Abstract
Herpesviruses are main sculptors of natural killer (NK) cell repertoires. While the β-herpesvirus human cytomegalovirus (CMV) drives the accumulation of adaptive NKG2C-positive NK cells, the human γ-herpesvirus Epstein–Barr virus (EBV) expands early differentiated NKG2A-positive NK cells. While adaptive NK cells support adaptive immunity by antibody-dependent cellular cytotoxicity, NKG2A-positive NK cells seem to preferentially target lytic EBV replicating B cells. The importance of this restriction of EBV replication during γ-herpesvirus pathogenesis will be discussed. Furthermore, the modification of EBV-driven NK cell expansion by coinfections, including by the other human γ-herpesvirus Kaposi sarcoma-associated herpesvirus (KSHV), will be summarized.
1. Human γ-Herpesvirus Infections and Associated Pathologies
The two human γ-herpesviruses Epstein–Barr virus (EBV) and Kaposi sarcoma-associated herpesvirus (KSHV) are associated with lymphomas and carcinomas [1,2]. They were discovered as human herpesvirus 4 and 8 in 1964 and 1994 in Burkitt’s lymphoma and Kaposi sarcoma, respectively [3,4,5]. Accordingly, these two oncogenic viruses encode proteins that either transform cells on their own or assist cellular oncogenes [6,7,8]. Despite this growth transforming capacity, both viruses are asymptomatically carried by more than 95% of the human adult population in the case of EBV and by more than 50% in Sub-Saharan Africa for KSHV [2,9]. B cells are the main host cells of both viruses. EBV and KSHV colonize these lymphocytes after transmission via saliva exchange. This process is better understood for EBV than for KSHV. B cell infection by EBV in submucosal secondary lymphoid tissues like the tonsils leads to the expression of six nuclear antigens (EBNA1, 2, 3A, 3B, 3C, -LP) and two latent membrane proteins (LMP1 and 2), as well as two Epstein–Barr virus-encoded small RNAs (EBERs) and more than 40 miRNAs in naïve B cells [10,11]. This so-called latency III program is thought to drive B cell differentiation into the germinal center reaction. The resulting centroblasts and -cytes only express EBNA1 and the two LMPs, as well as nontranslated RNAs [11]. This expression program is called latency II and is thought to help the infected B cell to survive the germinal center reaction. This allows EBV-infected B cells to enter the memory B cell pool for long-term persistence [12]. In memory B cells, EBV expresses no viral proteins (latency 0) or transiently EBNA1 during homeostatic proliferation [13]. The expression of EBNA1 and nontranslated viral RNAs is called latency I. Upon B cell activation and plasma cell differentiation from latency 0 or I, EBV switches into lytic replication with infectious virus production [14]. It can then infect polarized epithelial cells from the basolateral side [15,16,17], presumably for an additional round of lytic replication and efficient virus shedding into the saliva. KSHV seems to benefit from this B cell infection by EBV. Efficient KSHV persistence was found to depend on EBV coinfection in mice with reconstituted human immune system compartments [18]. EBV also supported the KSHV infection of human B cells in cell culture [19,20]. In these instances, KSHV infection can be primarily found in EBV-coinfected B cells. Such coinfection can also be observed in primary effusion lymphoma (PEL), a B cell tumor in which KSHV genome maintenance is compromised when EBV is deleted [21,22]. Furthermore, epidemiologically, KSHV infection is associated with EBV infection in African children [23,24]. It remains unclear under which circumstances KSHV infects the endothelial cells from which Kaposi sarcoma originates [2,25]. However, KSHV, similar to EBV, uses the ephrin A2 receptor for epithelial cell infection [16,17,26], presumably prior to shedding into saliva for transmission.
The above outlined evidence suggests that all EBV latency programs that can also be found in EBV-associated malignancies, such as latency I in Burkitt’s lymphoma and gastric carcinoma, latency II in Hodgkin’s lymphoma and nasopharyngeal carcinoma, and latency III in diffuse large B cell lymphoma (DLBCL), are already present in healthy EBV carriers [1,27]. Moreover, EBV-associated B cell lymphomas, PEL and Kaposi sarcoma are increased in patients whose immune system has been weakened by coinfection with the human immunodeficiency virus (HIV) [28]. Therefore, immune control seems to prevent pathology in individuals that are persistently infected with EBV and KSHV. In this review I will discuss the evidence that natural killer (NK) cells are part of this immune control and might be harnessed against EBV- and KSHV-associated pathologies.
2. Genetic Evidence for Immune Control of Human γ-Herpesviruses by NK Cells
NK cells seem to play a crucial role in the immune control of herpesvirus infections. Even in the first patient that was described with NK cell deficiency, severe α- (herpes simplex and varicella zoster virus) and β-herpesvirus (cytomegalovirus [CMV]) infections were observed [29]. Later-on mutations in the transcription factor GATA2 that prevent early differentiated CD56bright NK cell development were identified in this and other patients [30]. Interestingly, primary immunodeficiencies that cause NK cell deficiencies, such as the GATA2 mutations, predispose either for CMV or EBV pathologies [31]. Among the NK cell deficiencies that are associated with EBV-positive lymphomas and EBV-driven immunopathologies such as hemophagocytic lymphohistiocytosis (HLH) are mutations in the minichromosome maintenance complex component 4 (MCM4) or interferon regulatory factor 8 (IRF8) [32,33,34]. These deficiencies compromise NK cell maturation to CD56dim cells with increased cytotoxicity. Indeed, primary immunodeficiencies that compromise the cytotoxic machinery of lymphocytes predispose for EBV-associated pathologies [35,36,37]. The respective mutations affect the pore-forming protein perforin or the adaptor proteins Munc13-4 and Munc18-2 that are required for cytotoxic granule fusion with the plasma membrane [38,39,40]. In addition to the cytotoxic machinery itself, receptors that mediate NK cell function are also affected by mutations in some patients with primary immunodeficiencies that predispose for EBV-associated pathologies. These include the activating receptor NKG2D and the activating coreceptors CD2, 2B4 and NTB-A on NK cells [41,42,43,44,45,46,47,48,49,50,51]. In this respect, XMEN (X-linked immunodeficiency with magnesium defect, Epstein–Barr virus (EBV) infection and neoplasia) disease is caused by mutations in the magnesium transporter MagT1 [41,42,43]. This compromises the surface expression of the activating NK cell receptor NKG2D, which is involved in the NK cell recognition of lytically EBV replicating B cells [52]. Furthermore, mutations in the Fc receptor CD16 compromise natural cytotoxicity, but not antibody-dependent cellular cytotoxicity (ADCC) by the affected NK cells [45,53]. In particular, they prevent the efficient recruitment of the activating coreceptor CD2 to the immunological synapse for efficient NK cell killing [45]. Finally, loss-of-function mutations in the SLAM (signalling lymphocytic activation molecule) receptor-associated protein SAP convert costimulatory signals of the two SLAM receptors 2B4 and NTB-A to inhibitory signals, thereby inhibiting lysis of EBV-transformed B cells by NK cells [54,55]. These mutations in patients with primary immunodeficiencies argue for an important role of cytotoxic CD56dimCD16+CD2+NKG2D+2B4+NTB-A+ NK cells in the immune control of EBV.
