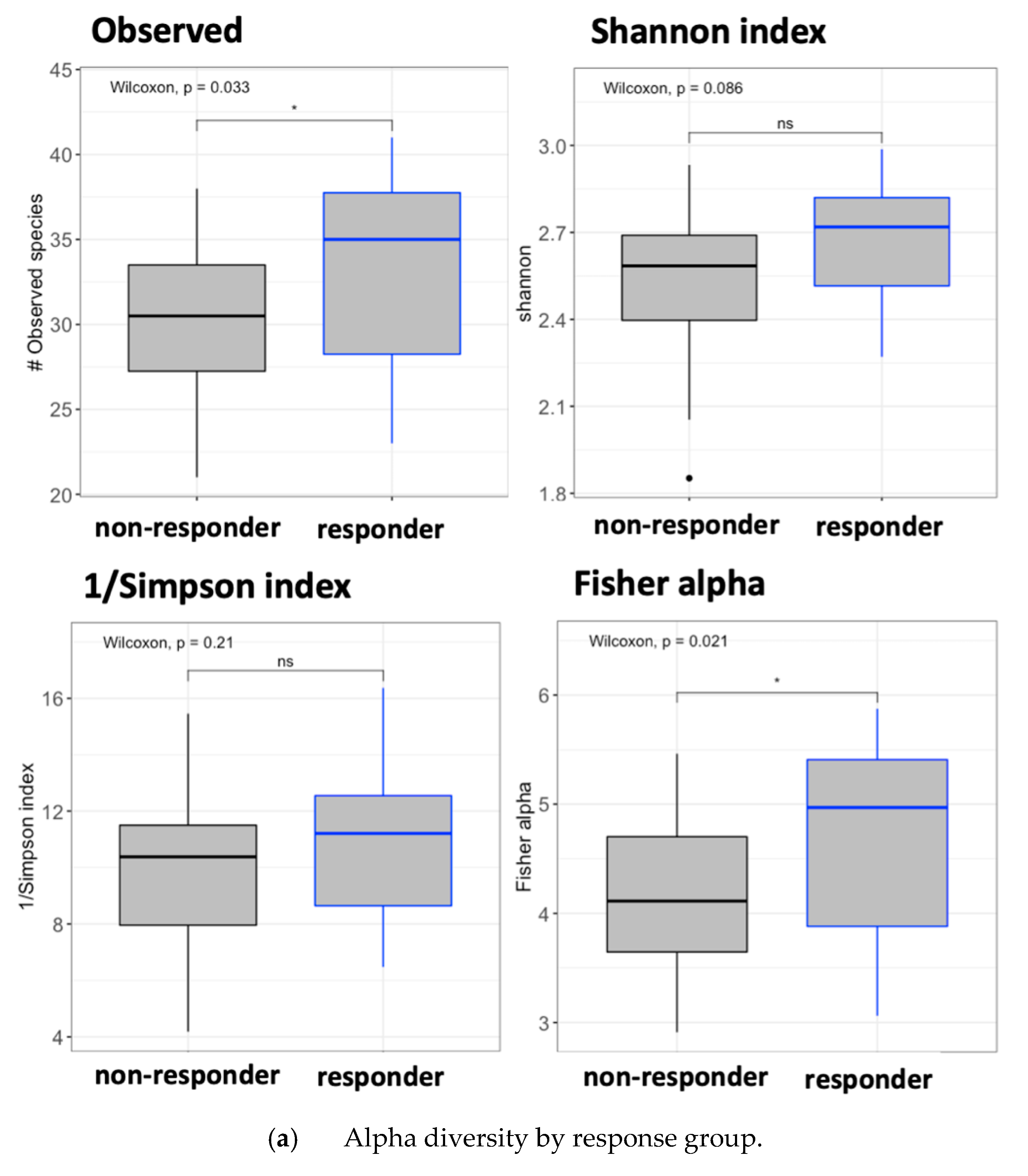
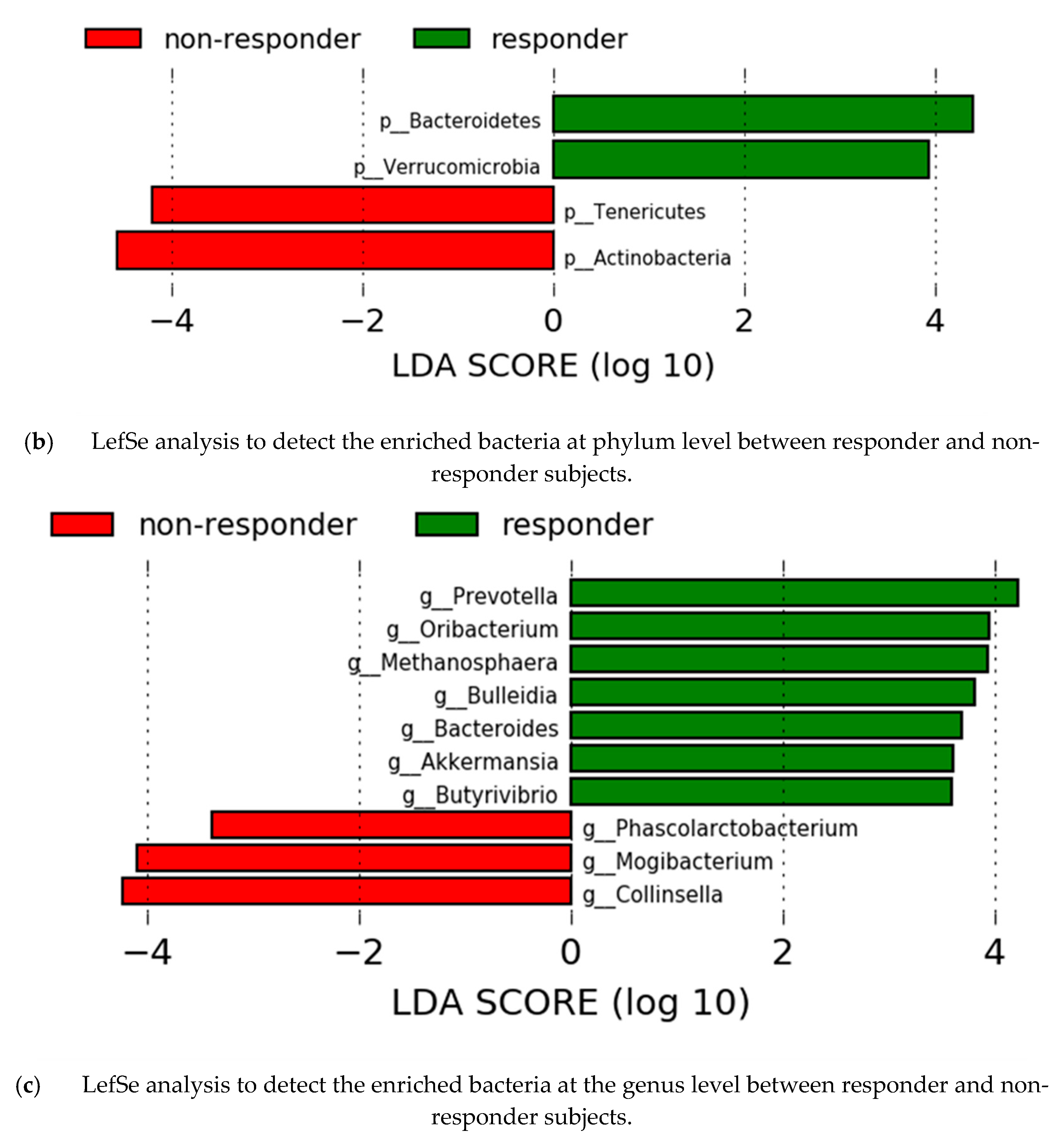
3.6. Gut Microbiota Analysis by Response Effect
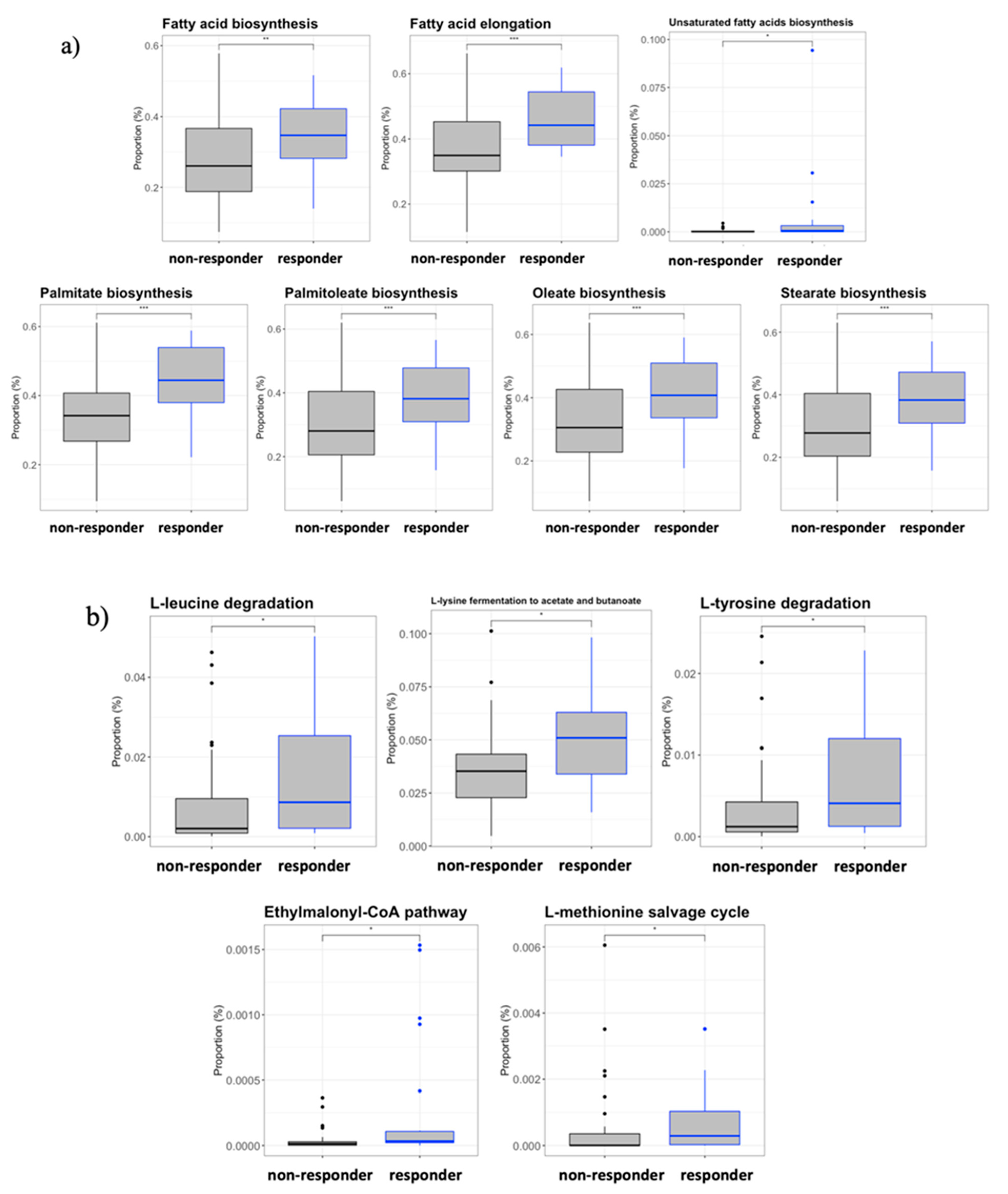
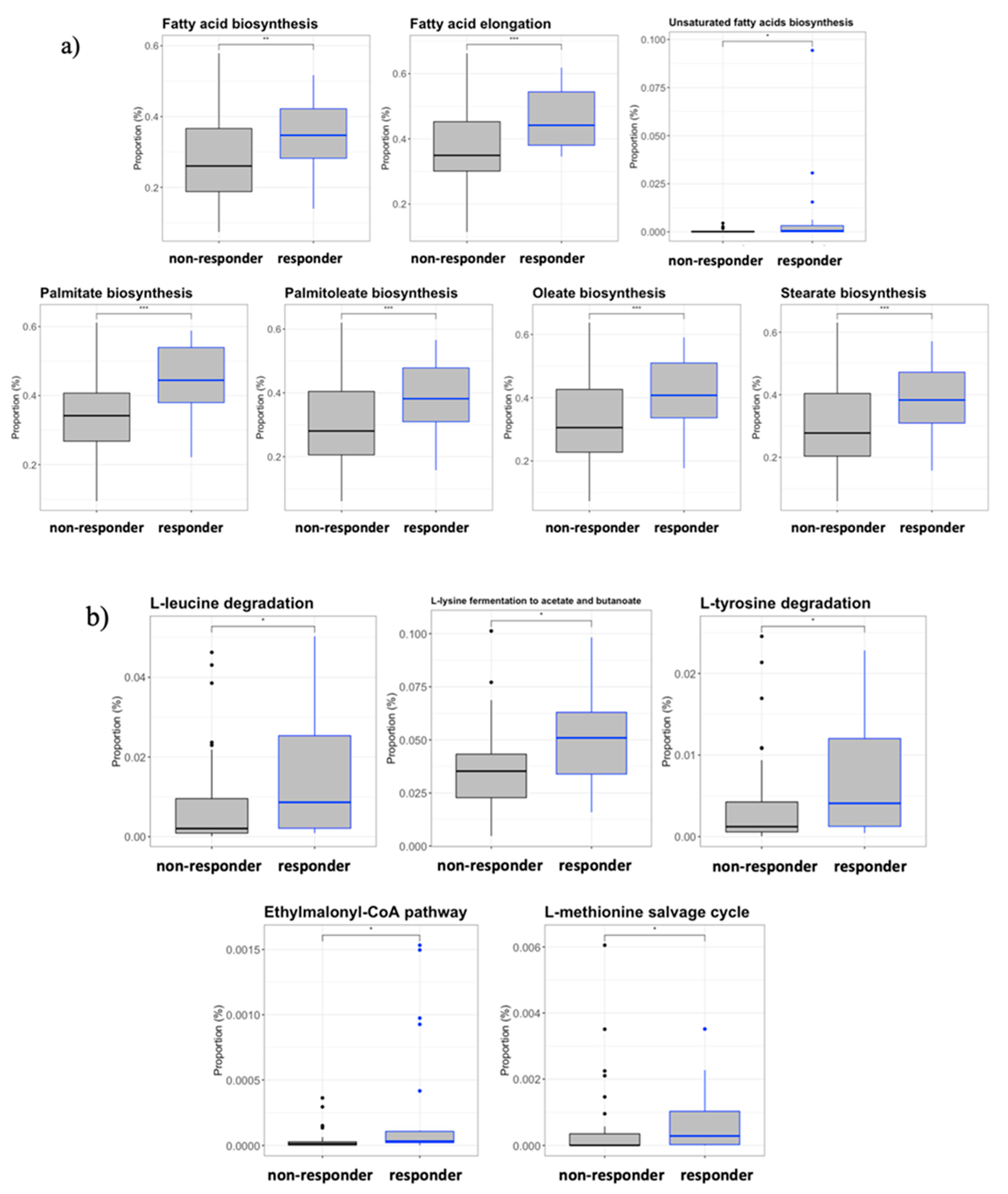
3.7. Inferred Microbiome Functionality by Response Effect
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- García, F.; Climent, N.; Assoumou, L.; Gil, C.; González, N.; Alcamí, J.; León, A.; Romeu, J.; Dalmau, J.; Martínez-Picado, J.; et al. A Therapeutic dendritic cell-based vaccine for HIV-1 infection. J. Infect. Dis. 2011, 203, 473–478. [Google Scholar] [CrossRef]
- Querec, T.; Akondy, R.; Lee, E.K.; Cao, W.; Nakaya, H.; Teuwen, D.; Pirani, A.; Gernert, K.; Deng, J.; Marzolf, B.; et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 2008, 10, 116–125. [Google Scholar] [CrossRef] [Green Version]
- de Goede, A.L.; Andeweg, A.C.; van den Ham, H.J.; Bijl, M.A.; Zaaraoui-Boutahar, F.; van IJcken, W.F.J.; Wilgenhof, S.; Aerts, J.L.; Gruters, R.A.; Osterhaus, A.D.M.E. DC immunotherapy in HIV-1 infection induces a major blood transcriptome shift. Vaccine 2015, 33, 2922–2929. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.C.; Kim, D.; Creegan, M.; Lal, K.; Ake, J.A.; Currier, J.R.; Streeck, H.; Robb, M.L.; Michael, N.L.; Bolton, D.L.; et al. Transcriptomic signatures of NK cells suggest impaired responsiveness in HIV-1 infection and increased activity post-vaccination. Nat. Commun. 2018, 9, 1212. [Google Scholar] [CrossRef] [Green Version]
