
Differentiation of Capripox Viruses by Nanopore Sequencing

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
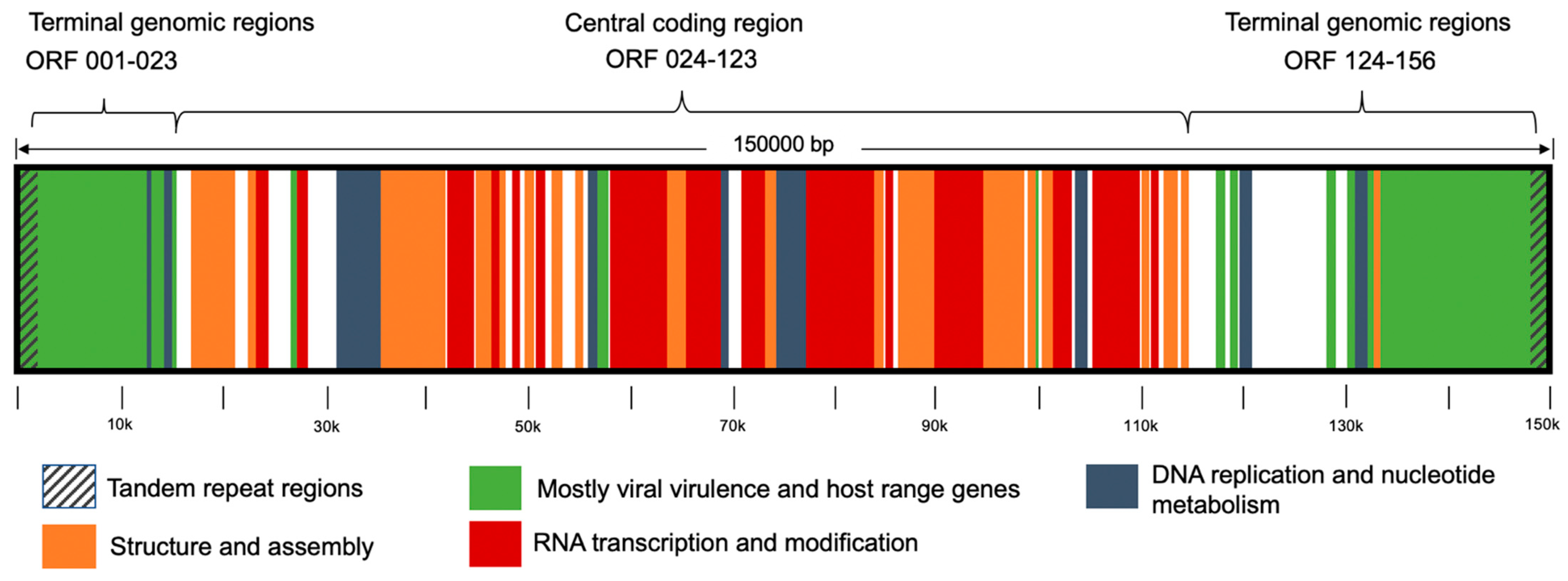
2.1. Viral DNA
2.2. Sample Preparation and Extraction
2.3. Library Preparation and Sequencing
2.4. Offline Database and Data Processing
2.5. Influence of Background
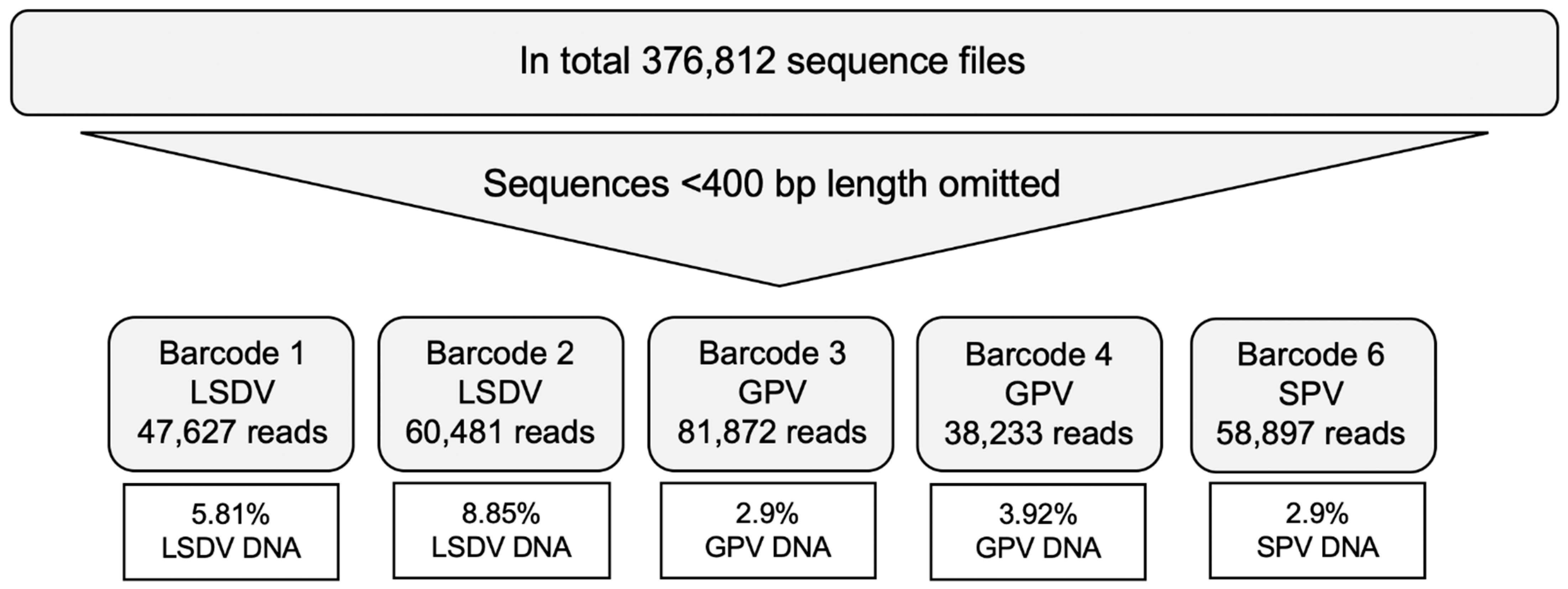
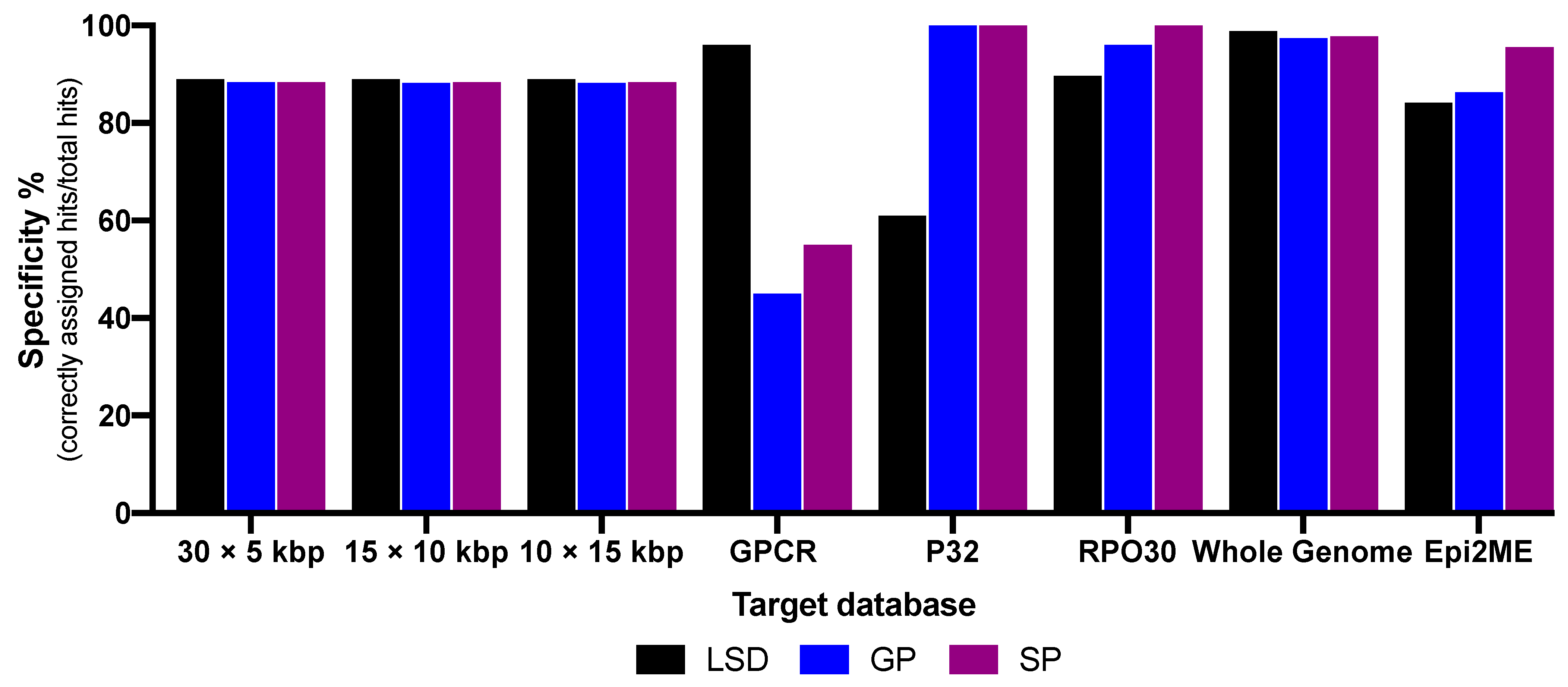
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedrich-Loeffler-Institut, F.L.I. National Reference Laboratory for Sheep and Goat Pox and National Reference Laboratory for Lumpy-skin-Disease (LSD). Available online: https://www.fli.de/en/institutes/institute-of-diagnostic-virology-ivd/reference-laboratories/nrl-for-sheep-and-goat-pox/ (accessed on 28 August 2020).
- World Organisation for Animal Health. Lumpy Skin Disease (LSD). Available online: https://www.oie.int/fileadmin/Home/eng/Media_Center/docs/pdf/Posters/EN_Poster_LSD_2016.pdf (accessed on 28 August 2020).
- Al-Salihi, K.A.; Hassan, I.Q. Lumpy Skin Disease in Iraq: Study of the Disease Emergence. Transbound Emerg. Dis. 2015, 62, 457–462. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. Scientific report on lumpy skin disease II. Data collection and analysis. EFSA J. 2018, 16, 5176. [Google Scholar]
- Services, G.O.F.V. Overview on Vector-borne diseases in Egypt. Available online: https://rr-africa.oie.int/wp-content/uploads/2020/12/15-egypt-overview-on-vector-borne-diseases-in-egypt.pdf (accessed on 8 January 2021).
- Kitching, R.P.; Mellor, P.S. Insect transmission of capripoxvirus. Res. Vet. Sci 1986, 40, 255–258. [Google Scholar] [CrossRef]
- Kahana-Sutin, E.; Klement, E.; Lensky, I.; Gottlieb, Y. High relative abundance of the stable fly Stomoxys calcitrans is associated with lumpy skin disease outbreaks in Israeli dairy farms. Med. Vet. Entomol. 2017, 31, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Chihota, C.M.; Rennie, L.F.; Kitching, R.P.; Mellor, P.S. Mechanical transmission of lumpy skin disease virus by Aedes aegypti (Diptera: Culicidae). Epidemiol. Infect. 