While an increased predisposition for Kaposi sarcoma was also described for patients with XMEN disease [56], KSHV-specific immune control seems to depend much more on the production of the antiviral cytokine IFN-γ than EBV-specific immune control. Accordingly, mutations in the IFN-γ receptor (IFN-γR1) and STAT4 that elicit IFN-γ transcription after IL-12 signaling increase the risk for Kaposi sarcoma [57,58]. This could indicate that, similar to CMV, early differentiated CD56bright NK cells with a preferential IFN-γ production but less pronounced natural cytotoxicity are more important in KSHV-specific immune control than in EBV-specific immune control.
3. NK Cell Phenotype and Function during EBV Infection
In addition to genetic lesions that compromise NK cell differentiation, effector function and stimulation, and thereby affect the immune control of EBV and KSHV infection [37], changes in the magnitude and composition of the NK cell compartment after the respective viral infections point towards their contribution to immune responses to these two oncogenic γ-herpesviruses. This has been primarily investigated during symptomatic primary EBV infection [9,59]. In the United States and Europe, two thirds of the population get infected with EBV before the age of 2, and a substantial proportion of the remaining one third experiences primary infection in the second decade of life [60,61,62]. In 30–50% of cases, delayed primary infections lead to infectious mononucleosis (IM) with a massive expansion of lytic EBV antigen-specific CD8+ T cells and the associated immunopathology [63,64]. In addition, NK cells expand during IM [61,65,66,67]. These are in particular CD56dimCD16+/− NK cells that are negative for the activating NKG2C receptor and express the inhibitory NKG2A receptor, both binding the nonclassical major histocompatibility complex (MHC) class I molecule HLA-E [66,68] (Figure 1). This NK cell differentiation stage is also still mostly negative for the expression of inhibitory killer immunoglobulin-like receptors (KIRs) that can distinguish haplotypes of classical MHC class I molecules. In good agreement with the phenotypic characteristics that can be deduced from the genetic predispositions for EBV pathologies, this phenotype represents an intermediate differentiation stage after CD56brightCD16− NK cells, but prior to CD56dimCD16+KIR+NKG2C+NKG2A− adaptive NK cells and terminally differentiated CD56−CD16+ NK cells [69,70,71] (Figure 1). In contrast to EBV, CMV seems to expand adaptive NKG2C+ NK cells [72,73] and even drive KIR+ NK cell accumulation in individuals with a genetic deletion of NKG2C [74,75,76] (Figure 1). NKG2A+KIR− NK cells that expand during IM preferentially target B cells that undergo lytic EBV replication [52,66,77,78]. In particular, the activating NK cell receptor NKG2D and the activating coreceptor DNAM-1 are involved in the recognition of lytically EBV replicating B cells by NK cells [52]. EBV compromises this recognition in part by the viral miRNA-mediated downregulation of the NKG2D ligand MICB [79]. Nevertheless, NK cells restrict primarily wild-type EBV infection but not lytic replication-deficient BZLF1 knock-out virus infection in mice with reconstituted human immune system components (humanized mice) [78]. Within three months after neonatal human CD34+ hematopoietic progenitor cell transfer, this preclinical model of the human immune system primarily reconstitutes CD56brightCD16− NK cells, unless IL-15 is supplied either by transgenic expression or improved myeloid cell reconstitution [80,81,82]. NK cells expand after EBV infection in this model and accumulate as CD56dimCD16+NKG2A+KIR− NK cells [78]. Upon NK cell depletion, EBV loads do not only increase, but so does the incidence of monoclonal B cell lymphomas as well as signs of lytic EBV replication in tissue sections [78]. In the absence of NK cell-mediated immune control of lytic EBV replication, CD8+ T cells expand dramatically after EBV infection of humanized mice [78]. This massive expansion and immune pathology, such as weight loss, is associated with an elevated proinflammatory cytokine production, reminiscent of IM. Indeed, the protective CD56dimCD16+NKG2A+KIR− NK cell population decreases in frequency during the first decade of a child’s life [66,83] and could thus predispose for IM upon delayed primary EBV infection in the second decade of life. These studies suggest that NKG2A+KIR− NK cells preferentially respond to EBV infection and restrict lytic EBV replication which might otherwise drive IM.
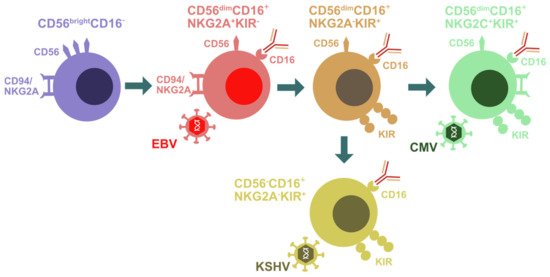
Figure 1.
Natural killer (NK) cell differentiation during herpesvirus infection. Early differentiated CD56brightCD16− NK cells are primarily found in secondary lymphoid tissues. They give rise to CD56dimCD16+NKG2A+KIR− NK cells that are preferentially expanded during EBV infection. Upon further differentiation, these cells acquire KIRs and lose NKGA. CD56dimCD16+NKG2A−KIR+ NK cells seem to then give rise to either CD56dimCD16+NKG2C+KIR+ adaptive NK cells that accumulate during CMV infection or to CD56−CD16+NKG2A−KIR+ terminally differentiated NK cells that are enriched in KSHV-coinfected individuals.