- Thiébaut, R.; Hejblum, B.; Hocini, H.; Bonnabau, H.; Skinner, J.; Montes, M.; Lacabaratz, C.; Richert, L.; Palucka, K.; Banchereau, J.; et al. Gene expression signatures associated with immune and virological responses to therapeutic vaccination with dendritic cells in HIV-infected individuals. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.; Belkaid, Y. Do the microbiota influence vaccines and protective immunity to pathogens? Engaging our endogenous adjuvants. Cold Spring Harb Perspect Biol. 2018, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Valdez, Y.; Brown, E.M.; Finlay, B.B. Influence of the microbiota on vaccine effectiveness. Trends Immunol. 2014, 35, 526–537. [Google Scholar] [CrossRef]
- Seekatz, A.M.; Panda, A.; Rasko, D.A.; Toapanta, F.R.; Eloe-Fadrosh, E.A.; Khan, A.Q.; Liu, Z.; Shipley, S.T.; DeTolla, L.J.; Sztein, M.B.; et al. Differential response of the cynomolgus macaque gut microbiota to shigella infection. PLoS ONE 2013, 8, e64212. [Google Scholar] [CrossRef]
- Eloe-Fadrosh, E.A.; McArthur, M.A.; Seekatz, A.M.; Drabek, E.F.; Rasko, D.A.; Sztein, M.B.; Fraser, C.M. Impact of oral typhoid vaccination on the human gut microbiota and correlations with S. Typhi-specific immunological responses. PLoS ONE 2013, 8, e62026. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Z.; Ravindran, R.; Chassaing, B.; Carvalho, F.A.; Maddur, S.; Bower, M.; Hakimpour, P.; Gill, K.P.; Nakaya, H.I.; Yarovinsky, F.; et al. TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza Vaccination Jason. Immunity 2014, 41, 478–492. [Google Scholar] [CrossRef] [Green Version]