2001, 126, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.; Lubinga, J.C.; Stoltsz, W.H.; Troskie, M.; Carpenter, S.T.; Coetzer, J.A.; Venter, E.H.; Oura, C.A. Mechanical transmission of lumpy skin disease virus by Rhipicephalus appendiculatus male ticks. Epidemiol. Infect. 2013, 141, 425–430. [Google Scholar] [CrossRef]
- Tuppurainen, E.S.; Lubinga, J.C.; Stoltsz, W.H.; Troskie, M.; Carpenter, S.T.; Coetzer, J.A.; Venter, E.H.; Oura, C.A. Evidence of vertical transmission of lumpy skin disease virus in Rhipicephalus decoloratus ticks. Ticks Tick Borne Dis. 2013, 4, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.G. Lumpy skin disease, an African capripox virus disease of cattle. Br. Vet. J. 1991, 147, 489–503. [Google Scholar] [CrossRef]
- Molla, W.; de Jong, M.C.M.; Gari, G.; Frankena, K. Economic impact of lumpy skin disease and cost effectiveness of vaccination for the control of outbreaks in Ethiopia. Prev. Vet. Med. 2017, 147, 100–107. [Google Scholar] [CrossRef]
- Garner, M.G.; Sawarkar, S.D.; Brett, E.K.; Edwards, J.R.; Kulkarni, V.B.; Boyle, D.B.; Singh, S.N. The extent and impact of sheep pox and goat pox in the state of Maharashtra, India. Trop Anim. Health Prod. 2000, 32, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Carn, V.M. Control of capripoxvirus infections. Vaccine 1993, 11, 1275–1279. [Google Scholar] [CrossRef]
- Tulman, E.R.; Afonso, C.L.; Lu, Z.; Zsak, L.; Kutish, G.F.; Rock, D.L. Genome of lumpy skin disease virus. J. Virol. 2001, 75, 7122–7130. [Google Scholar] [CrossRef]
- Tulman, E.R.; Afonso, C.L.; Lu, Z.; Zsak, L.; Sur, J.H.; Sandybaev, N.T.; Kerembekova, U.Z.; Zaitsev, V.L.; Kutish, G.F.; Rock, D.L. The genomes of sheeppox and goatpox viruses. J. Virol. 2002, 76, 6054–6061. [Google Scholar] [CrossRef] [PubMed]
- Capstick, P.B. Lumpy skin disease-experimental infection. Bul. Epiz Dis. Afr. 1959, 7, 51–62. [Google Scholar]