4. Modulation of NK Cell Responses by KSHV
In certain EBV-associated disease settings and upon coinfections, the composition of the NK cell compartment can change and thereby presumably fail to mediate EBV-specific immune control. Along these lines, it was noted that children with endemic Burkitt’s lymphoma accumulated CD56−CD16+ NK cells [84]. These are thought to be a further differentiation stage of CD56dimCD16+ NK cells and share their transcriptional profile to a large extent [85]. Since coinfection with the malaria parasite Plasmodium falciparum (Pf) is associated with an increased risk of developing Burkitt’s lymphoma [86] and since certain KIR receptor-HLA ligand pairs are associated with the immune control of this parasite [87,88], coinfection with Pf might be in part responsible for the observed accumulation of CD56−CD16+ NK cells. This could also occur via the Pf-mediated activation of another coinfection, such as KSHV infection. Indeed, antimalarial parasite antibodies were found to be associated with KSHV seropositivity [89], and KSHV-seropositive African children presented with a higher frequency of CD56−CD16+ NK cells [90]. Similarly transplant patients with EBV-associated lymphoproliferative disease (PTLD) present with diminished CD56dimCD16+NKG2A+KIR− NK cells, despite similarly high EBV loads as those observed in IM patients [67]. In CMV-positive patients, CD56dimCD16+NKG2C+NKG2A−KIR+ NK cells accumulate at the expense of this earlier NK cell differentiation stage. Possibly, CMV reactivation after immune suppressive treatment following transplantation drives this NK cell differentiation, away from the NK cell phenotype that protects against lytic EBV replication.
Such NK cell differentiation could also be observed upon EBV-plus-KSHV coinfection of humanized mice [90]. CD56−CD16+ NK cells accumulate upon KSHV coinfection at the expense of CD56brightCD16− NK cells (Figure 1). KSHV, but not EBV viral loads correlate with this accumulation. The cytokines IL-15, IL-18 and IL-27 that are increased during EBV-plus-KSHV coinfection might be at least partially responsible for the observed NK cell differentiation. As previously described in African children with endemic Burkitt’s lymphoma, these CD56−CD16+ NK cells share many similarities with CD56dimCD16+ NK cells [85,90]. However, they are even further enriched in the expression of the cytotoxic machinery, containing perforin and granzyme B [90]. KSHV coinfection also drives the expression of CXCR6 and CD69, which are often associated with tissue residency. Despite the high expression of perforin and granzyme B in the CD56−CD16+ NK cell population, they exercise less natural and antibody-dependent cytotoxicity. Furthermore, they produce less IFN-γ and do not proliferate. Instead, their expression of the ATPase CD39 might rather point to an immune suppressive function [90,91]. Furthermore, at least in Burkitt’s lymphoma patients, CD56−CD16+ NK cells seem to retain MIP1β/CCL4 production, which can attract CCR5+ regulatory T and suppressive myeloid cell populations [92,93]. Patients with Kaposi sarcoma also present with a diminished NK cell cytotoxicity [94,95]. Thus, KSHV coinfection drives the expansion of CD56−CD16+CXCR6+CD39+ NK cells, which might suppress immune responses in tissues by ATP hydrolysis and suppressive leucocyte attraction via MIP1β/CCL4 to avoid immune pathology.
In addition, KSHV also employs the direct modulation of NK cell recognition by inhibition of ligand upregulation for the activation of NK cell receptors, of migration of NK cells and by exploitation of certain KIR haplotypes. Along these lines, KSHV downregulates ligands for activating NK cell receptors, such as NKG2D, NKp44, NKp80 and DNAM-1 [96,97,98,99]. The viral ubiquitin ligase K5 downregulates the NKG2D ligands MICA and MICB, the NKp80 ligand AICL, and the DNAM-1 ligands Nectin-2 and CD155 [96,98]. Furthermore, viral ORF54 downregulates NKp44 ligands of an unknown identity independently of ORF54′s UTPase activity [97]. MICB and NKG2D ligands inducing AID upregulation are also targeted by KSHV-encoded miRNA [79,100]. In addition to inhibiting the NK cell recognition of infected cells, the KSHV-encoded viral MIP-II blocks NK cell migration [101]. Finally, Kaposi sarcoma is enriched in patients that encode the activating KIRs KIR3DS1 and KIR2DS1 with their MHC class I ligands [102,103]. How these receptors support Kaposi sarcoma development, however, remains unclear. Nevertheless, these studies suggest that KSHV inhibits NK cell responses by both driving NK cell differentiation to diminish antiviral effector functions and by compromising the NK cell recognition of KSHV-infected cells.
5. Harnessing NK Cells against γ-Herpesvirus-Associated Pathologies
These previous studies indicate that some coinfections, possibly including malaria, CMV and KSHV, differentiate NK cells and thereby diminish the immune control of lytic EBV infection by NKG2A+KIR− NK cells. Indeed, the control of lytic EBV infection seems important to prevent virus-associated pathologies; not only IM, but abortive early lytic replication also seems required, at least for some EBV-associated malignancies [10,18,104,105,106,107,108,109]. Furthermore, some primary immunodeficiencies that predispose for EBV-associated lymphomas seem to also preferentially affect the T cell-mediated immune control of lytic EBV infection [110]. Thus, the inherent reactivity of NK cells against lytic EBV replication could be harnessed against EBV-associated pathologies.
Along these lines, KIR mismatching could recruit further differentiated NK cell subpopulations, in addition to NKG2A+KIR− NK cells, to the immune response against EBV-associated lymphomas [111,112,113]. In humanized mice, KIR ligand (HLA-C1, -C2 and -Bw4) mismatched mixed human immune compartment reconstitution generates an environment for EBV infection, in which some EBV-infected B cells cannot efficiently engage inhibitory KIRs on NK cells [114]. Despite diminished NK cell licensing in trans, this leads to an improved immune control of inflammatory IM like EBV infection. Antibody-mediated NK cell depletion abolishes this improved immune control of mixed hematopoietic lineage reconstitution [114]. These findings suggest that KIR mismatched NK cells can target EBV-infected B cells, possibly via the engagement of the activating receptors NKG2D and DNAM-1 and in the absence of inhibitory KIR signaling.
The recognition of EBV-infected B cells by NK cells beyond lytic replication can be strengthened by providing additional activating receptors to these innate killers. Along these lines, chimeric antigen receptors (CARs) have been explored, which target surface molecules of B cells via a variable antibody fragment linked to T cell receptor- and costimulatory receptor-signaling domains [115,116,117,118]. Initial preclinical and clinical studies have focused on CD19-targeting CARs. In a phase I clinical trial, four of six patients with lymphoma experience completed remission without any signs of cytokine release syndrome (CRS) which is often common to CAR therapies [118]. Beyond CD19, CD22 and CD30 are now also explored for similar treatments [115,119]. Therefore, KIR mismatching might allow one to diminish inhibitory signaling, and CAR expression could enable NK cell-mediated targeting of EBV-infected B cells beyond lytic replication, in order to harness NK cell responses against EBV-associated malignancies.
