1. Introduction
Upon reinfection, the immune system faces a number of challenges. It needs to provide a rapid response via an adequate number of cells with high-affinity antigen receptors, and to further develop an immune arsenal against upcoming infections, even against variable pathogens. Immunological memory is divided into many levels to counteract such provocations. The innate immune system can retain epigenetic and transcriptional changes, resulting in altered immune responses on subsequent encounters [
1]. This phenomenon is called
trained innate immunity [
2], which is remarkably different from conventional immunological memory (
Table 1). Conventional immunological memory affects the repertoire of lymphocyte antigen receptors and defines our immune profile, as determined by our lifetime encounters with antigens. However, most of these encounters are silent and go unnoticed, triggering only the so-called natural immunity which we call
natural memory in this review. Cells responsible for this type of immunity are usually antigen experienced persistent cells with antigen receptors recognizing common environmental antigens. While
natural memory can provide a broad general defense, for survival, it is necessary to be prepared for pathogens that surpass this protection level. More focused protection is achieved by the generation of
effector memory cells that are more potent in the elimination of targets, and by
anticipatory memory cells that are more sensitive in the detection of specific dangerous targets. These goals are realized by increasing the frequency of antigen specific lymphocyte clones and by the polarization of the response via differentiation of lymphocytes. In the following sections, we briefly summarize the main aspects of trained immunity, but shall focus on the cells that contribute to the lymphocyte-based conventional memory, presenting the role of different B and T cell subsets and emphasizing the differences between the primary and secondary responses.
Active immunization seeks to activate immunological memory without causing pathological damage. Optimally, vaccines induce multiple levels of immunological memory. First, to provide rapid antibody response to directly block access to the pathogen and then if the pathogen breaks through the first line of defense, to activate anticipating memory T and B cells to protect against disease development. While we summarize the overlapping and collaborative steps of immune memory, we shall highlight the role of these different cell types and events in vaccination, taking into consideration the age-dependence of the immunological memory generation as well.
2. Trained Immunity
The term trained immunity refers to the phenomenon that occurs when, after exposure to a pathogen, the cells of the innate immune system (monocytes, macrophages, NK cells, etc.) are capable of mounting a heightened, nonspecific response against a broad range of pathogens in the case of secondary exposure [
3,
4]. In the initiation of trained immune phenotype, stimulation by various pattern recognition receptors results in genome wide epigenetic changes in these cells. This process leads to an altered transcriptional profile and the rewiring of the cellular metabolism [
4], shifting from oxidative phosphorylation towards glycolytic processes (Warburg effect) [
3]. Compared to their unexposed counterparts, these primed cells are more effective at eliminating infections both by direct (phagocytosis) and indirect (IL-1β, IL-6, TNF-α, and other cytokine production) mechanisms [
5]. The phenomena of trained immune responses were first described in monocytes, macrophages and NK cells; however, more recent studies have further expanded antigen nonspecific immune memory to other innate immune cells, such as polymorphonuclear leukocytes or innate lymphoid cells (ILCs) [
6]. The epigenetic and transcriptional changes associated with trained immunity can be detected up to one year [
3]. Since the half-life of leukocytes in the circulation is much shorter [
7], it is hypothesized that not only mature immune cells but also bone marrow stem- and progenitor cells can undergo “training” [
8].
Consequences on Vaccination
While there is no vaccine currently that directly aims to elicit a trained immune phenotype, it seems that we have been reaping the benefits of “training” through vaccination for decades. Indeed, the first evidence for the memory like properties of the innate immune system comes from vaccine studies [
3]. The Bacillus Calmette-Guerin (BCG) or oral polio vaccines, which contain live attenuated microbes, given at young age, have been shown to reduce infant mortality and morbidity to a greater extent than would be expected from vaccination with these pathogens alone [
9,
10]. Harnessing trained immunity may lead to the development of a new generation of prophylactic vaccines. Modulation of innate immune cells can enhance both specific and nonspecific immune responses. These responses may provide protection against infections with bystander pathogens during a functional period of trained immunity [
11].
3. Natural Memory
Specialized lymphocyte subsets, B1 cells and gamma-delta T cells (T
γδ), are responsible for natural memory. These cells appear early in ontogenesis, are capable of self-renewal, and are promiscuous in reactivity. These poly-specific cells preferentially recognize self-antigens and conserved microbial antigens and typically elicit a rapid or even an immediate response against a limited set of antigens. B1 cells secrete cross-reactive antibodies produced even prior to the first appearance of the pathogen (
Table 2).
Of the B lymphocytes B1b cells are the source of T-independent (TI) memory B lymphocytes in mice, producing low affinity antibodies in a constant, but inducible way. Nonmutated B1b lymphocytes may provide long lasting TI memory responses against the polysaccharide capsule of
Streptococcus pneumoniae [
12] and to
Borrelia hermsii [
13]. B1 cells can differentiate into both memory B cells [
12] and plasma cells [
14] and these memory responses can provide protection upon the transfer of these cells to immunodeficient hosts [
13].
A special population of the T lymphocytes, T
γδ, is programmed for effector functions very early in the thymus [
15]. There are two major groups of T
γδ, which produce IFNγ and IL-17A, and contribute to the protection against intracellular and extracellular pathogens, respectively. The skin, the gut and the reproductive tract are seeded by these cells early on in the developing fetus, thereby providing the first line of defense. Human T
γδ cells have limited specificity and generally use only two variable gene segments for their delta chains and are classified into Vδ1 and Vδ2 cells. Vδ1 T cells are found primarily at epithelial sites, while Vδ2 T cells are dominant among circulating Tγδ cells [
16]. Importantly, all naive Vδ2 cells disappear from the blood by 1 year of age and the non-naive cells show potent effector functions allowing rapid reactions to a limited group of recognized antigens [
17]. The restricted specificity and oligoclonality in the intestinal Vδ repertoire also indicates that these cells represent memory in the intestinal tract against various recurrent ligands [
18].
Consequences on Vaccination
Cells responsible for maintaining a natural, baseline protectivity establish threshold immunity against all available antigens. These cells possess self-renewing capability and are in a continuous minimal activations state, which also allows them to bypass conventional lymphocyte differentiation pathways. Thus, vaccination ideally induces the generation of memory cells that exceed this threshold immunity with regard to antigen elimination efficiency. Importantly, owing to the unique antigen-receptor signaling mechanisms in these cells, natural memory remains functional, even when effector memory cells appear and eliminate that particular antigen [
19,
20].
4. Overview of B Cell-Mediated Immune Response
Antibodies produced by plasma cells provide B-cell-mediated immune protection. First, short-lived plasma cells are formed upon contact with antigens and secrete low-affinity immunoglobulin M (IgM) antibodies for a few days, followed by apoptosis [
21,
22]. Not only can these cells play an important role against primary infections, but they also prevent reinfections in a short period of time. Different levels of B cell-mediated immunity are involved in the memory response. Soluble antibodies, produced by long-lived plasma cells (LLPC) as
effector memory cells, ensure the first lines of immune defense. Immunoglobulin A (IgA) secreted into mucosa can prevent the pathogen from entering the human body, while the appearance of other immunoglobulin isotypes (IgG, IgM) of antibodies in tissues or in the circulation can prevent pathogens from entering human cells, or mark invaders for destruction. LLPC producing immunoglobulin E (IgE) responses have evolved for protection against helminth parasites infections, but IgE is also a potent mediator of allergic diseases [
23]. Optimally high affinity, specific antibodies are secreted by LLPC even before the pathogen enters. This type of protection usually prevents infections (called sterilizing immunity) unless an extreme dose of the pathogen invades the host that goes beyond the effects of protective antibodies.
Memory B cells also contribute to B cell-mediated defense in reinfections, as
anticipatory memory cells. In contrast to LLPC, memory B cells express BCR on their cell surface and are therefore able to detect pathogens. Once encountered with antigens, these cells undergo clonal proliferation and mostly differentiate into short-lived plasma cells. This memory B cell-mediated response does not provide immediate protection, but is still significantly faster than the response of naive B cells. Memory B cells support plasma cell-mediated protection on at least three levels after reinfection. (1) The ability of these cells to proliferate and differentiate provides unlimited replenishment for the terminally differentiated, nondividing LLPC pool. (2) Memory B cells can undergo affinity maturation and this allows a further increase of the quality of the response. (3) These cells also provide flexibility in the B cell memory response, since low-affinity memory B cells may also be able to recognize altered pathogens. Both memory B cells and LLPC cells have a long lifespan, even without reinfection. The survival of the two cell populations is an independent process, LLPC may persist after elimination of peripheral B cells and are not sequentially replaced from the memory B cell pool [
24]. The adoptive transfer of any of these populations generates protection in the host upon reinfection [
25].
As most pathogens carry both protein and nonprotein epitopes, memory cells and LLPCs can be produced in three parallel ways: in a T-independent manner, and in a T-dependent manner either outside B-cell follicles or in the GCs of B-cell follicles. Theoretically, the B-cell response consists of successive stages: B1 cell-mediated innate immunity, marginal zone (MZ) B cells, extrafollicular and intrafollicular B cells, sequentially from lower affinity to higher affinity and faster to slower response. While these events are anatomically and functionally different, all of these cell types may contribute to both the production of antibodies and the formation of memory B cells. All of these effects can be considered during vaccination, it is presumably rational to find a balance between the induction of shorter and less focused responses versus long-lasting and sharply defined responses, taking into consideration the properties of the pathogen, of the host and also the epidemiology.
