
Detection of Alpha- and Betacoronaviruses in Miniopterus fuliginosus and Rousettus leschenaultii, two species of Sri Lankan Bats


 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
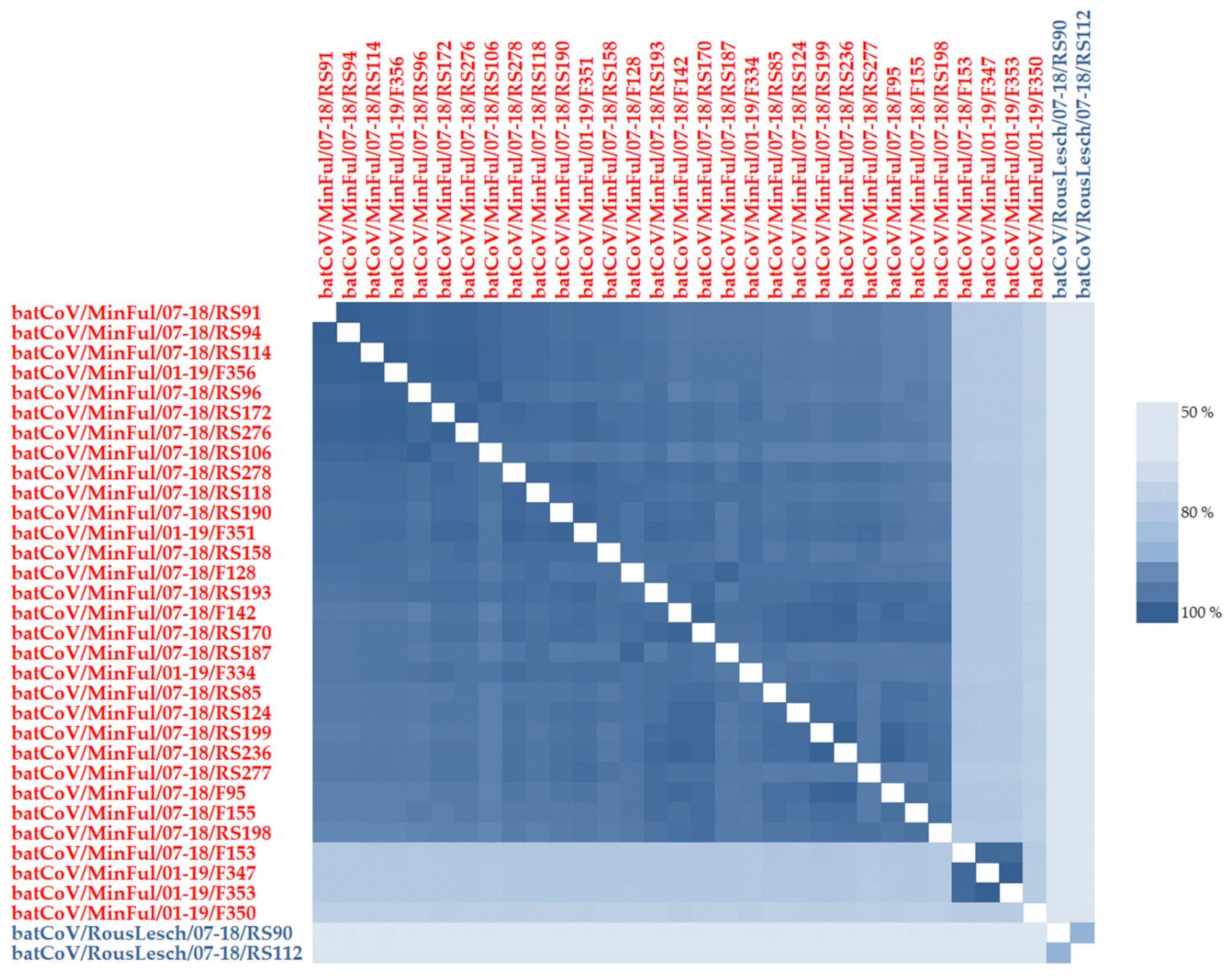
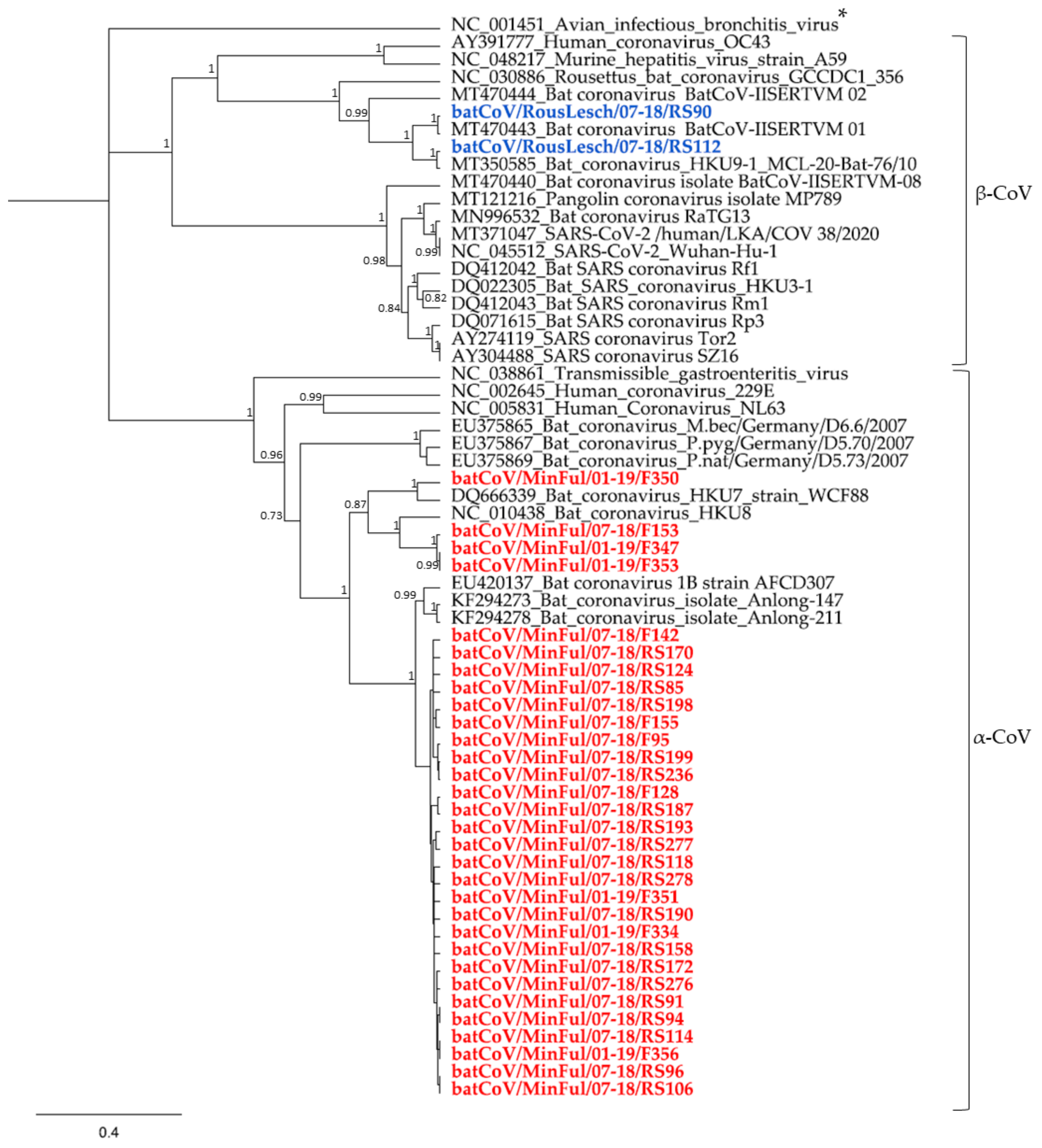
3. Results
4. Discussion
4.1. α-CoVs in Miniopterus fuliginosus Bat Samples
4.2. β-CoVs in Rousettus leschenaultii Bat Samples
4.3. Presumed Host Specificity of Bat CoVs
4.4. Evaluating the Risk of Viral Spillover to Humans
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, H.J.; Wen, H.L.; Zhou, C.M.; Chen, F.F.; Luo, L.M.; Liu, J.W.; Yu, X.J. Bats as Reservoirs of Severe Emerging Infectious Diseases. Virus Res. 2015, 205, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yapa, W.B.; Ratnasooriya, W.D. Ecology and Biology of Sri Lankan Bats. Univ. Colombo Rev. 2006, 1, 63–85. [Google Scholar]
- Saéz, A.M.; Weiss, S.; Nowak, K.; Lapeyre, V.; Zimmermann, F.; Düx, A.; Kühl, H.S.; Kaba, M.; Regnaut, S.; Merkel, K.; et al. Investigating the Zoonotic Origin of the West African Ebola Epidemic. EMBO Mol. Med. 2015, 7, 17–23. [Google Scholar] [CrossRef]
- Martini, G.A.; Knauff, H.G.; Schmidt, H.A.; Mayer, G.; Baltzer, G. A Hitherto Unknown Infectious Disease Contracted from Monkeys. “Marburg-Virus” Disease. Ger. Med. Mon. 1968, 13, 457–470. [Google Scholar]
- Wild, T.F. Henipaviruses: A New Family of Emerging Paramyxoviruses. Pathol. Biol. 2009, 57, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global Epidemiology of Bat Coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Poudel, U.; Subedi, D.; Pantha, S.; Dhakal, S. Animal Coronaviruses and Coronavirus Disease 2019: Lesson for One Health Approach. Open Vet. J. 2020, 10, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Valitutto, M.T.; Aung, O.; Tun, K.Y.N.; Vodzak, M.E.; Zimmerman, D.; Yu, J.H.; Win, Y.T.; Maw, M.T.; Thein, W.Z.; Win, H.H.; et al. Detection of Novel Coronaviruses in Bats in Myanmar. PLoS ONE 2020, 15, e0230802. [Google Scholar] [CrossRef]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and Cross-Species Transmission of Bat Coronaviruses in China. Nat. Commun. 2020, 11, 4235. [Google Scholar] [CrossRef]