In contrast to EBV and associated lymphomas, NK cells have not yet been harnessed for clinical use against KSHV-associated malignancies. However, NK cells have been described as killing KSHV-infected cells [120,121,122,123,124,125]. KSHV-mediated downregulation of MHC class I renders infected cells susceptible to NK cell recognition, but virus-induced ICAM-1 and B7-2 (CD86) suppression limits this recognition of PEL cells [124,125]. IFN-γ-mediated upregulation of these molecules might partially restore this recognition [126]. Therefore, IFN-γ-producing cytotoxic NK cells might prove useful for the therapeutic targeting of KSHV-associated malignancies, including PEL.
6. Conclusions and Outlook
Studying the human γ-herpesviruses EBV and KSHV reveals that they drive NK cells into distinct differentiation stages. While EBV expands early-differentiated CD56dimCD16+NKG2A+KIR− NK cells, KSHV coinfection leads to the accumulation of terminally differentiated CD56−CD16+NKG2A−KIR+CD39+CXCR6+ NK cells [66,90]. The mechanisms of this differential NK cell repertoire shaping should be further investigated in the future. They can teach us how to expand optimal NK cell populations for therapeutic applications, as is currently proposed for CMV-driven adaptive NK cell expansions [76,127]. They can then be further improved by transgenically expressing additional activations and deletions of inhibitory receptors to target malignancies that are caused by human γ-herpesviruses and tumors in general.
Funding
Research in the author’s laboratory is funded by Cancer Research Switzerland (KFS-4091-02-2017 and KFS-4962-02-2020), KFSP-PrecisionMS and HMZ ImmunoTargET of the University of Zurich, the Cancer Research Center Zurich, the Vontobel Foundation, the Sobek Foundation, the Swiss Vaccine Research Institute, Roche, Novartis, and the Swiss National Science Foundation (310030B_182827, 310030L_197952/1 and CRSII5_180323).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable. No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The author declares no conflict of interest.
References
- Shannon-Lowe, C.; Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and biological studies on a virus in cultured lymphoblasts from Burkitt’s lymphoma. J. Exp. Med. 1964, 121, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Kulwichit, W.; Edwards, R.H.; Davenport, E.M.; Baskar, J.F.; Godfrey, V.; Raab-Traub, N. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 11963–11968. [Google Scholar] [CrossRef]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. Embo. J. 1996, 15, 3117–3126. [Google Scholar] [CrossRef]
- Sin, S.H.; Kim, Y.; Eason, A.; Dittmer, D.P. KSHV latency locus cooperates with myc to drive lymphoma in mice. PLoS Pathog. 2015, 11, e1005135. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92. [Google Scholar] [CrossRef]
- Münz, C. Latency and lytic replication in the oncogenesis of the Epstein Barr virus. Nat. Rev. Micobiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Babcock, J.G.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez White, B.E.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Wang, H.B.; Zhang, A.; Chen, M.L.; Fang, Z.X.; Dong, X.D.; Li, S.B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol 2018, 3, 1–8. [Google Scholar] [CrossRef]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Rämer, P.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV infection increases EBV-associated tumor formation in vivo via enhanced EBV lytic gene expression. Cell Host Microbe 2017, 22, 61–73.e7. [Google Scholar] [CrossRef] [PubMed]
- Blackbourn, D.J.; Lennette, E.; Klencke, B.; Moses, A.; Chandran, B.; Weinstein, M.; Glogau, R.G.; Witte, M.H.; Way, D.L.; Kutzkey, T.; et al. The restricted cellular host range of human herpesvirus 8. Aids 2000, 14, 1123–1133. [Google Scholar] [CrossRef]
- Faure, A.; Hayes, M.; Sugden, B. How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16519–16528. [Google Scholar] [CrossRef]
- Bigi, R.; Landis, J.T.; An, H.; Caro-Vegas, C.; Raab-Traub, N.; Dittmer, D.P. Epstein-Barr virus enhances genome maintenance of Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 2018, 115, E11379–E11387. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef]
- Labo, N.; Marshall, V.; Miley, W.; Davis, E.; McCann, B.; Stolka, K.B.; Ndom, P.; Hemingway-Foday, J.J.; Abassora, M.; Newton, R.; et al. Mutual detection of Kaposi’s sarcoma-associated herpesvirus and Epstein-Barr virus in blood and saliva of Cameroonians with and without Kaposi’s sarcoma. Int. J. Cancer 2019, 145, 2468–2477. [Google Scholar] [CrossRef]
- Sallah, N.; Miley, W.; Labo, N.; Carstensen, T.; Fatumo, S.; Gurdasani, D.; Pollard, M.O.; Dilthey, A.T.; Mentzer, A.J.; Marshall, V.; et al. Distinct genetic architectures and environmental factors associate with host response to the gamma2-herpesvirus infections. Nat. Commun. 2020, 11, 3849. [Google Scholar] [CrossRef]
- Mariggio, G.; Koch, S.; Schulz, T.F. Kaposi sarcoma herpesvirus pathogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160275. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.S.; Kaufmann, J.K.; Wies, E.; Naschberger, E.; Panteleev-Ivlev, J.; Schmidt, K.; Holzer, A.; Schmidt, M.; Chen, J.; Konig, S.; et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 2012, 18, 961–966. [Google Scholar] [CrossRef]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef]
- Totonchy, J.; Cesarman, E. Does persistent HIV replication explain continued lymphoma incidence in the era of effective antiretroviral therapy? Curr. Opin. Virol. 2016, 20, 71–77. [Google Scholar] [CrossRef]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [CrossRef]