5. MZ B Cells and T-Independent B Cell Memory
B cells are able to recognize various antigen substances, but T cell activation is mostly restricted to peptide recognition. The TI B cellular response is mostly against saccharide or lipid antigens and is characterized by the rapid emergence of low-affinity antibodies. Optimal B cell memory response relies on the supportive signals of cognate helper T cells. Consequently, B cell memory develops primarily against protein antigens, but various evidence suggests the existence of effective memory responses even after the T-independent activation of B cells [
26,
27]. The formation of both TI long lived memory cells [
28] and plasma cells have been published [
14]. Hapten-specific memory and plasma cells survived for up to 100 days for both saccharides [
28,
29] and LPS [
30] antigens, even in T cell-deficient, and germinal center (GC)-deficient mice [
30]. Polysaccharide antigens of encapsulated bacteria are also used successfully in human vaccination. PNEUMOVAX® 23, the 23-valent anti-pneumococcal vaccine, provides protection in adults and children more than 2 years of age [
31,
32] and the protection against
Haemophilus influenzae type b is also effective due to antibodies against polysaccharide-type capsules [
33].
MZ B cells are complementary actors with B-1b cells in the humoral response to TI Ag. Vaccine-induced TI B-cell memory is likely to be mostly derived from MZ B-cells. The expansion of MZ B cells has been shown following immunization with pneumococcal polysaccharide vaccine [
34] and the lack of circulating MZ B cells makes humans highly susceptible to infection with
H. influenzae type b [
35]. Without T cell help, somatic hypermutation cannot take place, consequently, the amount of responding cells grows, instead of their affinity increasing in B1 and MZ cell-mediated memory response [
27].
Conventional B2 cells may also contribute to TI Ab responses, as B cell specificity develops in a random procedure. However, the function and survival of these classical B cells may be deterministically more dependent on T cell help. It is known that T-cell-derived signals, such as CD40 activation or IL-21 production are generally required for class switching, somatic hypermutation (SHM), and enhancement of B-cell proliferation. Therefore, the TI B cell response generally does not produce high affinity antibodies, is less intense, and is likely to be transient [
26,
28,
36].
An additional limitation of the T-independent response is that these reactions are more age-dependent. Development of MZ B cells does not occur under 2 years of age. Polysaccharide-based vaccines have had limited use in newborns and because the response of MZ B cells is primarily spleen-specific it was also missing in spleen-deficient individuals [
37], until the introduction of conjugate vaccines that circumvent T cell incompetence against saccharide antigens.
Consequences on Vaccination
Although most T-independent plasma cells are short-lived, protein-free antigens can also induce memory B cells and long-lived plasma cell formation such as it was evidenced by the usage of
S. pneumoniae [
38],
H. influenzae type b, or
Neisseria meningitidis polysaccharide vaccines [
39].
In the absence of protein epitopes or in immunocompromised individuals, this response may be of significant importance. However, after reinfection with complex antigens (which express both protein and nonprotein epitopes), high-affinity T-dependent memory B cells can outcompete T-independent memory B cells. Consequently, T-independent B cells generally have no or only a marginal role in the secondary response. In addition, negative regulatory feedback of antigen-specific immunoglobulins keeps TI memory B cells under control [
28]. Under very specific conditions, when the first immunization activates only saccharide-specific B cells (such as it can be in the case of
S. pneumoniae polysaccharide vaccines, PPSV23) the number of saccharide-specific B cells increases and thus these cells may dominate the secondary response even after immunization with complex antigens [
40].
Age related structural changes in the spleen and lymph nodes [
41] results in a substantial loss of marginal zone [
42] also in the elderly. The decline in the function of MZ B cells [
43] with age is less intense than the downregulation of T dependent response, so a polysaccharide vaccine PPSV23 is still used in the elderly over the age of 65 years [
44].
6. T-Dependent, Extrafollicular B Cell Activation
Adaptive immunity plays a dual role in the immune response, providing protection against current infections and creating memory cells to prepare the immune system for upcoming infections. Accordingly, T-dependent B cell activation occurs by two main mechanisms. The principal purpose of extrafollicular B cell activation is to generate a rapid response to eliminate the current pathogens, while intrafollicular, GC-related B cell responses primarily serve for long-term protection [
45,
46,
47,
48].
The first phase of T and B cell interaction begins at the junction of the T cell-zone and B cell follicles, where the activated B cells present the specific antigen to the T cells already activated by dendritic cells (DCs) [
47]. A few hundred short-lived plasma cells (plasmablasts) are typically formed in these foci within a few days after the first encounter with the antigen [
45]. Characteristically, high-affinity B cells from the available repertoire prefer extrafollicular plasmablasts differentiation [
49,
50] to provide protection rapidly, while the originally lower-affinity, but still antigen specific, B cells migrate to the follicles [
51,
52].
The interaction with T cells is also important for extrafollicular B cells, as it mediates support and induces the expression of activation-induced deaminase (AID) [
53], the enzyme regulates antibody class switching and SHM. Although these processes are not completely ruled out [
48], extrafollicular plasmablasts typically do not undergo SHM and affinity maturation [
45,
54]. In contrast, antibody class switching frequently occurs in these cells during the perifollicular proliferative phase [
49,
55].
Extrafollicular B cells are predominantly short-lived, their half-life is only a few days [
48], however, these B cells can also induce early memory B cell [
56] and LLPC differentiation [
49,
57]. These memory B cells develop independently of GC within a few days of the response [
58]. Like all conventional memory B cells, these cells are quiescent, long-lived cells and are able to produce larger amounts of antibodies during recall than naive B cells [
59]. Once differentiated, their survival is independent of T cells and the continuous presence of related antigens [
45,
60,
61].
Consequences on Vaccination
Since active immunization focuses only on future infections, extrafollicular reactions which aim to serve actual protection, have a limited role in vaccination. We have already mentioned in the T-independent memory section, that in the secondary response, high-affinity T-dependent memory B cells are able to outcompete lower affinity counterparts, including extrafollicular memory B cells. In general, extrafollicular memory B cells do not contribute significantly to the long term secondary antibody response [
45] unless GC formation is intensely blocked. Some infections (such as
Borrelia burgdorferi, and
Ehrlichia muris) have been reported to elicit an intense extrafollicular response while suppressing GCs in the meantime. SARS-CoV-2 infection also blocks T cell function in the GCs and consequently reduces GC formation with the accumulation of non-germinal-center-derived activated B cells [
62].
7. T Dependent B Cell Memory, Intrafollicular B Cell Activation
The follicular GC reaction is initiated by antigen-specific B cells and the infiltrating antigen-specific follicular helper T cells (T
FH) [
49,
50]. B cells here undergo affinity maturation during time-consuming reactions, resulting in a drastic increase in their affinity in the following weeks (
Figure 1).
In the primary response, natural immunity provides minimal protection within hours, which in turn determines the threshold of response. Marginal zone B cells (MZ) are able to react in a few days to produce low-affinity antibodies. The intense proliferation of naive T and B cells and their differentiation into effector cells, short-lived plasma cells (SLPC) and effector T cells (TEF), eliminates infections in about a week. Development of antigen specific long-lived cells (with white rim), memory B cells (BM), long-lived plasma cells (LLPC), central (TCM) and tissue-resident (TRM), and effector memory T cells (TEM) will be completed within weeks, and provides long-term protection.
In the secondary response, an increased number of antigen-specific cells are ready to initiate the response. High-affinity antibodies produced by LLPC induce an immediate response that may result in sterilizing immunity. Due to the optimal localization of TRM, an efficient T cell response develops immediately after antigen presentation. A relatively larger number of TEM cells enter from the circulation at the site of infection even in a day. High numbers of BM and TCM begin to proliferate within a few days in secondary lymphatic organs, resulting in quick replenishment of effector cells. The efficiency of the response is increased due to the higher affinity antibodies and their optimal isotype, as well as the increased reactivity of T cells. A new generation of long-lived cells may also develop, even in larger numbers and with higher affinity antibody production.
The absolute threshold of the B cell affinity for selection is unknown, selection for antigen binding depends rather on the relative affinity of competing clones [
63,
64]. Antigens in the form of immune complexes are localized on the surface of the FDC in the follicles [
65,
66], but antigen presentation to T cells requires BCR-mediated internalization of foreign antigens, processing into peptide fragments, and binding to MHC-class molecules on the B cell surface [
47,
67]. B cells with relatively higher affinity may consume all antigens or at least block access to antigen-rich sites of FDCs from other B cells. B cell–B cell interactions cannot be ruled out in GC, and as a result, higher-affinity B cells can rip off the antigen from those with lower affinity. The presentation of more MHC-peptide complexes to T
FH cells provides a competitive advantage with stronger feedback or a higher probability of survival [
63,
68] and these T-cell-mediated supportive signals are dominant in the positive selection of B cells [
66,
69]. The BCR stimulus alone, without successful antigen presentation, rather induces GC B cell apoptosis, whereas B cells capable of presenting antigen-derived peptides to T
FH receive survival and proliferation signals [
47]. Although T cell-derived signals determine the fate of competing clones, it should be noted that the number of antigen presented is directly proportional to the affinity of the BCR [
70,
71].