- Amaratunga, D.; Fernando, N.; Haigh, R.; Jayasinghe, N. The COVID-19 Outbreak in Sri Lanka: A synoptic analysis focusing on trends, impacts, risks and science-policy interaction processes. Prog. Disaster Sci. 2020, 8, 100133. [Google Scholar] [CrossRef]
- Yapa, W.B. A Field Guide to the Bats of Sri Lanka, 1st ed.; Dilmah Conservation: Nawalapitiya, Sri Lanka, 2017; pp. 8, 55, 109. [Google Scholar]
- Kudagammana, H.D.W.S.; Thevanesam, V.; Chu, D.K.W.; Eriyagama, N.B.; Peiris, J.S.M.; Noordeen, F. Coronaviruses in Guano from Pteropus medius Bats in Peradeniya, Sri Lanka. Transbound. Emerg. Dis. 2018, 65, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Wibbelt, G.; Kurth, A.; Yasmum, N.; Bannert, M.; Nagel, S.; Nitsche, A.; Ehlers, B. Discovery of Herpesviruses in Bats. J. Gen. Virol. 2007, 88, 2651–2655. [Google Scholar] [CrossRef] [PubMed]
- De Souza Luna, L.K.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; et al. Generic Detection of Coronaviruses and Differentiation at the Prototype Strain Level by Reverse Transcription-PCR and Nonfluorescent Low-Density Microarray. J. Clin. Microbiol. 2007, 45, 1049–1052. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian Inference of Phylogenetic Trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Peiris, J.S.M.; Chen, H.; Guan, Y.; Poon, L.L.M. Genomic Characterizations of Bat Coronaviruses (1A, 1B and HKU8) and Evidence for Co-Infections in Miniopterus Bats. J. Gen. Virol. 2008, 89, 1282–1287. [Google Scholar] [CrossRef]
- Lin, X.-D.; Wang, W.; Hao, Z.-Y.; Wang, Z.-X.; Guo, W.-P.; Guan, X.-Q.; Wang, M.-R.; Wang, H.-W.; Zhou, R.-H.; Li, M.-H.; et al. Extensive Diversity of Coronaviruses in Bats from China. Virology 2017, 507, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.D.; Shete-Aich, A.; Nyayanit, D.A.; Pardeshi, P.; Majumdar, T.; Balasubramanian, R.; Ullas, P.T.; Mohandas, S.; Dighe, H.; Sawant, P.; et al. Detection of Coronaviruses in Pteropus & Rousettus Species of Bats from Different States of India. Indian J. Med. Res. 2020, 151, 226–235. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A Bat-Derived Putative Cross-Family Recombinant Coronavirus with a Reovirus Gene. PLoS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef]
- Obameso, J.O.; Li, H.; Jia, H.; Han, M.; Zhu, S.; Huang, C.; Zhao, Y.; Zhao, M.; Bai, Y.; Yuan, F.; et al. The Persistent Prevalence and Evolution of Cross-Family Recombinant Coronavirus GCCDC1 among a Bat Population: A Two-Year Follow-Up. Sci. China Life Sci. 2017, 60, 1357–1363. [Google Scholar] [CrossRef]