- Mace, E.M.; Hsu, A.P.; Monaco-Shawver, L.; Makedonas, G.; Rosen, J.B.; Dropulic, L.; Cohen, J.I.; Frenkel, E.P.; Bagwell, J.C.; Sullivan, J.L.; et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56bright subset. Blood 2013, 121, 2669–2677. [Google Scholar] [CrossRef]
- Mace, E.M.; Orange, J.S. Emerging insights into human health and NK cell biology from the study of NK cell deficiencies. Immunol. Rev. 2019, 287, 202–225. [Google Scholar] [CrossRef] [PubMed]
- Eidenschenk, C.; Dunne, J.; Jouanguy, E.; Fourlinnie, C.; Gineau, L.; Bacq, D.; McMahon, C.; Smith, O.; Casanova, J.L.; Abel, L.; et al. A novel primary immunodeficiency with specific natural-killer cell deficiency maps to the centromeric region of chromosome 8. Am. J. Hum. Genet. 2006, 78, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef]
- Mace, E.M.; Bigley, V.; Gunesch, J.T.; Chinn, I.K.; Angelo, L.S.; Care, M.A.; Maisuria, S.; Keller, M.D.; Togi, S.; Watkin, L.B.; et al. Biallelic mutations in IRF8 impair human NK cell maturation and function. J. Clin. Investig. 2017, 127, 306–320. [Google Scholar] [CrossRef]
- Latour, S.; Fischer, A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: Lessons from genetic diseases. Immunol. Rev. 2019, 291, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Latour, S. Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood 2020, 135, 644–655. [Google Scholar] [CrossRef]
- Damania, B.; Münz, C. Immunodeficiencies that predispose to pathologies by human oncogenic gamma-herpesviruses. FEMS Microbiol. Rev. 2019, 43, 181–192. [Google Scholar] [CrossRef]
- Katano, H.; Ali, M.A.; Patera, A.C.; Catalfamo, M.; Jaffe, E.S.; Kimura, H.; Dale, J.K.; Straus, S.E.; Cohen, J.I. Chronic active Epstein-Barr virus infection associated with mutations in perforin that impair its maturation. Blood 2004, 103, 1244–1252. [Google Scholar] [CrossRef]
- Rohr, J.; Beutel, K.; Maul-Pavicic, A.; Vraetz, T.; Thiel, J.; Warnatz, K.; Bondzio, I.; Gross-Wieltsch, U.; Schundeln, M.; Schutz, B.; et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica 2010, 95, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Niemela, J.E.; Stoddard, J.L.; Pittaluga, S.; Heslop, H.; Jaffe, E.S.; Dowdell, K. Late-onset severe chronic active EBV in a patient for five years with mutations in STXBP2 (MUNC18-2) and prf1 (perforin 1). J. Clin. Immunol. 2015, 35, 445–448. [Google Scholar] [CrossRef]
- Li, F.Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef]
- Chaigne-Delalande, B.; Li, F.Y.; O’Connor, G.M.; Lukacs, M.J.; Jiang, P.; Zheng, L.; Shatzer, A.; Biancalana, M.; Pittaluga, S.; Matthews, H.F.; et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science 2013, 341, 186–191. [Google Scholar] [CrossRef]
- Dhalla, F.; Murray, S.; Sadler, R.; Chaigne-Delalande, B.; Sadaoka, T.; Soilleux, E.; Uzel, G.; Miller, J.; Collins, G.P.; Hatton, C.S.; et al. Identification of a novel mutation in MAGT1 and progressive multifocal leucoencephalopathy in a 58-year-old man with XMEN disease. J. Clin. Immunol. 2015, 35, 112–118. [Google Scholar] [CrossRef]
- De Vries, E.; Koene, H.R.; Vossen, J.M.; Gratama, J.W.; von dem Borne, A.E.; Waaijer, J.L.; Haraldsson, A.; de Haas, M.; van Tol, M.J. Identification of an unusual Fc gamma receptor IIIa (cd16) on natural killer cells in a patient with recurrent infections. Blood 1996, 88, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Grier, J.T.; Forbes, L.R.; Monaco-Shawver, L.; Oshinsky, J.; Atkinson, T.P.; Moody, C.; Pandey, R.; Campbell, K.S.; Orange, J.S. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J. Clin. Investig. 2012, 122, 3769–3780. [Google Scholar] [CrossRef] [PubMed]
- Coffey, A.J.; Brooksbank, R.A.; Brandau, O.; Oohashi, T.; Howell, G.R.; Bye, J.M.; Cahn, A.P.; Durham, J.; Heath, P.; Wray, P.; et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat. Genet. 1998, 20, 129–135. [Google Scholar] [CrossRef]
- Nichols, K.E.; Harkin, D.P.; Levitz, S.; Krainer, M.; Kolquist, K.A.; Genovese, C.; Bernard, A.; Ferguson, M.; Zuo, L.; Snyder, E.; et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-linked lymphoproliferative-disease gene product sap regulates signals induced through the co-receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Sumegi, J.; Huang, D.; Lanyi, A.; Davis, J.D.; Seemayer, T.A.; Maeda, A.; Klein, G.; Seri, M.; Wakiguchi, H.; Purtilo, D.T.; et al. Correlation of mutations of the SH2D1A gene and Epstein-Barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood 2000, 96, 3118–3125. [Google Scholar]
- Booth, C.; Gilmour, K.C.; Veys, P.; Gennery, A.R.; Slatter, M.A.; Chapel, H.; Heath, P.T.; Steward, C.G.; Smith, O.; O’Meara, A.; et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: A multicenter study on the manifestations, management and outcome of the disease. Blood 2011, 117, 53–62. [Google Scholar] [CrossRef]
- Pachlopnik Schmid, J.; Canioni, D.; Moshous, D.; Touzot, F.; Mahlaoui, N.; Hauck, F.; Kanegane, H.; Lopez-Granados, E.; Mejstrikova, E.; Pellier, I.; et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood 2011, 117, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The switch from latent to productive infection in Epstein-Barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Jawahar, S.; Moody, C.; Chan, M.; Finberg, R.; Geha, R.; Chatila, T. Natural killer (NK) cell deficiency associated with an epitope-deficient Fc receptor type IIIa (CD16-II). Clin. Exp. Immunol. 1996, 103, 408–413. [Google Scholar] [CrossRef]
- Parolini, S.; Bottino, C.; Falco, M.; Augugliaro, R.; Giliani, S.; Franceschini, R.; Ochs, H.D.; Wolf, H.; Bonnefoy, J.Y.; Biassoni, R.; et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J. Exp. Med. 2000, 192, 337–346. [Google Scholar] [CrossRef]
- Bottino, C.; Falco, M.; Parolini, S.; Marcenaro, E.; Augugliaro, R.; Sivori, S.; Landi, E.; Biassoni, R.; Notarangelo, L.D.; Moretta, L.; et al. NTB-A, a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J. Exp. Med. 2001, 194, 235–246. [Google Scholar] [CrossRef]