- Rocafort, M.; Noguera-Julian, M.; Rivera, J.; Pastor, L.; Guillén, Y.; Langhorst, J.; Parera, M.; Mandomando, I.; Carrillo, J.; Urrea, V.; et al. Evolution of the gut microbiome following acute HIV-1 infection. Microbiome 2019, 7, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguera-Julian, M.; Rocafort, M.; Guillén, Y.; Rivera, J.; Casadellà, M.; Nowak, P.; Hildebrand, F.; Zeller, G.; Parera, M.; Bellido, R.; et al. Gut microbiota linked to sexual preference and HIV infection. EBioMedicine 2016, 5, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesterbacka, J.; Rivera, J.; Noyan, K.; Parera, M.; Neogi, U.; Calle, M.; Paredes, R.; Sönnerborg, A.; Noguera-Julian, M.; Nowak, P. Richer gut microbiota with distinct metabolic profile in HIV infected Elite Controllers. Sci. Rep. 2017, 7, 6269. [Google Scholar] [CrossRef] [PubMed]
- Nowak, P.; Troseid, M.; Avershina, E.; Barqasho, B.; Neogi, U.; Holm, K.; Hov, J.; Noyan, K.; Vesterbacka, J.; Svärd, J.; et al. Gut microbiota diversity predicts immune status in HIV-1 infection. AIDS 2015, 29, 2409–2418. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- McKay, P.F.; Cizmeci, D.; Aldon, Y.; Maertzdorf, J.; Weiner, J.; Kaufmann, S.H.; Lewis, D.J.; Berg, R.A.V.D.; Del Giudice, G.; Shattock, R.J. Author response: Identification of potential biomarkers of vaccine inflammation in mice. Elife 2019, 8, e46149. [Google Scholar] [CrossRef]