- Tuppurainen, E.S.; Pearson, C.R.; Bachanek-Bankowska, K.; Knowles, N.J.; Amareen, S.; Frost, L.; Henstock, M.R.; Lamien, C.E.; Diallo, A.; Mertens, P.P. Characterization of sheep pox virus vaccine for cattle against lumpy skin disease virus. Antivir. Res. 2014, 109, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.M.; Venter, E.H.; Shisler, J.L.; Gari, G.; Mekonnen, G.A.; Juleff, N.; Lyons, N.A.; De Clercq, K.; Upton, C.; Bowden, T.R.; et al. Review: Capripoxvirus Diseases: Current Status and Opportunities for Control. Transbound Emerg. Dis. 2017, 64, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Kitching, R.P.; Taylor, W.P. Clinical and antigenic relationship between isolates of sheep and goat pox viruses. Trop Anim. Health Prod. 1985, 17, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Santhamani, R.Y.R.; Venkatesan, G.; Shivachandra, S.B.; Pandey, A.B.; Ramakrishnan, M.A. Detection and differentiation of sheeppox virus and goatpox virus from clinical samples using 30 kDa RNA polymerase subunit (RPO30) gene based PCR. Vet. World 2013, 6, 923–925. [Google Scholar] [CrossRef]
- Mahmoud, M.A.; Khafagi, M.H. Detection, identification, and differentiation of sheep pox virus and goat pox virus from clinical cases in Giza Governorate, Egypt. Vet. World 2016, 9, 1445–1449. [Google Scholar] [CrossRef]
- Orlova, E.S.; Shcherbakova, A.V.; Diev, V.I.; Zakharov, V.M. Differentiation of capripoxvirus species and strains by polymerase chain reaction. Mol. Biol. 2006, 40, 158–164. [Google Scholar] [CrossRef]
- Lamien, C.E.; Lelenta, M.; Goger, W.; Silber, R.; Tuppurainen, E.; Matijevic, M.; Luckins, A.G.; Diallo, A. Real time PCR method for simultaneous detection, quantitation and differentiation of capripoxviruses. J. Virol. Methods 2011, 171, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, G.; Balamurugan, V.; Yogisharadhya, R.; Kumar, A.; Bhanuprakash, V. Differentiation of sheeppox and goatpox viruses by polymerase Chain reaction-restriction fragment length polymorphism. Virol. Sin. 2012, 27, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Hosamani, M.; Mondal, B.; Tembhurne, P.A.; Bandyopadhyay, S.K.; Singh, R.K.; Rasool, T.J. Differentiation of sheep pox and goat poxviruses by sequence