- Brigida, I.; Chiriaco, M.; Di Cesare, S.; Cittaro, D.; Di Matteo, G.; Giannelli, S.; Lazarevic, D.; Zoccolillo, M.; Stupka, E.; Jenkner, A.; et al. Large deletion of MAGT1 gene in a patient with classic Kaposi sarcoma, CD4 lymphopenia, and EBV infection. J. Clin. Immunol. 2017, 37, 32–35. [Google Scholar] [CrossRef]
- Camcioglu, Y.; Picard, C.; Lacoste, V.; Dupuis, S.; Akcakaya, N.; Cokura, H.; Kaner, G.; Demirkesen, C.; Plancoulaine, S.; Emile, J.F.; et al. HHV-8-associated Kaposi sarcoma in a child with IFNgammaR1 deficiency. J. Pediatr. 2004, 144, 519–523. [Google Scholar] [CrossRef]
- Aavikko, M.; Kaasinen, E.; Nieminen, J.K.; Byun, M.; Donner, I.; Mancuso, R.; Ferrante, P.; Clerici, M.; Brambilla, L.; Tourlaki, A.; et al. Whole-genome sequencing identifies STAT4 as a putative susceptibility gene in classic Kaposi sarcoma. J. Infect. Dis. 2015, 211, 1842–1851. [Google Scholar] [CrossRef]
- Luzuriaga, K.; Sullivan, J.L. Infectious mononucleosis. N. Engl. J. Med. 2010, 362, 1993–2000. [Google Scholar] [CrossRef]
- Rostgaard, K.; Balfour, H.H., Jr.; Jarrett, R.; Erikstrup, C.; Pedersen, O.; Ullum, H.; Nielsen, L.P.; Voldstedlund, M.; Hjalgrim, H. Primary Epstein-Barr virus infection with and without infectious mononucleosis. PLoS ONE 2019, 14, e0226436. [Google Scholar] [CrossRef] [PubMed]
- Balfour, H.H., Jr.; Odumade, O.A.; Schmeling, D.O.; Mullan, B.D.; Ed, J.A.; Knight, J.A.; Vezina, H.E.; Thomas, W.; Hogquist, K.A. Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary Epstein-Barr virus infection in university students. J. Infect. Dis. 2013, 207, 80–88. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Grimm, J.M.; Schmeling, D.O.; Balfour, H.H., Jr.; Hogquist, K.A. The incubation period of primary Epstein-Barr virus infection: Viral dynamics and immunologic events. PLoS Pathog. 2015, 11, e1005286. [Google Scholar] [CrossRef] [PubMed]
- Callan, M.F.; Steven, N.; Krausa, P.; Wilson, J.D.; Moss, P.A.; Gillespie, G.M.; Bell, J.I.; Rickinson, A.B.; McMichael, A.J. Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat. Med. 1996, 2, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Callan, M.F.; Tan, L.; Annels, N.; Ogg, G.S.; Wilson, J.D.; O’Callaghan, C.A.; Steven, N.; McMichael, A.J.; Rickinson, A.B. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J. Exp. Med. 1998, 187, 1395–1402. [Google Scholar] [CrossRef]
- Williams, H.; McAulay, K.; Macsween, K.F.; Gallacher, N.J.; Higgins, C.D.; Harrison, N.; Swerdlow, A.J.; Crawford, D.H. The immune response to primary EBV infection: A role for natural killer cells. Br. J. Haematol. 2005, 129, 266–274. [Google Scholar] [CrossRef]
- Azzi, T.; Lunemann, A.; Murer, A.; Ueda, S.; Beziat, V.; Malmberg, K.J.; Staubli, G.; Gysin, C.; Berger, C.; Münz, C.; et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood 2014, 124, 2533–2543. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.P.; Azzi, T.; Hui, K.F.; Wong, A.M.G.; McHugh, D.; Caduff, N.; Chan, K.H.; Münz, C.; Chiang, A.K.S. Co-infection of cytomegalovirus and Epstein-Barr virus diminishes the frequency of CD56dimNKG2A+KIR− NK cells and contributes to suboptimal control of EBV in immunosuppressed children with post-transplant lymphoproliferative disorder. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Hendricks, D.W.; Balfour, H.H., Jr.; Dunmire, S.K.; Schmeling, D.O.; Hogquist, K.A.; Lanier, L.L. Cutting edge: NKG2ChiCD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein-Barr virus. J. Immunol. 2014, 192, 4492–4496. [Google Scholar] [CrossRef]
- Bjorkstrom, N.K.; Riese, P.; Heuts, F.; Andersson, S.; Fauriat, C.; Ivarsson, M.A.; Bjorklund, A.T.; Flodstrom-Tullberg, M.; Michaelsson, J.; Rottenberg, M.E.; et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 2010, 116, 3853–3864. [Google Scholar] [CrossRef]
- Ferlazzo, G.; Thomas, D.; Lin, S.L.; Goodman, K.; Morandi, B.; Muller, W.A.; Moretta, A.; Münz, C. The abundant NK cells in human lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J. Immunol. 2004, 172, 1455–1462. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 102, 3052–3057. [Google Scholar] [CrossRef]
- Guma, M.; Angulo, A.; Vilches, C.; Gomez-Lozano, N.; Malats, N.; Lopez-Botet, M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Budt, M.; Saez, A.; Brckalo, T.; Hengel, H.; Angulo, A.; Lopez-Botet, M. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 2006, 107, 3624–3631. [Google Scholar] [CrossRef] [PubMed]
- Beziat, V.; Dalgard, O.; Asselah, T.; Halfon, P.; Bedossa, P.; Boudifa, A.; Hervier, B.; Theodorou, I.; Martinot, M.; Debre, P.; et al. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur. J. Immunol. 2012, 42, 447–457. [Google Scholar] [CrossRef]
- Beziat, V.; Liu, L.L.; Malmberg, J.A.; Ivarsson, M.A.; Sohlberg, E.; Bjorklund, A.T.; Retiere, C.; Sverremark-Ekstrom, E.; Traherne, J.; Ljungman, P.; et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 2013, 121, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Landskron, J.; Ask, E.H.; Enqvist, M.; Sohlberg, E.; Traherne, J.A.; Hammer, Q.; Goodridge, J.P.; Larsson, S.; Jayaraman, J.; et al. Critical role of CD2 co-stimulation in adaptive natural killer cell responses revealed in NKG2C-deficient humans. Cell Rep. 2016, 15, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.R.; Quinn, L.L.; Rowe, M.; Zuo, J.M. Induction of the lytic cycle sensitizes Epstein-Barr virus-infected B cells to NK cell killing that is counteracted by virus-mediated NK cell evasion mechanisms in the late lytic cycle. J. Virol. 2016, 90, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Chijioke, O.; Muller, A.; Feederle, R.; Barros, M.H.; Krieg, C.; Emmel, V.; Marcenaro, E.; Leung, C.S.; Antsiferova, O.; Landtwing, V.; et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep. 2013, 5, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef]