Interaction with cognate T cells activates CD40-induced signaling, which stimulates AID that controls somatic mutation and isotype switching. Selected high-affinity B cells undergo clonal proliferation while SHM occurs, resulting in different affinities of developing clones. New clones with partially different affinities may return and compete again for antigens and T cell help. While cells with higher affinity are selectively enriched in each cycle, cells with lower affinity are eliminated by apoptosis, consequently, B cell affinity increases in a stepwise fashion in each additional cycle in the GCs. At the end of the process, the affinity of GC B cells far exceeds the affinity of extrafollicularly activated B cells [
47]. The number of cycles and how they are controlled is currently unknown. Positively selected B cells can differentiate in three ways after interaction with T cells at the end of each cycle. These cells can return to start a new selection cycle for further diversification or can be exported from GC as LLPC or as B memory cells.
The B cells with the highest affinity differentiate primarily into LLPCs in the follicles, but it is still not clear what factors determine whether a B cell stays in the GC or exits as a memory cell. Class-switched memory cells do not differ from GCs B cells either in affinity or in the number of somatic mutations [
50].
Relatively low affinity clones of memory B cells may also survive to provide flexibility in the immune response. As a result, the sensitivity of memory B cells against a given antigen covers a broad spectrum of affinity from low affinity clones, typically expressing IgM, to cells expressing higher affinity IgG, thereby preparing the body to respond to mutations in pathogens [
72,
73].
After reinfection, basically, the same process takes place as in the primary response, but with higher speed and improved quality. The antibodies produced by LLPCs not only inactivate pathogens, but also facilitate the formation of immune complexes and thus the delivery of antigens to the FDCs [
74]. A large number of B memory cells have a competitive advantage over naive cells, and higher affinity clones (primarily expressing IgG) initiate short lived plasma cell differentiation for rapid protection. Lower affinity memory cells (typically with IgM expression) initiate cyclic responses in the newly formed GC, which results in sequentially higher affinity long-lived memory B cells and LLPCs after each repeated infection [
75,
76,
77].
Consequences on Vaccination
The majority of responding memory cells and LLPCs against T-dependent antigens are likely to be derived from GCs, and express BCR with higher affinity than naive B cells. Most vaccines use exclusively LLPC as correlates (for example toxoid induced immunization or conjugated vaccines, HPV, etc.) or LLPC together with memory B cells or memory T cells (among many others smallpox, influenza, varicella-zoster,
Bordetella pertussis, BCG) [
78,
79]. If the amount, effectiveness and localization of antibodies (mostly IgG and IgA) are sufficient to prevent the colonization of pathogens, vaccines result in sterilizing immunity such as vaccines against polio, rubella,
H. influenzae type b,
S. pneumoniae and others [
80].
It must be noted that elevated specific antibody levels do not automatically provide protection. While the neutralizing effect of antibodies is undoubtedly beneficial for vaccination, when an antibody associates with a pathogen without its functional inactivation, it can facilitate phagocytosis as an opsonin and consequently, the entry of the microbe into human cells, a phenomenon called “Antibody-Dependent Enhancement” or ADE. It has been found that antibodies and even vaccine-induced antibody responses, can have an adverse effect in the case of various infections [
81], such as influenza [
82] or SARS vaccinations [
83].
It is important to note that the rate of pathogen spreading can be deterministic in deciding whether memory B cells can be considered as effective correlates. The protection of fast-dividing pathogens relies on LLPC, while the spread of slower pathogens gives enough time for memory B cells to proliferate and differentiate, such as in the case of hepatitis B and probably of SARS-CoV-2 viruses [
84]. In the case of an extremely slow pathogen spread (such as rabies), there is sufficient time for a vaccine to elicit a primary response and in these cases, the active immune response may be protective even when given after infection [
85] (called post-exposure prophylaxis).
The memory B cells not only outcompete any naive cells, but reduce the minimal antigen requirements for initiating a response [
86]. These additional advantages of memory B cells over naive B cells are also exploited in booster injection used in most immunization procedures [
87]. The limited number of antigens in the vaccine does not necessarily trigger the formation of LLPC during the first exposure, since the generation of these cells requires high affinity interactions, but memory B cells formed in the meantime may sensitize the secondary response [
47].
Although the relationship between B cell affinity and the lifespan of memory B cells or LLPCs is not exactly known, we can assume that a response initiated with higher affinity clones results in a longer lifespan. Both the activation of the transcription factors required for the development of LLPC and the chance of achieving an optimal tissue environment (i.e., bone marrow) depend on BCR affinity [
88]. If this correlation is relevant in determining longevity, then after primary infection with a lower dose of the antigen, when only available naive B cells respond, relatively short-lived cells are formed. However secondary infection activates memory cells with already higher affinity, thus creating long-lived memory and plasma cells. As the selection process to create high affinity cells in GCs lasts for weeks, antigen persistence may be deterministic in affinity maturation, highlighting the importance of the depot effect of the adjuvant. However, chronic reactions can lead to lymphocyte depletion, so determining the optimal time window may be important for the development of ideal B-cell responses [
89,
90].
We must consider that most antigens express a number of different epitopes. Immunization with complex antigens leads to the activation and competition of B cells of different specificities [
63]. Repeated B cell responses against quickly changing pathogens such as influenza virus, result in a restriction to act only to unmodified epitopes, as B memory cells outcompete naive B cells that attack newly emerging epitopes. Accordingly, B-cell clones activated during the first infection may also dominate the response to upcoming influenza infections [
91]. By sequentially limiting the recognized targets, modified pathogen strains can eventually avoid B-cell memory response, a phenomenon called original antigenic sin [
91]. The emergence of new variations observed with SARS-CoV-2 raises concerns that similar processes may occur with this pathogen, especially for vaccines that focus on only one protein to induce protection. If different epitopes were chosen as dominant epitopes in different individuals, it might limit the spread of pathogens in the human population, but in the meantime might make it difficult to establish a widely effective vaccine.
8. Overview of T Cell-Mediated Immune Response
In addition to B cells, the other type of lymphocyte that expresses antigen receptors and contributes to memory responses is the T cell. Most T cells recognize the antigens presented by other cells. Glycolipids are represented by a class of CD1 molecules, while conventional T cells, which belong to either the CD8 or CD4 population, recognize MHCI and MHCII peptides of the major histocompatibility complex. Following activation, T cells act on host cells through direct cell–cell contact or secreted cytokines.
Antigen-specific naive T cells require antigen transport from the site of infection into the secondary lymphatic organs (SLO) and antigen presentation by activated/mature DCs. Consequently, the activation of innate immunity is a prerequisite for naive T cell activation, and T-dependent B cell activation. This initial contact between DCs and T cells depends on the expression of costimulatory molecules on stimulated DCs. Subsequently, naive T cells differentiate into effector T cells during clonal proliferation. It is important to note that costimulation is not required to activate effector T cells, so the effector phase of the response is not necessarily dependent on innate immune cells.
An important aspect of T cell activation for optimal vaccination is that intracellular pathogens must also be transported and presented in SLO, a process that is predominantly performed by DCs. Dead cells phagocytosed by DCs are the source of antigen, as direct infections of DCs are very rare. Following the phagocytosis of destroyed human cells, DCs are able to present the absorbed antigen by MHCI and MHCII molecules during so-called cross-presentation, which results in the activation of both cytotoxic and helper T cells.
Helper T cells are classified according to their functional characteristics. TH1, TH2, TH17 cells direct the immune response mainly against intracellular pathogens, multicellular parasites, and extracellular pathogens (bacteria and fungi), respectively, with distinctive cytokine production in each class. Certain helper T cell subsets have special functions, such as follicular helper T (T
FH) cells to interact with B cells [
92] or regulatory T cells to induce immune tolerance. Effective vaccination should mimic the proper TH subtype response generated by the target pathogen. In non-live vaccines, this is primarily determined by adjuvant-mediated activation of innate immunity.
As compared to the primary immune response, T cells show quantitative and qualitative differences during these recall responses. Memory T cells will remain overrepresented in the repertoire and this quantitative difference enhances the probability of an encounter with the antigen. During the primary response, chromatin modifications render activation-inducible genes ready for transcription in memory cells [
93,
94]. Thus, upon the secondary response, the differentiation of the cells is accompanied by polarization and changes in homing patterns, the cells will have preferentially accumulated in tissues where they are needed and will deploy cytokines and cytotoxic measures as needed, all leading towards faster and more effective immunity.
The development of memory cells is similar in each T cell type, so unless otherwise indicated, reference is made below to both CD4 subtypes and CD8 cells. T cell memory relies on many different T memory cell types. Gamma-delta T cells and effector T memory cells provide a faster rate of memory response compared to primary immunization, while central memory T cells, following clonal proliferation are the source of newly formed effector T cells during reinfection and also provide a degree of flexibility in the T cell memory response (
Figure 1).
9. The Effector Cells in T Cell Memory
The first line of defense provided by memory T cells is the immediate action towards the removal of target cells. This is brought about by cells that are continuously looking for targets and can instantly release cytokines or induce cell death: tissue resident memory T cells (T
RM) (systematically reviewed [
95,
96]), and the somewhat later acting effector memory T cells (T
EM).
9.1. Tissue Resident Memory T Cells
T
RM, unlike circulating memory cells, remains localized in peripheral tissues defined by the previous infections, by downregulating tissue egress receptors [
97] and expressing tissue specific adhesion molecules [
98]. T
RM cells have the same TCR repertoire as central memory T cells (T
CM), so it is not specificity but localization that makes this cell population unique [
96]. This is a heterogeneous population of cells owing to differences in localization (skin, mucosal sites, brain, and liver) and may show plasticity in their differentiation.