- Ferreira, R.; Antunes, L.C.M.; Finlay, B.B. Should the human microbiome be considered when developing vaccines? PLOS Pathog. 2010, 6, e1001190. [Google Scholar] [CrossRef] [Green Version]
- Lamousé-Smith, E.S.; Tzeng, A.; Starnbach, M.N. The Intestinal flora is required to support antibody responses to systemic immunization in infant and germ free mice. PLoS ONE 2011, 6, e27662. [Google Scholar] [CrossRef] [Green Version]
- Vlasova, A.N.; Takanashi, S.; Miyazaki, A.; Rajashekara, G.; Saif, L.J. How the gut microbiome regulates host immune responses to viral vaccines. Curr. Opin. Virol. 2019, 37, 16–25. [Google Scholar] [CrossRef]
- Nozza, S.; Ferrarese, R.; Poli, A.; Galli, L.; Sampaolo, M.; Bigoloni, A.; Galli, A.; Muccini, C.; Spagnuolo, V.; Lazzarin, A.; et al. Analysis of the faecal microbiome during analytical treatment interruption in people with chronic HIV infection and long-lasting virological suppression (APACHE study). J. Antimicrob. Chemother. 2020, 75, 2700–2702. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Sakaguchi, S.; Nakajima, C.; Watanabe, A.; Yanai, H.; Matsumoto, M.; Ohteki, T.; Kaisho, T.; Takaoka, A.; Akira, S.; et al. Selective contribution of IFN-α/β signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 10872–10877. [Google Scholar] [CrossRef] [Green Version]
- Bradley, K.C.; Finsterbusch, K.; Schnepf, D.; Crotta, S.; Llorian, M.; Davidson, S.; Fuchs, S.Y.; Staeheli, P.; Wack, A. Microbiota-driven tonic interferon signals in lung stromal cells protect from influenza virus infection. Cell Rep. 2019, 28, 245–256.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprooten, J.; Agostinis, P.; Garg, A.D. Type I interferons and dendritic cells in cancer immunotherapy. Int. Rev. Cell Mol. Biol. 2019, 348, 217–262. [Google Scholar] [CrossRef]