analysis and PCR-RFLP of P32 gene. Virus Genes 2004, 29, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Lamien, C.E.; Lelenta, M.; Silber, R.; Goff, C.L.; Wallace, D.; Gulyaz, V.; Tuppurainen, E.; Luckins, A.G.; Albina, E.; Diallo, A. Phylogenetic analysis of the Capripox virus RPO30 gene and its use for development of a PCR for differentiating SPPV from GTPV. In Proceedings of the FAO/IAEA International Symposium on Sustainable Improvement of Animal Production and Health, Vienna, Austria, 8–11 June 2009; pp. 323–326. [Google Scholar]
- Agianniotaki, E.I.; Mathijs, E.; Vandenbussche, F.; Tasioudi, K.E.; Haegeman, A.; Iliadou, P.; Chaintoutis, S.C.; Dovas, C.I.; Van Borm, S.; Chondrokouki, E.D.; et al. Complete Genome Sequence of the Lumpy Skin Disease Virus Isolated from the First Reported Case in Greece. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Noyce, R.S.; Babiuk, L.A.; Lung, O.; Bulach, D.M.; Bowden, T.R.; Boyle, D.B.; Babiuk, S.; Evans, D.H. Extended sequencing of vaccine and wild-type capripoxvirus isolates provides insights into genes modulating virulence and host range. Transbound Emerg. Dis. 2020, 67, 80–97. [Google Scholar] [CrossRef]
- Lahens, N.F.; Ricciotti, E.; Smirnova, O.; Toorens, E.; Kim, E.J.; Baruzzo, G.; Hayer, K.E.; Ganguly, T.; Schug, J.; Grant, G.R. A comparison of Illumina and Ion Torrent sequencing platforms in the context of differential gene expression. BMC Genomics 2017, 18, 602. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Oxford-Nanopore-Technologies. Rapid Barcoding Sequencing (SQK-RBK004). Available online: https://store.nanoporetech.com/rapid-barcoding-kit.html (accessed on 30 January 2020).
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Abd El Wahed, A.; Weidmann, M.; Hufert, F.T. Diagnostics-in-a-Suitcase: Development of a portable and rapid assay for the detection of the emerging avian influenza A (H7N9) virus. J. Clin. Virol. 2015, 69, 16–21. [Google Scholar] [CrossRef]