T
RM expresses effector molecules like granzyme B, TNFα and IFNγ thereby providing quick protection against invading pathogens [
99]. In general, the CD4
+ T
RM response has a lag-phase because secreted cytokines need time to take effect, while a CD8
+ T
RM response may represent quick cytotoxic action, if antigen specific cells are present in sufficiently large numbers. Low random migration of T
RM cells may considerably enhance the ability of the T
RM cells to scan their environment [
96].
T
RM cells can persist for long periods in the absence of the cognate antigen. The number of T
RM is limited in a given tissue, and T
RM cell populations of different specificities may also compete for niche, survival and growth factors [
96]. Repetitive stimulation may provide an advantage for antigen specific clones in this competition [
100]. Repeated infection can induce a local response where the in situ proliferation of T
RM generates a secondary T
RM population besides the cells differentiating from recruited cells [
101,
102], thus the regulation of local memory independently of central memory is possible. However, the overall limited number of T
RM cells may be critical, mainly against rapidly replicating pathogens in terms of direct effects. It can be hypothesized that the main function of T
RM is not to eliminate pathogens directly, but this accelerated antigen detection can facilitate the recruitment of circulating T
EM and T
CM cells by the induced cytokine production. Killing some infected cells may also accelerate cross-presentation by producing dead cells for DC-mediated phagocytosis, facilitating the antigen presentation of intracellular pathogens by DCs [
96,
103]. Parenterally administered vaccines, which dominate in current formulations, should be ideally enhanced in their ability to induce T
RM.
9.2. Effector Memory T Cells
The other population of memory T cells, which circulates in the blood and is present in nonlymphoid, peripheral tissues is the T
EM [
104]. T
EM cells, which are present in relatively large numbers but do not have proliferative potential, can exit into tissues (T
RM can also be important in determining the location of an exit) and survey peripheral tissues for a specific antigen, where they can quickly exert effector functions or return to the blood via lymphatics [
99]. T
EM cells lack the lymph node homing receptors [
105] and constitutively display effector functions such as IFNγ production and cytotoxic activity [
106].
TEM and TRM are equipped with the arsenal of cytokines and cytolytic mechanisms necessary to maintain a balance between the host and pathogens. Once an invasion has been averted and the immune response has contracted, these cells continue to survey the tissues and eliminate remaining or newly encountered pathogens. Should the target breach this barrier again, destroy tissues and multiply, these cells are not able to proliferate. This is when cells anticipating such events step in.
10. The Anticipatory Memory Cells in T Cell Memory
10.1. Central Memory T Cells
The second line of defense is formed by memory T cells that can quickly divide and differentiate into powerful effector cells. These cells possess a central phenotype, localizing to and recirculating between secondary lymphoid organs. T
CM are comparable to naive T cells with respect to their homing pattern and proliferative burst upon antigenic stimulation [
107]. However, T
CM are much more sensitive to activation stimuli owing to having been selected with high affinity TCR, changes in the composition and clustering of signaling molecules and epigenetic differences allowing rapid transcription of genes [
108]. Accordingly, it provides replenishment for T
EM cells relatively quickly following reinfection.
Another type of T cell with central phenotype is the follicular helper T cell (T
FH) (systematic reviews in [
109,
110]). Though these cells reside in the follicles of secondary lymphoid organs, where they provide signals to differentiating B cells, T
FH orchestrates GC reactions and are indispensable for TD memory B-cell responses. Like conventional memory cells, T
FH cells have a long lifespan and when transferred between mice in in vivo experiments, also provide memory for the recipients [
109,
110]. Two cooperating mechanisms provide long-term protection by these cells. First, T
FH cells are able to survive for up to 400 days after infection and also have self-renewing potential [
111,
112]. This subset is rapidly activated after reinfection and their differentiation into effector cells does not require antigen presentation by DCs, only by memory B cells [
109]. Second, T
FH cells may have a central memory phenotype and at certain stages of development they are also present in the circulation [
113,
114]. Circulating memory T
FH cells have a less T
FH-polarized phenotype than local memory T
FH cells [
114]. The T helper memory response is substantially plastic, with TH subtypes repolarizing to other populations upon antigen recall [
115]. Overall, T
FH cells maintain anticipatory immunological memory by helping the development of secondary B-cell response [
112], as discussed above.
10.2. Memory Stem T Cells
T
SCM are minimally differentiated, self-renewing, long-lived cells with significant reconstitution potential even after the disappearance of the antigen. These cells show phenotypic, distribution and recirculation pattern similarity to naive T cells, yet they are clonally expanded, also express several characteristic cell surface markers of memory cells and create a rapid response to antigen [
116]. T
SCM can be differentiated directly from naive cells by omitting the effector phase [
100]. T
SCM are generated during the immune response against various pathogens and are precursors of other memory T cells. T
SCM seems to have critical importance in maintaining life-long cellular immunity [
117,
118], thus ideally clinical vaccine formulations should be able to induce T
SCM along with other memory compartments, in order to induce long-lasting T-cell memory [
119].
10.3. Consequences on Vaccination
Most vaccines focus on the plasma cell response and the optimal production of neutralizing antibody. Unilateral overrepresentation of plasma cells during vaccination may obscure the beneficial or critical functions of other memory cells (
Table 3). The reaction of memory T cells as well as memory B cells is unable to provide immediate protection and cannot prevent infections, but these mechanisms are fast and effective enough to prevent the development of the disease, thereby protecting individuals and also protecting the population by reducing the duration of infection. T cell mediated protection at least partially correlates with vaccine efficacy in various vaccines such as in BCG, measles, varicella-zoster virus, rabies, and in many others [
78,
79].
Cytotoxic T cells are essential for immune protection against intracellular pathogens, and accordingly are important correlates in vaccination. For example, T-cell deficiency leads to severe and fatal disease in measles, while B-cell deficient people recover [
80]. As the cytotoxic T cell response begins with cross-presentation, vaccine derived antigen must be intracellular (as live, DNA or RNA vaccine) to be engulfed and properly presented by DCs. Among others, considerable long lasting cytotoxic T cell memory has been published following SARS-Cov-2 infection [
84], and at least in rhesus macaques T cells may contribute to the protection in collaboration with antibodies [
120]. Vaccine antigens that are not expressed intracellularly (e.g., killed pathogens or some subunit vaccines) are unable to activate cytotoxic T cells, and these cells cannot differentiate to memory cells during such vaccinations.
The dose of the antigen delivered during immunization is critical for both cytotoxic and helper T cell activation. High antigen concentrations can result in T cell exhaustion or clonal deletion of highly reactive T cells through activation-induced cell death (AICD). In contrast, a low antigen dose primarily leads to the expansion of high-affinity CD8 T cells [
121], while relatively weaker TCR signals also efficiently produce memory CD8 T cells [
122]. The selection, activation, and polarization of helper T cells also depend on the intensity of immunization [
123,
124].
The role of T
RM in initiating a secondary response highlights the importance of finding the optimal route of vaccine administration that results in the proliferation of T
RM cells in tissues where an upcoming infection may start. The longevity and reactivity of T
FH cells directly regulate the response of memory B cells, which may thus affect the development of the immune response during reinfection, but even during booster vaccination [
125]. The polarization of TH cells to different populations is mostly influenced by adjuvants, which in some cases may reduce the efficiency of the immune response, such as that reported for the acellular pertussis vaccine, where the mixed TH1/TH2-directed immune response is activated instead of the optimal TH17 [
126].
Effective T cell memory is particularly important against highly variable pathogens. While epitopes available for B cells (often on the surface of the pathogen) are generally frequently changed, peptides involved in antigen presentation may be derived from more conservative structural elements. Reinfection results in all T
RM, T
EM and T
CM activity recognizing mutation-free, conserved epitopes. These cells may also be reactive even after infection with newly emerging, antigen-shifted virus strains as was highlighted for example in influenza epidemics [
127].
T cell-related reactions, in parallel with or in combination with ADE, may contribute to vaccine-associated disease enhancement (VADE). Live vaccines such as dengue, RSV vaccines, or vaccines against coronaviruses may also increase susceptibility to viral infection by a mechanism that has not yet been fully elucidated. Vaccine-induced altered cytokine profile, inflammatory damage, TH2 activation, and subsequent eosinophil infiltration can be responsible for aberrant viral pathogenesis [
128,
129].
11. Age-Dependence of Immunological Memory
The development of the immune system starts already in utero, but it is after birth that exposure to the abundance of environmental antigens and danger signals initiates immunological memory formation. This cumulative phase of memory corresponds to the diversification and tuning of immune responses and goes on until early adulthood. Following decades of maintenance of immune function in general, memory efficacy and diversity start to wane, typically at the age of 65–70 years. Due to the distinct characteristics of early and late years of life, we briefly summarize the critical changes that characterize the immune memory response in neonates and in the elderly (
Figure 2).
Early in life the maternal immune memory provides protection by IgG and IgA transport and transfer. Memory cell levels are very low in neonates and only reach the intensity typical of adults around the age of 10–15 years. Weaker antigen receptor and costimulatory signals in the first years of life favor the development of anticipatory rather than effector memory. Memory efficacy and diversity start to wane, typically at the age of 65–70 years. After this age, the elderly respond poorly to new infections and memory response is also limited. The restricted repertoire of antigen specific cells primarily limits protection to recurrent infections.