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and interferons: Who’s interfering with whom? Nat. Rev. Microbiol. 2015, 13, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Cirión, A.; Sereti, I. Immunometabolism and HIV-1 pathogenesis: Food for thought. Nat. Rev. Immunol. 2021, 21, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Mikhailova, A.; Valle-Casuso, J.C.; David, A.; Monceaux, V.; Volant, S.; Passaes, C.; Elfidha, A.; Müller-Trutwin, M.; Poyet, J.-L.; Sáez-Cirión, A. Antiapoptotic clone 11-derived peptides induce in vitro death of CD4 + T cells susceptible to HIV-1 infection. J. Virol. 2020, 94, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Castellano, P.; Prevedel, L.; Valdebenito, S.; Eugenin, E.A. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 2019, 9, 3941. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Hang, S.; Mao, S.; Zhu, W. Diversity of Butyrivibrio group bacteria in the rumen of goats and its response to the supplementation of garlic oil. Asian-Australas. J. Anim. Sci. 2014, 27, 179–186. [Google Scholar] [CrossRef]
- Zheng, L.; Kelly, C.J.; Battista, K.D.; Schaefer, R.; Lanis, J.M.; Alexeev, E.E.; Wang, R.X.; Onyiah, J.C.; Kominsky, D.J.; Colgan, S.P. Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor–dependent repression of claudin-2. J. Immunol. 2017, 199, 2976–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earley, H.; Lennon, G.; Balfe, Á.; Coffey, J.C.; Winter, D.C.; O’Connell, P.R. The abundance of Akkermansia muciniphila and its relationship with sulphated colonic mucins in health and ulcerative colitis. Sci Rep. 2019, 9, 15683. [Google Scholar] [CrossRef] [Green Version]
- Zhai, R.; Xue, X.; Zhang, L.; Yang, X.; Zhao, L.; Zhang, C. Strain-specific anti-inflammatory properties of two Akkermansia muciniphila strains on chronic colitis in mice. Front. Cell. Infect. Microbiol. 2019, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, J.; Lin, J.; Isnard, S.; Fombuena, B.; Peng, X.; Marette, A.; Routy, B.; Messaoudene, M.; Chen, Y.; Routy, J.-P. The bacterium Akkermansia muciniphila: A sentinel for gut permeability and its relevance to HIV-related inflammation. Front. Immunol. 2020, 11, 645. [Google Scholar] [CrossRef]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef]
- Pinto-Cardoso, S.; Klatt, N.R.; Reyes-Terán, G. Impact of antiretroviral drugs on the microbiome: Unknown answers to important questions. Curr. Opin. HIV AIDS. 2018, 13, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Dillon, S.M.; Kibbie, J.; Lee, E.J.; Guo, K.; Santiago, M.L.; Austin, G.; Gianella, S.; Landay, A.L.; Donovan, A.M.; Frank, D.N.; et al. Low abundance of colonic butyrate-producing bacteria in HIV infection is associated with microbial translocation and immune activation. AIDS 2017, 31, 511–521. [Google Scholar] [CrossRef]