- Moller, J.; Moritz, T.; Schlottau, K.; Krstevski, K.; Hoffmann, D.; Beer, M.; Hoffmann, B. Experimental lumpy skin disease virus infection of cattle: Comparison of a field strain and a vaccine strain. Arch. Virol. 2019, 164, 2931–2941. [Google Scholar] [CrossRef]
- Wolff, J.; King, J.; Moritz, T.; Pohlmann, A.; Hoffmann, D.; Beer, M.; Hoffmann, B. Experimental Infection and Genetic Characterization of Two Different Capripox Virus Isolates in Small Ruminants. Viruses 2020, 12, 1098. [Google Scholar] [CrossRef]
- Shalaby, M.A.; El-Deeb, A.; El-Tholoth, M.; Hoffmann, D.; Czerny, C.P.; Hufert, F.T.; Weidmann, M.; Abd El Wahed, A. Recombinase polymerase amplification assay for rapid detection of lumpy skin disease virus. BMC Vet. Res. 2016, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Kononov, A.; Byadovskaya, O.; Kononova, S.; Yashin, R.; Zinyakov, N.; Mischenko, V.; Perevozchikova, N.; Sprygin, A. Detection of vaccine-like strains of lumpy skin disease virus in outbreaks in Russia. Arch. Virol. 2019, 164, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Menasherow, S.; Rubinstein-Giuni, M.; Kovtunenko, A.; Eyngor, Y.; Fridgut, O.; Rotenberg, D.; Khinich, Y.; Stram, Y. Development of an assay to differentiate between virulent and vaccine strains of lumpy skin disease virus (LSDV). J. Virol. Methods 2014, 199, 95–101. [Google Scholar] [CrossRef]
- Sprygin, A.; Pestova, Y.; Bjadovskaya, O.; Prutnikov, P.; Zinyakov, N.; Kononova, S.; Ruchnova, O.; Lozovoy, D.; Chvala, I.; Kononov, A. Evidence of recombination of vaccine strains of lumpy skin disease virus with field strains, causing disease. PLoS ONE 2020, 15, e0232584. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Protocol Time | Kit/Program | |
|---|---|---|---|
| Short | Long | ||
| Extraction | 1 h | QIAamp DNA Blood Mini Kit | |
| Library Preparation | 20 min | Rapid Barcoding Sequencing Kit | |
| Sequencing | 25 min | 12 h | MinION Flow Cell 9.4 |
| Data Processing | 15 min | 8 h | MinKNOW |
| Analysis | 5 min | 30 min | GENEIOUS |
| Total | 2 h 5 min | 22 h | |