12. Neonatal Immunity
Immune maturation continues into adolescence, but the most significant changes occur in the first years of life. The immune system of newborns is typically tolerogenic, which influences memory cell formation and vaccination at several points. Basically, the number of lymphocytes at birth is high [
130], but with unique characteristics in functionality in neonates. As there is no pathogenicity in the uterus, memory cell levels are very low and remain limited for years [
131].
Maternal immunological experience is transferred to a newborn by two means. IgG transfer from the mother to the fetus occurs during pregnancy across the placenta, while IgA is transferred to the neonatal gastrointestinal tract by breastfeeding. These antibodies not only protect but also direct the developing immune system towards target molecules, and are therefore expected to have a strong impact on the establishment of immunological memory of the offspring [
132].
Humoral immunity is impaired in neonates. Although B-cell levels are high at birth [
130] and the repertoire is quite diverse, most of the cells are still immature B cells (approximately 95% of the B cells) that only slowly transform into peripheral mature B cells [
133] and do not reach the adult level till childhood (5–10 years). Isotype switched and memory B cells are very rare in newborns and at 1 year of age, infant IgG levels are 70%, while IgA levels are even lower, only 30% of the adults [
133]. The T-independent B cell response is severely limited, among other things, because the peripheral zone of the spleen is not fully developed by the age of 2 years and the level of CD21 receptor (complement receptor 2) on B cells is reduced [
133,
134]. Germinal center reactions, which are critical for the long-term memory response, are highly compromised. Weaker B cell receptor-mediated signaling in naive B cells, incomplete help provided by follicular dendritic cells, and limited expansion of T
FH cells together result in suboptimal B cell activation in GCs [
135,
136].
The number of memory B cells increases slowly with age and reaches adult levels only in children aged 10–15 years [
137]. Thus, higher sensitivity of memory cells, i.e., higher affinity of memory B cells or lower dependence on costimulation of memory T cells, cannot be exploited in neonate vaccination. Weaker BCR and TCR dependent signaling results in the dominance of memory B cells formation versus plasma cells and insufficient survival factors available LLPC further exacerbate this phenomenon [
135].
At birth, the number of T cells is high and increases steadily for years, and then begins to decline until it reaches adult levels in older childhood [
130]. Although the T cell number is high, their function is inadequate. The functional activity of neonatal APCs is impaired, and lower expression of costimulatory molecules complicates the activation of naive T cells, rather enhances the possibility of an anergic T cell response [
138]. In addition, functional exhaustion is also characteristic of neonatal effector T cells [
139]. In neonates CD8 + T cells are less responsive and require a greater degree of antigen specific stimuli [
140]. In contrast, the activity of regulatory T cells is upregulated. These cells are present in large amounts in human cord blood and are highly tolerogenic in their function [
141,
142,
143]. This overall decrease in antigen-specific response complicates the formation of both memory B cells and memory T cells. Not only does the strength of the T cell response decrease, but the balance of the response is also strongly skewed toward Th2 polarization [
144]. Overall reduced IFN and IL-12 production leads to decreased TH1 differentiation, highlighting the importance of using adjuvants capable of regulating T cell polarization during vaccination [
131].
Consequences on Vaccination
Newborns are highly vulnerable to infectious diseases. It is therefore important to develop the most effective and safest vaccines possible for them. The highly tolerogenic immune system, decreased strength of antigen receptor signaling, lack of pre-existing memory cells, severely impaired GC function, TH imbalance, and the presence of the maternal neutralizing antibody all counteract successful vaccination. The need for early intervention and the suboptimal circumstances for vaccination require thoughtful strategies [
145].
Nonetheless, some vaccines are already suitable for newborns, and some, moreover, are the most effective in this age. An example is the BCG vaccine, which is one of the first and most widely used vaccines [
146]. BCG does not need to contain any exogenous adjuvants because the live attenuated strain of
Mycobacterium bovis induces immune responses via many PRRs [
147]. BCG induces a strong TH1 response demonstrating that decreased TH1 capacity in neonates is not absolute [
148]. There is growing evidence that BCG perhaps induces maturation of dendritic cells, affects TH cytokine balance and influences the heterologous trained immunity [
10]. Many studies suggest that early immunization with BCG may protect against, inter alia, leukemia, allergies, and childhood diabetes and that BCG elevates cytokine and antibody responses to other vaccine antigens (HBV and OPV) in neonates [
149,
150,
151]. The BCG vaccine is not only an example of a vaccine that can function under the tolerogenic conditions, typically present in newborns, but also of vaccines that can direct the maturation of the immune system.
A special challenge in the vaccination of newborns is the prevention of the transmission of pathogens from mother to infant [
152]. The HBV vaccine induces at least as much antibody production in neonates as in adults and gives a lifetime protection after a minimum of three doses. The HBV vaccine represents that subunit vaccines with adjuvant (aluminum salts) can effectively function in newborns while oral polio vaccine demonstrates active intestinal immune response and the potential success of mucosal vaccination [
145]. Extending maternal immunization, which has already been successful with various vaccines among others tetanus-diphtheria-pertussis, influenza, hepatitis B, could be a promising strategy to solve the problem of early infections [
153].
The special characteristic of newborn immunity has to be considered in vaccination, for example in T-cell independent B-cell response to polysaccharide antigens. The delay in the development of the peripheral zone of the spleen, decreased CD21 receptor levels on B cells, lower complement activity [
154], unbalanced cytokine profile [
155], and increased susceptibility to tolerance induction in neonates may also play a role in suboptimal reactions to encapsulated bacteria [
154,
155]. These mechanisms can be circumvented with conjugate vaccines that switch the reactions to T-dependent response.
Experience with these existing vaccines suggests that in spite of the restricted memory response neonates are able to develop efficient immune response, especially to live attenuated vaccines, but these immune responses depend on several factors, such as maternal Ab at the time of administration, breastfeeding, timing of postpartum vaccination, and geographical location [
133].
13. Immunity in the Elderly
The adaptive immune system undergoes numerous alterations with age and the last stage of life is characterized by relevant changes in the immune response. When the first exposure to an antigen occurs at an older age, a less effective immune response is induced. As a result, a weaker memory response is generated to new antigens and the success of vaccination is reduced in elderly. Long-lived memory cells (both T and B cells) do not just persist, but even protect the elderly, however, the diversity of the repertoire of memory cells narrows and their responsiveness also deteriorates [
122,
156,
157,
158,
159,
160].
In the bone marrow, the production of B cells decreases as internal changes appear that preclude commitment to the lymphoid line, IL-7 survival factor production decreases, the proliferation rate regresses and apoptotic capacity increases [
161]. The number of naive B cells in the periphery is also declining in older adults [
162]. In addition to the decrease in number, the function of aging B cells is also impaired. Antigen-induced plasma cell differentiation and antigen-specific antibody production are reduced [
163,
164], although total levels of serum antibodies are rather increased in the elderly [
165]. Internal defects in germinal centers, including decreased AID production studied in mice, have been identified, resulting in impaired mechanisms of both class-switching recombination CSR and SHM [
166,
167]. All of these processes limit the way memory B cells and LLPC are formed. As these changes have a lesser effect on TI B cells, the decrease in TI responses with age is less severe than that of TD responses [
44].
B memory cells accumulate in the periphery and the population of B cells in the periphery is saturated with memory B-cells over time. However, even in the presence of increased autoantibody levels, these memory B cells have limited repertoire diversity and exhibit paralyzed functions. A part of memory B cells differentiate to exhausted memory cells with poor ability to replicate and likely prone to apoptosis [
168]. Reduced function of T
FH cells, decreased production of AID and impaired immune-complex retention of FDC compromise memory B cell functions [
41,
122] and consequently limits optimal plasma cell formation [
165]. Further exacerbating the problem, the imperfect bone marrow environment is less conducive to LLPC survival [
169], accordingly, BM stores fewer plasma cells at old age [
163].
While the number of circulating T cells remains almost the same throughout life, there are decisive changes in their composition and reactivity. Due to the involution of the thymus, the development of T cells becomes independent of the thymus and a general decrease in the production and diversity of naive T cells occurs [
170]. The functionality of naive T cells was also impaired, including weaker TCR and costimulatory signals, reduced proliferative capacity and decreased IL-2 production [
171]. Three mechanisms compensate for the decrease in naive T cell production: (1) increased antigen-independent clonal expansion of naive T cells [
172]; (2) more naive T cells (both CD4 + and CD8 +) differentiate into “virtual” memory cells [
173,
174], which are cells with a longer lifespan but reduced antigen reactivity; (3) memory T cells are activated from time to time by recurrent viral infections (average of 8-12 recurrent pathogens). Intermittent activation and proliferation of pathogen-specific memory T cells impairs the balance of naive and memory T cells and can account for 50% of all CD4 T cells turning memory T cells [
175,
176]. All of these mechanisms result in a decrease in the diversity of T cells in the elderly, even a hundred-fold decrease in the diversity of CD4 T cells has been observed after the age of 70 years [
177]. Although the number of memory T cells increases with age, the diversity and reactivity of these cells also decrease, limiting the possibility of an effective response to some well-known antigens. It further limits the maintenance of T cell memory intensity that both primary and memory T cell responses generate short-lived effector T cells rather than memory precursor cells [
122]. In addition to cytokine production, the cytokine balance also changes with age. Th1 responses decrease, shifting toward Th2 cytokine predominance [
178]. In summary, the reactivity and plasticity of naive and memory T cells are reduced, limiting the possibility of developing a stronger immune response to an upcoming infection.