- Strowig, T.; Chijioke, O.; Carrega, P.; Arrey, F.; Meixlsperger, S.; Ramer, P.C.; Ferlazzo, G.; Münz, C. Human NK cells of mice with reconstituted human immune system components require preactivation to acquire functional competence. Blood 2010, 116, 4158–4167. [Google Scholar] [CrossRef]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]
- Herndler-Brandstetter, D.; Shan, L.; Yao, Y.; Stecher, C.; Plajer, V.; Lietzenmayer, M.; Strowig, T.; de Zoete, M.R.; Palm, N.W.; Chen, J.; et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9626–E9634. [Google Scholar] [CrossRef]
- Sundstrom, Y.; Nilsson, C.; Lilja, G.; Karre, K.; Troye-Blomberg, M.; Berg, L. The expression of human natural killer cell receptors in early life. Scand. J. Immunol. 2007, 66, 335–344. [Google Scholar] [CrossRef]
- Forconi, C.S.; Cosgrove, C.P.; Saikumar-Lakshmi, P.; Nixon, C.E.; Foley, J.; Ong’echa, J.M.; Otieno, J.A.; Alter, G.; Münz, C.; Moormann, A.M. Poorly cytotoxic terminally differentiated CD56negCD16pos NK cells accumulate in Kenyan children with Burkitt lymphomas. Blood Adv. 2018, 2, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Forconi, C.S.; Oduor, C.I.; Oluoch, P.O.; Ong’echa, J.M.; Münz, C.; Bailey, J.A.; Moormann, A.M. A new hope for CD56negCD16pos NK cells as unconventional cytotoxic mediators: An adaptation to chronic diseases. Front. Cell Infect. Microbiol 2020, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Quintana, M.D.P.; Smith-Togobo, C.; Moormann, A.; Hviid, L. Endemic Burkitt lymphoma—An aggressive childhood cancer linked to Plasmodium falciparum exposure, but not to exposure to other malaria parasites. APMIS 2020, 128, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Digitale, J.C.; Callaway, P.C.; Martin, M.; Nelson, G.; Viard, M.; Rek, J.; Arinaitwe, E.; Dorsey, G.; Kamya, M.; Carrington, M.; et al. HLA alleles B*53:01 and C*06:02 are associated with higher risk of P. falciparum parasitemia in a cohort in Uganda. Front. Immunol. 2021, 12, 650028. [Google Scholar] [CrossRef]
- Digitale, J.C.; Callaway, P.C.; Martin, M.; Nelson, G.; Viard, M.; Rek, J.; Arinaitwe, E.; Dorsey, G.; Kamya, M.; Carrington, M.; et al. Inhibitory KIR ligands are associated with higher P. falciparum parasite prevalence. J. Infect. Dis. 2020, 12, 650028. [Google Scholar] [CrossRef]
- Oluoch, P.O.; Oduor, C.I.; Forconi, C.S.; Ong’echa, J.M.; Münz, C.; Dittmer, D.P.; Bailey, J.A.; Moormann, A.M. Kaposi sarcoma-associated herpesvirus infection and endemic Burkitt’s lymphoma. J. Infect. Dis. 2020, 222, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Caduff, N.; McHugh, D.; Rieble, L.; Forconi, C.S.; Ong’echa, J.M.; Oluoch, P.O.; Raykova, A.; Murer, A.; Böni, M.; Zuppiger, L.; et al. KSHV infection drives poorly cytotoxic cd56 negative natural killer cell differentiation in vivo upon KSHV/EBV dual infection. Cell Rep. 2021, 35, 109056. [Google Scholar] [CrossRef]
- Moesta, A.K.; Li, X.Y.; Smyth, M.J. Targeting CD39 in cancer. Nat. Rev. Immunol. 2020, 20, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Hemmatazad, H.; Berger, M.D. CCR5 is a potential therapeutic target for cancer. Expert Opin. Ther. Targets 2021. [Google Scholar] [CrossRef]
- Mukaida, N.; Sasaki, S.I.; Baba, T. CCL4 signaling in the tumor microenvironment. Adv. Exp. Med. Biol. 2020, 1231, 23–32. [Google Scholar]
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vely, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbe, C.; et al. PD-1 mediates functional exhaustion of activated nk cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, S.; Lambert, M.; Zucman, D.; Choukem, S.P.; Tognarelli, S.; Pages, C.; Lebbe, C.; Caillat-Zucman, S. Human herpesvirus 8 (HHV8) sequentially shapes the NK cell repertoire during the course of asymptomatic infection and Kaposi sarcoma. PLoS Pathog. 2012, 8, e1002486. [Google Scholar] [CrossRef]
- Gabaev, I.; Williamson, J.C.; Crozier, T.W.M.; Schulz, T.F.; Lehner, P.J. Quantitative proteomics analysis of lytic KSHV infection in human endothelial cells reveals targets of viral immune modulation. Cell Rep. 2020, 33, 108249. [Google Scholar] [CrossRef] [PubMed]
- Madrid, A.S.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus ORF54/dUTPase downregulates a ligand for the NK activating receptor NKp44. J. Virol. 2012, 86, 8693–8704. [Google Scholar] [CrossRef]
- Thomas, M.; Boname, J.M.; Field, S.; Nejentsev, S.; Salio, M.; Cerundolo, V.; Wills, M.; Lehner, P.J. Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 1656–1661. [Google Scholar] [CrossRef]
- Ishido, S.; Choi, J.K.; Lee, B.S.; Wang, C.; DeMaria, M.; Johnson, R.P.; Cohen, G.B.; Jung, J.U. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity 2000, 13, 365–374. [Google Scholar] [CrossRef]
- Bekerman, E.; Jeon, D.; Ardolino, M.; Coscoy, L. A role for host activation-induced cytidine deaminase in innate immune defense against KSHV. PLoS Pathog. 2013, 9, e1003748. [Google Scholar] [CrossRef]
- Yamin, R.; Kaynan, N.S.; Glasner, A.; Vitenshtein, A.; Tsukerman, P.; Bauman, Y.; Ophir, Y.; Elias, S.; Bar-On, Y.; Gur, C.; et al. The viral KSHV chemokine vMIP-II nhibits the migration of naive and activated human NK cells by antagonizing two distinct chemokine receptors. PLoS Pathog. 2013, 9, e1003568. [Google Scholar] [CrossRef]
- Guerini, F.R.; Mancuso, R.; Agostini, S.; Agliardi, C.; Zanzottera, M.; Hernis, A.; Tourlaki, A.; Calvo, M.G.; Bellinvia, M.; Brambilla, L.; et al. Activating KIR/HLA complexes in classic Kaposi’s sarcoma. Infect. Agent Cancer 2012, 7, 9. [Google Scholar] [CrossRef]