- Lim, X.F.; Lee, C.B.; Pascoe, S.M.; How, C.B.; Chan, S.; Tan, J.H.; Yang, X.; Zhou, P.; Shi, Z.; Sessions, O.M.; et al. Detection and Characterization of a Novel Bat-Borne Coronavirus in Singapore Using Multiple Molecular Approaches. J. Gen. Virol. 2019, 100, 1363–1374. [Google Scholar] [CrossRef]
- Subudhi, S.; Rapin, N.; Misra, V. Immune System Modulation and Viral Persistence in Bats: Understanding Viral Spillover. Viruses 2019, 11, 192. [Google Scholar] [CrossRef]
- Plowright, R.K.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; McFarlane, R.A.; Martin, G.; et al. Ecological Dynamics of Emerging Bat Virus Spillover. Proc. Biol. Sci. 2015, 282, 20142124. [Google Scholar] [CrossRef]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an Ancestral Association of Human Coronavirus 229E with Bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Li, K.S.M.; Tsang, A.K.L.; Shek, C.-T.; Wang, M.; Choi, G.K.Y.; Guo, R.; Wong, B.H.L.; Poon, R.W.S.; Lam, C.S.F.; et al. Recent Transmission of a Novel Alphacoronavirus, Bat Coronavirus HKU10, from Leschenault’s Rousettes to Pomona Leaf-Nosed Bats: First Evidence of Interspecies Transmission of Coronavirus between Bats of Different Suborders. J. Virol. 2012, 86, 11906–11918. [Google Scholar] [CrossRef] [PubMed]
- Grange, Z.L.; Goldstein, T.; Johnson, C.K.; Anthony, S.; Gilardi, K.; Daszak, P.; Olival, K.J.; O’Rourke, T.; Murray, S.; Olson, S.H.; et al. Ranking the Risk of Animal-to-Human Spillover for Newly Discovered Viruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2002324118. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Ge, X.; Wang, L.-F.; Shi, Z. Bat Origin of Human Coronaviruses. Virol. J. 2015, 12, 221. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Genus | March 2018 | June 2018 | January 2019 | Sampled Bats in Total | |||
|---|---|---|---|---|---|---|---|
| Rectal swabs | Feces | Rectal swabs | Feces | Rectal swabs | Feces | ||
| Miniopterus | 0/3 | 0/0 | 20/115 | 5/76 | 0/4 | 6/27 | 31/225 |
| Rousettus | 0/8 | 0/2 | 2/11 | 0/0 | 0/16 | 0/3 | 2/40 |
| Hipposideros | 0/3 | 0/0 | 0/1 | 0/0 | 0/16 | 0/7 | 0/27 |
| Rhinolophus | 0/62 | 0/9 | 0/0 | 0/0 | 0/16 | 0/17 | 0/104 |