Consequences on Vaccination
Due to the aging of the immune system, the elderly are more vulnerable to infections, resulting in higher mortality and morbidity by pathogens. Changes in memory response also result in moderate vaccine efficacy in the elderly. For example knowing that long-lived memory T and B cells can provide effective protection against influenza infection that recurs after a long time, following influenza vaccination seroconversion (a fourfold increase in antibody titer) and protective hemagglutination inhibitor antibody titers is successfully produced in only 10–30% of the elderly, compared with 50–75% of the younger [
179]. Even when antibody levels rise, it has been observed that elevated levels do not persist for even a year [
180]. Crippled T cell response also manifested for example in suboptimal Th1 cells response in the lung [
181].
Changes not only in the intensity but also in the composition of the immune system may require new vaccination strategies, for example to avoid
S. pneumoniae infection [
181]. However, pneumococcal conjugate vaccine (PCV13) stimulates T cells, via conjugating the pneumococcus glycans to CRM197 (nontoxic mutant of diphtheria toxin) and accordingly this vaccine is used for people under 65 years of age [
182], but for adults ≥65 years of age the PPV23 is also recommended because the effectiveness of the T-independent response decreases less with age [
44].
Due to age-related immune system defects, the control of pathogen reactivation is also weakened, such as a latent varicella zoster infection can lead to herpes zoster disease. Limited susceptibility to vaccines makes it even more difficult to control such diseases at this age. Live attenuated virus vaccination against Varicella zoster virus, which is quite effective at age 60 (51%), is almost ineffective in older populations [
183]. Studies showed that the immune responses to the vaccine waned in 6-8 years [
184]. In summary, vaccines need to be improved to protect this highly vulnerable population through regular booster vaccinations, finding optimal doses and routes of administration or by innovative adjuvants and inventing new technologies.
14. Conclusions
Social alterations in humanity increase the global risk of pandemics, which demand more effective vaccination. As the scope of the article highlights, the memory response relies on a wide variety of cell populations, with their different localizations, affinities, reaction times, and flexibility. Although neutralizing antibody production is the only way to generate sterilizing immunity, other cells and other mechanisms of immunological memory can/should be considered during vaccination. The variety and variability of pathogens requires the plasticity of the responses used against them. In addition, the heterogeneity of the human population, in terms of age, immune status, and comorbidities, may necessitate the development of several vaccines against the same pathogen. These challenges require a more accurate understanding of the complex processes of immunological memory, all of which can make targeted approaches in vaccination.
Author Contributions
Conceptualization, J.P. and G.K.; Writing—Original Draft Preparation, Z.B., T.S., J.P., G.K.; Writing—Review and Editing, É.N., A.B., J.P. and G.K.; Visualization, J.P. and G.K.; Supervision, G.K. Funding Acquisition, A.B., J.P. and G.K. All authors have read and agreed to the published version of the manuscript.
Funding
The work/publication is supported by the GINOP-2.3.2-15-2016-00050 project. The project is cofinanced by the European Union and the European Regional Development Fund, the National Research, Development and Innovation Office—NKFIH K 125224 and VEKOP-2.1.1-15-2016-00098 are acknowledged for financial support of this work.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dominguez-Andres, J.; Netea, M.G. The specifics of innate immune memory. Science 2020, 368, 1052–1053. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P. Innate Immune Memory: Time for Adopting a Correct Terminology. Front. Immunol. 2018, 9, 799. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Riksen, N.P.; Netea, M.G. Be aware, innate immune cells remember. Aging 2018, 10, 2218–2219. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Benn, C.S.; Joosten, L.A.; Jacobs, C.; van Loenhout, J.; Xavier, R.J.; Aaby, P.; van der Meer, J.W.; et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J. Innate Immun. 2014, 6, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Latz, E.; Mills, K.H.; O’Neill, L.A. Innate immune memory: A paradigm shift in understanding host defense. Nat. Immunol. 2015, 16, 675–679. [Google Scholar] [CrossRef]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161.e12. [Google Scholar] [CrossRef]
- Garly, M.L.; Martins, C.L.; Bale, C.; Balde, M.A.; Hedegaard, K.L.; Gustafson, P.; Lisse, I.M.; Whittle, H.C.; Aaby, P. BCG scar and positive tuberculin reaction associated with reduced child mortality in West Africa. A non-specific beneficial effect of BCG? Vaccine 2003, 21, 2782–2790. [Google Scholar] [CrossRef]
- Pollard, A.J.; Finn, A.; Curtis, N. Non-specific effects of vaccines: Plausible and potentially important, but implications uncertain. Arch. Dis. Child. 2017, 102, 1077–1081. [Google Scholar] [CrossRef]
- Sanchez-Ramon, S.; Conejero, L.; Netea, M.G.; Sancho, D.; Palomares, O.; Subiza, J.L. Trained Immunity-Based Vaccines: A New Paradigm for the Development of Broad-Spectrum Anti-infectious Formulations. Front. Immunol. 2018, 9, 2936. [Google Scholar] [CrossRef]
- Haas, K.M.; Poe, J.C.; Steeber, D.A.; Tedder, T.F. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 2005, 23, 7–18. [Google Scholar] [CrossRef]
- Alugupalli, K.R.; Leong, J.M.; Woodland, R.T.; Muramatsu, M.; Honjo, T.; Gerstein, R.M. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity 2004, 21, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Taillardet, M.; Haffar, G.; Mondiere, P.; Asensio, M.J.; Gheit, H.; Burdin, N.; Defrance, T.; Genestier, L. The thymus-independent immunity conferred by a pneumococcal polysaccharide is mediated by long-lived plasma cells. Blood 2009, 114, 4432–4440. [Google Scholar] [CrossRef]
- Parker, M.E.; Ciofani, M. Regulation of gammadelta T Cell Effector Diversification in the Thymus. Front. Immunol. 2020, 11, 42. [Google Scholar] [CrossRef]
- Deusch, K.; Luling, F.; Reich, K.; Classen, M.; Wagner, H.; Pfeffer, K. A major fraction of human intraepithelial lymphocytes simultaneously expresses the gamma/delta T cell receptor, the CD8 accessory molecule and preferentially uses the V delta 1 gene segment. Eur. J. Immunol. 1991, 21, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, S.C.; Andrus, J.P.; Perfetto, S.P.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A.; Roederer, M. Ontogeny of gamma delta T cells in humans. J. Immunol. 2004, 172, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Holtmeier, W.; Witthoft, T.; Hennemann, A.; Winter, H.S.; Kagnoff, M.F. The TCR-delta repertoire in human intestine undergoes characteristic changes during fetal to adult development. J. Immunol. 1997, 158, 5632–5641. [Google Scholar] [PubMed]
- Ochsenbein, A.F.; Fehr, T.; Lutz, C.; Suter, M.; Brombacher, F.; Hengartner, H.; Zinkernagel, R.M. Control of early viral and bacterial distribution and disease by natural antibodies. Science 1999, 286, 2156–2159. [Google Scholar] [CrossRef] [PubMed]
- Jayasekera, J.P.; Moseman, E.A.; Carroll, M.C. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol. 2007, 81, 3487–3494. [Google Scholar] [CrossRef]
- Smith, K.G.; Hewitson, T.D.; Nossal, G.J.; Tarlinton, D.M. The phenotype and fate of the antibody-forming cells of the splenic foci. Eur. J. Immunol. 1996, 26, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Auner, H.W.; Beham-Schmid, C.; Dillon, N.; Sabbattini, P. The life span of short-lived plasma cells is partly determined by a block on activation of apoptotic caspases acting in combination with endoplasmic reticulum stress. Blood 2010, 116, 3445–3455. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.P.; Ma, E.G.M.; Aranda, C.J.; Curotto de Lafaille, M.A. Non-classical B Cell Memory of Allergic IgE Responses. Front. Immunol. 2019, 10, 715. [Google Scholar] [CrossRef]
- Hammarlund, E.; Thomas, A.; Amanna, I.J.; Holden, L.A.; Slayden, O.D.; Park, B.; Gao, L.; Slifka, M.K. Plasma cell survival in the absence of B cell memory. Nat. Commun. 2017, 8, 1781. [Google Scholar] [CrossRef]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral immunity due to long-lived plasma cells. Immunity 1998, 8, 363–372. [Google Scholar] [CrossRef]