| Barcode | 1 (LSD) | 2 (LSD) | 3 (GP) | 4 (GP) | 6 (SP) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Run time | Short (~2 h) | Long (22 h) | Short (~2 h) | Long (22 h) | Short (~2 h) | Long (22 h) | Short (~2 h) | Long (22 h) | Short (~2 h) | Long (22 h) |
| Total number of Reads | 1854 | 47,627 | 2174 | 60,481 | 3104 | 81,872 | 2158 | 38,233 | 2202 | 58,897 |
| Specificity in % Whole genome BLAST | 98.68 | 98.8 | 98.8 | 98.8 | 96.69 | 96.7 | 97.79 | 98 | 97.59 | 97.7 |
| Sample (Total Reads) | Egypt #1 (17,912) | Egypt #2 (10,652) | Egypt #3 (11,268) | Sudan #1 (30,529) | Sudan #2 (6396) |
|---|---|---|---|---|---|
| Whole genome | 95.08% LSDV | 90.67% LSDV | 74.19% LSDV 17.2% SPV | No results | |
| RPO30 | No results | No results | SPV (1 Hit) | ||
| WIMP | 95.88% LSDV | 84.54% LSDV | 77.58% LSDV 19.82% SPV | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eltom, K.H.; Althoff, A.C.; Hansen, S.; Böhlken-Fascher, S.; Yousif, A.; El-Sheikh, H.A.; ElWakeel, A.A.; Elgamal, M.A.; Mossa, H.M.; Aboul-Soud, E.A.; et al. Differentiation of Capripox Viruses by Nanopore Sequencing. Vaccines 2021, 9, 351. https://doi.org/10.3390/vaccines9040351
Eltom KH, Althoff AC, Hansen S, Böhlken-Fascher S, Yousif A, El-Sheikh HA, ElWakeel AA, Elgamal MA, Mossa HM, Aboul-Soud EA, et al. Differentiation of Capripox Viruses by Nanopore Sequencing. Vaccines. 2021; 9(4):351. https://doi.org/10.3390/vaccines9040351
Chicago/Turabian StyleEltom, Kamal H., Anna Christina Althoff, Sören Hansen, Susanne Böhlken-Fascher, Ausama Yousif, Hussein A. El-Sheikh, Ahmed A. ElWakeel, Mahmoud A. Elgamal, Hadeer M. Mossa, Emad A. Aboul-Soud, and et al. 2021. "Differentiation of Capripox Viruses by Nanopore Sequencing" Vaccines 9, no. 4: 351. https://doi.org/10.3390/vaccines9040351
APA StyleEltom, K. H., Althoff, A. C., Hansen, S., Böhlken-Fascher, S., Yousif, A., El-Sheikh, H. A., ElWakeel, A. A., Elgamal, M. A., Mossa, H. M., Aboul-Soud, E. A., Wolff, J., Korthase, C., Hoffmann, B., Adam, N. M., Abdelaziz, S. A., Shalaby, M. A., & Abd El Wahed, A. (2021). Differentiation of Capripox Viruses by Nanopore Sequencing. Vaccines, 9(4), 351. https://doi.org/10.3390/vaccines9040351