| Total samples per session | 76 | 11 | 127 | 76 | 52 | 54 | |
| Sample | Species | Date | Sex | Forearm Length (cm) | Bat CoV Description | Accession Number |
|---|---|---|---|---|---|---|
| RS85 | M. fuliginosus | 07/07/18 | m | 4.76 | batCoV/MinFul/07-18/RS85 | MW987547 |
| RS90 | R. leschenaultii | 07/07/18 | f | 5.67 | batCoV/RousLesch/07-18/RS90 | MW987539 |
| RS91 | M. fuliginosus | 07/07/18 | f | 4.55 | batCoV/MinFul/07-18/RS91 | MW987548 |
| RS94 | M. fuliginosus | 08/07/18 | m | 4.53 | batCoV/MinFul/07-18/RS94 | MW987549 |
| RS96 | M. fuliginosus | 07/07/18 | f | 4.49 | batCoV/MinFul/07-18/RS96 | MW987554 |
| RS106 | M. fuliginosus | 07/07/18 | f | 4.54 | batCoV/MinFul/07-18/RS106 | MW987555 |
| RS112 | R. leschenaultii | 07/07/18 | f | n.a. | batCoV/RousLesch/07-18/RS112 | MW987540 |
| RS114 | M. fuliginosus | 07/07/18 | f | 4.65 | batCoV/MinFul/07-18/RS114 | MW987550 |
| RS118 | M. fuliginosus | 07/07/18 | f | 4.65 | batCoV/MinFul/07-18/RS118 | MW987556 |
| RS124 | M. fuliginosus | 07/07/18 | f | 4.65 | batCoV/MinFul/07-18/RS124 | MW987546 |
| RS158 | M. fuliginosus | 08/07/18 | f | 4.41 | batCoV/MinFul/07-18/RS158 | MW987566 |
| RS170 | M. fuliginosus | 08/07/18 | f | 4.91 | batCoV/MinFul/07-18/RS170 | MW987545 |
| RS172 | M. fuliginosus | 08/07/18 | f | 4.64 | batCoV/MinFul/07-18/RS172 | MW987552 |
| RS187 | M. fuliginosus | 08/07/18 | f | 4.61 | batCoV/MinFul/07-18/RS187 | MW987563 |
| RS190 | M. fuliginosus | 08/07/18 | m | 4.65 | batCoV/MinFul/07-18/RS190 | MW987559 |
| RS193 | M. fuliginosus | 08/07/18 | f | 4.58 | batCoV/MinFul/07-18/RS193 | MW987560 |
| RS198 | M. fuliginosus | 08/07/18 | f | 4.54 | batCoV/MinFul/07-18/RS198 | MW987564 |
| RS199 | M. fuliginosus | 08/07/18 | f | 4.51 | batCoV/MinFul/07-18/RS199 | MW987542 |
| RS236 | M. fuliginosus | 09/07/18 | f | 4.67 | batCoV/MinFul/07-18/RS236 | MW987543 |
| RS276 | M. fuliginosus | 10/07/18 | f | 4.45 | batCoV/MinFul/07-18/RS276 | MW987553 |
| RS277 | M. fuliginosus | 10/07/18 | m | 4.65 | batCoV/MinFul/07-18/RS277 | MW987561 |
| RS278 | M. fuliginosus | 10/07/18 | f | 4.41 | batCoV/MinFul/07-18/RS278 | MW987557 |
| F95 | M. fuliginosus | 07/07/18 | f | 4.38 | batCoV/MinFul/07-18/F95 | MW987541 |
| F128 | M. fuliginosus | 07/07/18 | m | 4.55 | batCoV/MinFul/07-18/F128 | MW987562 |