- Bortnick, A.; Allman, D. What is and what should always have been: Long-lived plasma cells induced by T cell-independent antigens. J. Immunol. 2013, 190, 5913–5918. [Google Scholar] [CrossRef]
- Defrance, T.; Taillardet, M.; Genestier, L. T cell-independent B cell memory. Curr. Opin. Immunol. 2011, 23, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Obukhanych, T.V.; Nussenzweig, M.C. T-independent type II immune responses generate memory B cells. J. Exp. Med. 2006, 203, 305–310. [Google Scholar] [CrossRef]
- Hsu, M.C.; Toellner, K.M.; Vinuesa, C.G.; Maclennan, I.C. B cell clones that sustain long-term plasmablast growth in T-independent extrafollicular antibody responses. Proc. Natl. Acad. Sci. USA 2006, 103, 5905–5910. [Google Scholar] [CrossRef]
- Bortnick, A.; Chernova, I.; Quinn, W.J., 3rd; Mugnier, M.; Cancro, M.P.; Allman, D. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J. Immunol. 2012, 188, 5389–5396. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.L.; Hochman, M.; Goldblatt, D. Combined schedules of pneumococcal conjugate and polysaccharide vaccines: Is hyporesponsiveness an issue? Lancet Infect. Dis. 2007, 7, 597–606. [Google Scholar] [CrossRef]
- Daniels, C.C.; Rogers, P.D.; Shelton, C.M. A Review of Pneumococcal Vaccines: Current Polysaccharide Vaccine Recommendations and Future Protein Antigens. J. Pediatr. Pharmacol. Ther. 2016, 21, 27–35. [Google Scholar] [CrossRef]
- Kayhty, H.; Peltola, H.; Karanko, V.; Makela, P.H. The protective level of serum antibodies to the capsular polysaccharide of Haemophilus influenzae type b. J. Infect. Dis. 1983, 147, 1100. [Google Scholar] [CrossRef]
- Weller, S.; Braun, M.C.; Tan, B.K.; Rosenwald, A.; Cordier, C.; Conley, M.E.; Plebani, A.; Kumararatne, D.S.; Bonnet, D.; Tournilhac, O.; et al. Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 2004, 104, 3647–3654. [Google Scholar] [CrossRef]
- Zouali, M.; Richard, Y. Marginal zone B-cells, a gatekeeper of innate immunity. Front. Immunol. 2011, 2, 63. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, D.A.; Stavnezer, J. Enhanced IgA class switching in marginal zone and B1 B cells relative to follicular/B2 B cells. J. Immunol. 2006, 177, 6025–6029. [Google Scholar] [CrossRef] [PubMed]
- Breukels, M.A.; Zandvoort, A.; van Den Dobbelsteen, G.P.; van Den Muijsenberg, A.; Lodewijk, M.E.; Beurret, M.; Klok, P.A.; Timens, W.; Rijkers, G.T. Pneumococcal conjugate vaccines overcome splenic dependency of antibody response to pneumococcal polysaccharides. Infect. Immun. 2001, 69, 7583–7587. [Google Scholar] [CrossRef]
- Moberley, S.A.; Holden, J.; Tatham, D.P.; Andrews, R.M. Vaccines for preventing pneumococcal infection in adults. Cochrane Database Syst. Rev. 2008, CD000422. [Google Scholar] [CrossRef]
- Patel, M.; Lee, C.K. Polysaccharide vaccines for preventing serogroup A meningococcal meningitis. Cochrane Database Syst. Rev. 2001, CD001093. [Google Scholar] [CrossRef]
- Papadatou, I.; Tzovara, I.; Licciardi, P.V. The Role of Serotype-Specific Immunological Memory in Pneumococcal Vaccination: Current Knowledge and Future Prospects. Vaccines 2019, 7, 13. [Google Scholar] [CrossRef]
- Turner, V.M.; Mabbott, N.A. Influence of ageing on the microarchitecture of the spleen and lymph nodes. Biogerontology 2017, 18, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Cortegano, I.; Rodriguez, M.; Martin, I.; Prado, M.C.; Ruiz, C.; Hortiguela, R.; Alia, M.; Vilar, M.; Mira, H.; Cano, E.; et al. Altered marginal zone and innate-like B cells in aged senescence-accelerated SAMP8 mice with defective IgG1 responses. Cell Death Dis. 2017, 8, e3000. [Google Scholar] [CrossRef] [PubMed]
- Turner, V.M.; Mabbott, N.A. Ageing adversely affects the migration and function of marginal zone B cells. Immunology 2017, 151, 349–362. [Google Scholar] [CrossRef]
- Shi, Y.; Yamazaki, T.; Okubo, Y.; Uehara, Y.; Sugane, K.; Agematsu, K. Regulation of aged humoral immune defense against pneumococcal bacteria by IgM memory B cell. J. Immunol. 2005, 175, 3262–3267. [Google Scholar] [CrossRef] [PubMed]
- Takemori, T.; Kaji, T.; Takahashi, Y.; Shimoda, M.; Rajewsky, K. Generation of memory B cells inside and outside germinal centers. Eur. J. Immunol. 2014, 44, 1258–1264. [Google Scholar] [CrossRef]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- McHeyzer-Williams, M.; Okitsu, S.; Wang, N.; McHeyzer-Williams, L. Molecular programming of B cell memory. Nat. Rev. Immunol. 2011, 12, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.D.; Gatto, D.; Wood, K.; Camidge, T.; Basten, A.; Brink, R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J. Immunol. 2009, 183, 3139–3149. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Vogel, L.A.; Zhang, W.; Loo, W.; Shnider, D.; Lind, E.F.; Ratliff, M.; Noelle, R.J.; Erickson, L.D. Imprinting the fate of antigen-reactive B cells through the affinity of the B cell receptor. J. Immunol. 2006, 177, 7723–7732. [Google Scholar] [CrossRef]
- Paus, D.; Phan, T.G.; Chan, T.D.; Gardam, S.; Basten, A.; Brink, R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 2006, 203, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, Y.R.; Batista, F.D. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 2007, 27, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Stuber, E.; Strober, W. The T cell-B cell interaction via OX40-OX40L is necessary for the T cell-dependent humoral immune response. J. Exp. Med. 1996, 183, 979–989. [Google Scholar] [CrossRef]
- MacLennan, I.C.; Toellner, K.M.; Cunningham, A.F.; Serre, K.; Sze, D.M.; Zuniga, E.; Cook, M.C.; Vinuesa, C.G. Extrafollicular antibody responses. Immunol. Rev. 2003, 194, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Pape, K.A.; Kouskoff, V.; Nemazee, D.; Tang, H.L.; Cyster, J.G.; Tze, L.E.; Hippen, K.L.; Behrens, T.W.; Jenkins, M.K. Visualization of the genesis and fate of isotype-switched B cells during a primary immune response. J. Exp. Med. 2003, 197, 1677–1687. [Google Scholar] [CrossRef]
- Toyama, H.; Okada, S.; Hatano, M.; Takahashi, Y.; Takeda, N.; Ichii, H.; Takemori, T.; Kuroda, Y.; Tokuhisa, T. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity 2002, 17, 329–339. [Google Scholar] [CrossRef]
- Malkiel, S.; Barlev, A.N.; Atisha-Fregoso, Y.; Suurmond, J.; Diamond, B. Plasma Cell Differentiation Pathways in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 427. [Google Scholar] [CrossRef]
- Kaji, T.; Ishige, A.; Hikida, M.; Taka, J.; Hijikata, A.; Kubo, M.; Nagashima, T.; Takahashi, Y.; Kurosaki, T.; Okada, M.; et al. Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. J. Exp. Med. 2012, 209, 2079–2097. [Google Scholar] [CrossRef] [PubMed]
- Inamine, A.; Takahashi, Y.; Baba, N.; Miyake, K.; Tokuhisa, T.; Takemori, T.; Abe, R. Two waves of memory B-cell generation in the primary immune response. Int. Immunol. 2005, 17, 581–589. [Google Scholar] [CrossRef]
- Vieira, P.; Rajewsky, K. Persistence of memory B cells in mice deprived of T cell help. Int. Immunol. 1990, 2, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Lam, K.P.; Rajewsky, K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature 2000, 407, 636–642. [Google Scholar] [CrossRef]
- Kaneko, N.; Kuo, H.H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183, 143–157.e13. [Google Scholar] [CrossRef] [PubMed]
- Finney, J.; Yeh, C.H.; Kelsoe, G.; Kuraoka, M. Germinal center responses to complex antigens. Immunol. Rev. 2018, 284, 42–50. [Google Scholar] [CrossRef]
- Dal Porto, J.M.; Haberman, A.M.; Kelsoe, G.; Shlomchik, M.J. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J. Exp. Med. 2002, 195, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Schwickert, T.A.; Lindquist, R.L.; Shakhar, G.; Livshits, G.; Skokos, D.; Kosco-Vilbois, M.H.; Dustin, M.L.; Nussenzweig, M.C. In vivo imaging of germinal centres reveals a dynamic open structure. Nature 2007, 446, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.D.; Okada, T.; Tang, H.L.; Cyster, J.G. Imaging of germinal center selection events during affinity maturation. Science 2007, 315, 528–531. [Google Scholar] [CrossRef]
- Batista, F.D.; Harwood, N.E. The who, how and where of antigen presentation to B cells. Nat. Rev. Immunol. 2009, 9, 15–27. [Google Scholar] [CrossRef]
- Good-Jacobson, K.L.; Szumilas, C.G.; Chen, L.; Sharpe, A.H.; Tomayko, M.M.; Shlomchik, M.J. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol. 2010, 11, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Schwickert, T.A.; Fooksman, D.R.; Kamphorst, A.O.; Meyer-Hermann, M.; Dustin, M.L.; Nussenzweig, M.C. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 2010, 143, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.S.; Ke, F.; Grigorova, I.L. B Cell Receptor Crosslinking Augments Germinal Center B Cell Selection when T Cell Help Is Limiting. Cell Rep. 2018, 25, 1395–1403.e4. [Google Scholar] [CrossRef]