- Pande, S.V. On rate-controlling factors of long chain fatty acid oxidation. J. Biol. Chem. 1971, 246, 5384–5390. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastor-Ibáñez, R.; Díez-Fuertes, F.; Sánchez-Palomino, S.; Alcamí, J.; Plana, M.; Torrents, D.; Leal, L.; García, F. Impact of Transcriptome and Gut Microbiome on the Response of HIV-1 Infected Individuals to a Dendritic Cell-Based HIV Therapeutic Vaccine. Vaccines 2021, 9, 694. https://doi.org/10.3390/vaccines9070694
Pastor-Ibáñez R, Díez-Fuertes F, Sánchez-Palomino S, Alcamí J, Plana M, Torrents D, Leal L, García F. Impact of Transcriptome and Gut Microbiome on the Response of HIV-1 Infected Individuals to a Dendritic Cell-Based HIV Therapeutic Vaccine. Vaccines. 2021; 9(7):694. https://doi.org/10.3390/vaccines9070694
Chicago/Turabian StylePastor-Ibáñez, Roque, Francisco Díez-Fuertes, Sonsoles Sánchez-Palomino, Jose Alcamí, Montserrat Plana, David Torrents, Lorna Leal, and Felipe García. 2021. "Impact of Transcriptome and Gut Microbiome on the Response of HIV-1 Infected Individuals to a Dendritic Cell-Based HIV Therapeutic Vaccine" Vaccines 9, no. 7: 694. https://doi.org/10.3390/vaccines9070694
APA StylePastor-Ibáñez, R., Díez-Fuertes, F., Sánchez-Palomino, S., Alcamí, J., Plana, M., Torrents, D., Leal, L., & García, F. (2021). Impact of Transcriptome and Gut Microbiome on the Response of HIV-1 Infected Individuals to a Dendritic Cell-Based HIV Therapeutic Vaccine. Vaccines, 9(7), 694. https://doi.org/10.3390/vaccines9070694