- Goedert, J.J.; Martin, M.P.; Vitale, F.; Lauria, C.; Whitby, D.; Qi, Y.; Gao, X.; Carrington, M. Risk of classic Kaposi sarcoma with combinations of killer immunoglobulin-like receptor and human leukocyte antigen loci: A population-based case-control study. J. Infect. Dis. 2016, 213, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef]
- Ma, S.D.; Yu, X.; Mertz, J.E.; Gumperz, J.E.; Reinheim, E.; Zhou, Y.; Tang, W.; Burlingham, W.J.; Gulley, M.L.; Kenney, S.C. An Epstein-Barr virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J. Virol. 2012, 86, 7976–7987. [Google Scholar] [CrossRef]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R.; et al. A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Lin, X.; Shumilov, A.; Bernhardt, K.; Feederle, R.; Poirey, R.; Kopp-Schneider, A.; Pereira, B.; Almeida, R.; Delecluse, H.J. The biological properties of different Epstein-Barr virus strains explain their association with various types of cancers. Oncotarget 2017, 8, 10238–10254. [Google Scholar] [CrossRef]
- Tsai, M.H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gartner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Münz, C.; Feederle, R.; et al. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef]
- Deng, Y.; Chatterjee, B.; Zens, K.; Zdimerova, H.; Müller, A.; Schuhmachers, P.; Ligeon, L.-A.; Bongiovanni, A.; Capaul, R.; Zbinden, A.; et al. CD27 is required for protective lytic EBV antigen specific CD8+ T cell expansion. Blood 2021, 137, 3225–3236. [Google Scholar] [CrossRef]
- Parham, P.; McQueen, K.L. Alloreactive killer cells: Hindrance and help for haematopoietic transplants. Nat. Rev. Immunol. 2003, 3, 108–122. [Google Scholar] [CrossRef]
- Rathmann, S.; Glatzel, S.; Schonberg, K.; Uhrberg, M.; Follo, M.; Schulz-Huotari, C.; Kaymer, M.; Veelken, H.; Finke, J.; Fisch, P. Expansion of NKG2A−LIR1− natural killer cells in HLA-matched, killer cell immunoglobulin-like receptors/HLA-ligand mismatched patients following hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2010, 16, 469–481. [Google Scholar] [CrossRef][Green Version]
- Bachanova, V.; Weisdorf, D.J.; Wang, T.; Marsh, S.G.E.; Trachtenberg, E.; Haagenson, M.D.; Spellman, S.R.; Ladner, M.; Guethlein, L.A.; Parham, P.; et al. Donor KIR B genotype improves progression-free survival of non-Hodgkin lymphoma patients receiving unrelated donor transplantation. Biol. Blood Marrow Transplant. 2016, 22, 1602–1607. [Google Scholar] [CrossRef]
- Landtwing, V.; Raykova, A.; Pezzino, G.; Beziat, V.; Marcenaro, E.; Graf, C.; Moretta, A.; Capaul, R.; Zbinden, A.; Ferlazzo, G.; et al. Cognate HLA absence in trans diminishes human NK cell education. J. Clin. Investig. 2016, 126, 3772–3782. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanaka, J. Recent advances in cellular therapy for malignant lymphoma. Cytotherapy 2021. [Google Scholar] [CrossRef]
- Chu, Y.; Yahr, A.; Huang, B.; Ayello, J.; Barth, M.; Cairo, M.S. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology 2017, 6, e1341031. [Google Scholar] [CrossRef]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Nunez Cortes, A.K.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Kerbauy, L.N.; Marin, N.D.; Kaplan, M.; Banerjee, P.P.; Berrien-Elliott, M.M.; Becker-Hapak, M.K.; Basar, R.; Foster, M.; Garcia Melo, L.; Neal, C.C.; et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated cord blood-derived NK cells facilitates CAR-like responses against CD30+ malignancies. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Matthews, N.C.; Goodier, M.R.; Robey, R.C.; Bower, M.; Gotch, F.M. Killing of Kaposi’s sarcoma-associated herpesvirus-infected fibroblasts during latent infection by activated natural killer cells. Eur. J. Immunol. 2011, 41, 1958–1968. [Google Scholar] [CrossRef]
- Sirianni, M.C.; Libi, F.; Campagna, M.; Rossi, D.; Capello, D.; Sciaranghella, G.; Carbone, A.; Simonelli, C.; Monini, P.; Gaidano, G.; et al. Downregulation of the major histocompatibility complex class I molecules by human herpesvirus type 8 and impaired natural killer cell activity in primary effusion lymphoma development. Br. J. Haematol. 2005, 130, 92–95. [Google Scholar] [CrossRef]
- Sirianni, M.C.; Vincenzi, L.; Topino, S.; Giovannetti, A.; Mazzetta, F.; Libi, F.; Scaramuzzi, D.; Andreoni, M.; Pinter, E.; Baccarini, S.; et al. NK cell activity controls human herpesvirus 8 latent infection and is restored upon highly active antiretroviral therapy in AIDS patients with regressing Kaposi’s sarcoma. Eur. J. Immunol. 2002, 32, 2711–2720. [Google Scholar] [CrossRef]
- Sirianni, M.C.; Campagna, M.; Scaramuzzi, D.; Carbonari, M.; Toschi, E.; Bacigalupo, I.; Monini, P.; Ensoli, B. Control of human herpes virus type 8-associated diseases by NK cells. Ann. N. Y. Acad. Sci. 2007, 1096, 37–43. [Google Scholar] [CrossRef]
- Davis, D.A.; Mishra, S.; Anagho, H.A.; Aisabor, A.I.; Shrestha, P.; Wang, V.; Takamatsu, Y.; Maeda, K.; Mitsuya, H.; Zeldis, J.B.; et al. Restoration of immune surface molecules in Kaposi sarcoma-associated herpes virus infected cells by lenalidomide and pomalidomide. Oncotarget 2017, 8, 50342–50358. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.; Davis, D.A.; Jaeger, H.K.; Stream, A.; Aisabor, A.I.; Yarchoan, R. Pomalidomide restores immune recognition of primary effusion lymphoma through upregulation of ICAM-1 and B7-2. PLoS Pathog. 2021, 17, e1009091. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, N.; Aoki, S.; Maruyama, S.; Kishi, K.; Takahashi, M.; Aizawa, Y. The heterogeneous expression of CD80, CD86 and other adhesion molecules on leukemia and lymphoma cells and their induction by interferon. J. Exp. Clin. Cancer Res. 1997, 16, 171–176. [Google Scholar]
- Malmberg, K.J.; Carlsten, M.; Bjorklund, A.; Sohlberg, E.; Bryceson, Y.T.; Ljunggren, H.G. Natural killer cell-mediated immunosurveillance of human cancer. Semin. Immunol. 2017, 31, 20–29. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).