| F142 | M. fuliginosus | 07/07/18 | f | 4.69 | batCoV/MinFul/07-18/F142 | MW987544 |
| F153 | M. fuliginosus | 07/07/18 | m | 4.59 | batCoV/MinFul/07-18/F153 | MW987568 |
| F155 | M. fuliginosus | 08/07/18 | f | n.a. | batCoV/MinFul/07-18/F155 | MW987567 |
| F334 | M. fuliginosus | 23/01/19 | m | 4.65 | batCoV/MinFul/01-19/F334 | MW987565 |
| F347 | M. fuliginosus | 23/01/19 | f | 4.76 | batCoV/MinFul/01-19/F347 | MW987569 |
| F350 | M. fuliginosus | 23/01/19 | f | 4.51 | batCoV/MinFul/01-19/F350 | MW987571 |
| F351 | M. fuliginosus | 23/01/19 | m | 4.55 | batCoV/MinFul/01-19/F351 | MW987558 |
| F353 | M. fuliginosus | 23/01/19 | m | 4.58 | batCoV/MinFul/01-19/F353 | MW987570 |
| F356 | M. fuliginosus | 23/01/19 | m | 4.56 | batCoV/MinFul/01-19/F356 | MW987551 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muzeniek, T.; Perera, T.; Siriwardana, S.; Bas, D.; Kaplan, F.; Öruc, M.; Becker-Ziaja, B.; Schwarz, F.; Premawansa, G.; Premawansa, S.; et al. Detection of Alpha- and Betacoronaviruses in Miniopterus fuliginosus and Rousettus leschenaultii, two species of Sri Lankan Bats. Vaccines 2021, 9, 650. https://doi.org/10.3390/vaccines9060650
Muzeniek T, Perera T, Siriwardana S, Bas D, Kaplan F, Öruc M, Becker-Ziaja B, Schwarz F, Premawansa G, Premawansa S, et al. Detection of Alpha- and Betacoronaviruses in Miniopterus fuliginosus and Rousettus leschenaultii, two species of Sri Lankan Bats. Vaccines. 2021; 9(6):650. https://doi.org/10.3390/vaccines9060650
Chicago/Turabian StyleMuzeniek, Therese, Thejanee Perera, Sahan Siriwardana, Dilara Bas, Fatimanur Kaplan, Mizgin Öruc, Beate Becker-Ziaja, Franziska Schwarz, Gayani Premawansa, Sunil Premawansa, and et al. 2021. "Detection of Alpha- and Betacoronaviruses in Miniopterus fuliginosus and Rousettus leschenaultii, two species of Sri Lankan Bats" Vaccines 9, no. 6: 650. https://doi.org/10.3390/vaccines9060650
APA StyleMuzeniek, T., Perera, T., Siriwardana, S., Bas, D., Kaplan, F., Öruc, M., Becker-Ziaja, B., Schwarz, F., Premawansa, G., Premawansa, S., Perera, I., Yapa, W., Nitsche, A., & Kohl, C. (2021). Detection of Alpha- and Betacoronaviruses in Miniopterus fuliginosus and Rousettus leschenaultii, two species of Sri Lankan Bats. Vaccines, 9(6), 650. https://doi.org/10.3390/vaccines9060650