- Akkaya, M.; Kwak, K.; Pierce, S.K. B cell memory: Building two walls of protection against pathogens. Nat. Rev. Immunol. 2020, 20, 229–238. [Google Scholar] [CrossRef]
- Inoue, T.; Moran, I.; Shinnakasu, R.; Phan, T.G.; Kurosaki, T. Generation of memory B cells and their reactivation. Immunol. Rev. 2018, 283, 138–149. [Google Scholar] [CrossRef]
- Pape, K.A.; Maul, R.W.; Dileepan, T.; Paustian, A.S.; Gearhart, P.J.; Jenkins, M.K. Naive B Cells with High-Avidity Germline-Encoded Antigen Receptors Produce Persistent IgM(+) and Transient IgG(+) Memory B Cells. Immunity 2018, 48, 1135–1143.e4. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.K.; Desikan, R.; Dixit, N.M. Preferential Presentation of High-Affinity Immune Complexes in Germinal Centers Can Explain How Passive Immunization Improves the Humoral Response. Cell Rep. 2019, 29, 3946–3957.e5. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Kometani, K.; Ise, W. Memory B cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Pape, K.A.; Taylor, J.J.; Maul, R.W.; Gearhart, P.J.; Jenkins, M.K. Different B cell populations mediate early and late memory during an endogenous immune response. Science 2011, 331, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Dogan, I.; Bertocci, B.; Vilmont, V.; Delbos, F.; Megret, J.; Storck, S.; Reynaud, C.A.; Weill, J.C. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 2009, 10, 1292–1299. [Google Scholar] [CrossRef]
- Plotkin, S.A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A. Updates on immunologic correlates of vaccine-induced protection. Vaccine 2020, 38, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A. Vaccines: Correlates of vaccine-induced immunity. Clin. Infect. Dis. 2008, 47, 401–409. [Google Scholar] [CrossRef]
- Taylor, A.; Foo, S.S.; Bruzzone, R.; Dinh, L.V.; King, N.J.; Mahalingam, S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol. Rev. 2015, 268, 340–364. [Google Scholar] [CrossRef]
- Winarski, K.L.; Tang, J.; Klenow, L.; Lee, J.; Coyle, E.M.; Manischewitz, J.; Turner, H.L.; Takeda, K.; Ward, A.B.; Golding, H.; et al. Antibody-dependent enhancement of influenza disease promoted by increase in hemagglutinin stem flexibility and virus fusion kinetics. Proc. Natl. Acad. Sci. USA 2019, 116, 15194–15199. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 2021, 371, eabf4063. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2020, 21, 83–100. [Google Scholar] [CrossRef]
- Phan, T.G.; Paus, D.; Chan, T.D.; Turner, M.L.; Nutt, S.L.; Basten, A.; Brink, R. High affinity germinal center B cells are actively selected into the plasma cell compartment. J. Exp. Med. 2006, 203, 2419–2424. [Google Scholar] [CrossRef]
- Blanchard-Rohner, G.; Pulickal, A.S.; Jol-van der Zijde, C.M.; Snape, M.D.; Pollard, A.J. Appearance of peripheral blood plasma cells and memory B cells in a primary and secondary immune response in humans. Blood 2009, 114, 4998–5002. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, I.; Rodriguez-Bayona, B.; Mora-Lopez, F.; Campos-Caro, A.; Brieva, J.A. Increased survival is a selective feature of human circulating antigen-induced plasma cells synthesizing high-affinity antibodies. Blood 2008, 111, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math. Methods Med. 2012, 2012, 842329. [Google Scholar] [CrossRef]
- Tam, H.H.; Melo, M.B.; Kang, M.; Pelet, J.M.; Ruda, V.M.; Foley, M.H.; Hu, J.K.; Kumari, S.; Crampton, J.; Baldeon, A.D.; et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc. Natl. Acad. Sci. USA 2016, 113, E6639–E6648. [Google Scholar] [CrossRef]
- Henry, C.; Palm, A.E.; Krammer, F.; Wilson, P.C. From Original Antigenic Sin to the Universal Influenza Virus Vaccine. Trends Immunol. 2018, 39, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Ma, C.S.; Brink, R.; Deenick, E.K. The good, the bad and the ugly—TFH cells in human health and disease. Nat. Rev. Immunol. 2013, 13, 412–426. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Kartashov, A.V.; Liu, C.; Imamichi, H.; Yang, W.; Peng, W.; Lane, H.C.; Zhao, K. Rapid Recall Ability of Memory T cells is Encoded in their Epigenome. Sci. Rep. 2017, 7, 39785. [Google Scholar] [CrossRef]
- Chen, Y.; Zander, R.; Khatun, A.; Schauder, D.M.; Cui, W. Transcriptional and Epigenetic Regulation of Effector and Memory CD8 T Cell Differentiation. Front. Immunol. 2018, 9, 2826. [Google Scholar] [CrossRef] [PubMed]
- Muruganandah, V.; Sathkumara, H.D.; Navarro, S.; Kupz, A. A Systematic Review: The Role of Resident Memory T Cells in Infectious Diseases and Their Relevance for Vaccine Development. Front. Immunol. 2018, 9, 1574. [Google Scholar] [CrossRef]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef]
- Jameson, S.C.; Masopust, D. Understanding Subset Diversity in T Cell Memory. Immunity 2018, 48, 214–226. [Google Scholar] [CrossRef]
- Park, S.L.; Zaid, A.; Hor, J.L.; Christo, S.N.; Prier, J.E.; Davies, B.; Alexandre, Y.O.; Gregory, J.L.; Russell, T.A.; Gebhardt, T.; et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018, 19, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Beura, L.K.; Mitchell, J.S.; Thompson, E.A.; Schenkel, J.M.; Mohammed, J.; Wijeyesinghe, S.; Fonseca, R.; Burbach, B.J.; Hickman, H.D.; Vezys, V.; et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol. 2018, 19, 173–182. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Vezys, V.; Marzo, A.L.; Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef]
- Bouneaud, C.; Garcia, Z.; Kourilsky, P.; Pannetier, C. Lineage relationships, homeostasis, and recall capacities of central- and effector-memory CD8 T cells in vivo. J. Exp. Med. 2005, 201, 579–590. [Google Scholar] [CrossRef]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef]
- Youngblood, B.; Hale, J.S.; Kissick, H.T.; Ahn, E.; Xu, X.; Wieland, A.; Araki, K.; West, E.E.; Ghoneim, H.E.; Fan, Y.; et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 2017, 552, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Ahmed, R. Memory T follicular helper CD4 T cells. Front. Immunol. 2015, 6, 16. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Luthje, K.; Kallies, A.; Shimohakamada, Y.; Belz, G.T.; Light, A.; Tarlinton, D.M.; Nutt, S.L. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat. Immunol. 2012, 13, 491–498. [Google Scholar] [CrossRef]
- Kunzli, M.; Schreiner, D.; Pereboom, T.C.; Swarnalekha, N.; Litzler, L.C.; Lotscher, J.; Ertuna, Y.I.; Roux, J.; Geier, F.; Jakob, R.P.; et al. Long-lived T follicular helper cells retain plasticity and help sustain humoral immunity. Sci. Immunol. 2020, 5, eaay5552. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Craft, J. T follicular helper cell heterogeneity: Time, space, and function. Immunol. Rev. 2019, 288, 85–96. [Google Scholar] [CrossRef]
- Asrir, A.; Aloulou, M.; Gador, M.; Perals, C.; Fazilleau, N. Interconnected subsets of memory follicular helper T cells have different effector functions. Nat. Commun. 2017, 8, 847. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.T.; Kanno, Y.; Cannons, J.L.; Handon, R.; Bible, P.; Elkahloun, A.G.; Anderson, S.M.; Wei, L.; Sun, H.; O’Shea, J.J.; et al. Functional and epigenetic studies reveal multistep differentiation and plasticity of in vitro-generated and in vivo-derived follicular T helper cells. Immunity 2011, 35, 622–632. [Google Scholar] [CrossRef]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Lugli, E.; Dominguez, M.H.; Gattinoni, L.; Chattopadhyay, P.K.; Bolton, D.L.; Song, K.; Klatt, N.R.; Brenchley, J.M.; Vaccari, M.; Gostick, E.; et al. Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Investig. 2013, 123, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Fuertes Marraco, S.A.; Soneson, C.; Delorenzi, M.; Speiser, D.E. Genome-wide RNA profiling of long-lasting stem cell-like memory CD8 T cells induced by Yellow Fever vaccination in humans. Genom. Data 2015, 5, 297–301. [Google Scholar] [CrossRef][Green Version]
- McMahan, K.; Yu, J.; Mercado, N.B.; Loos, C.; Tostanoski, L.H.; Chandrashekar, A.; Liu, J.; Peter, L.; Atyeo, C.; Zhu, A.; et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 2020. [Google Scholar] [CrossRef]
- Billeskov, R.; Beikzadeh, B.; Berzofsky, J.A. The effect of antigen dose on T cell-targeting vaccine outcome. Hum. Vaccine Immunother. 2019, 15, 407–411. [Google Scholar] [CrossRef]
- Gustafson, C.E.; Kim, C.; Weyand, C.M.; Goronzy, J.J. Influence of immune aging on vaccine responses. J. Allergy Clin. Immunol. 2020, 145, 1309–1321. [Google Scholar] [CrossRef]
- Keck, S.; Schmaler, M.; Ganter, S.; Wyss, L.; Oberle, S.; Huseby, E.S.; Zehn, D.; King, C.G. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 14852–14857. [Google Scholar] [CrossRef]
- Fazilleau, N.; McHeyzer-Williams, L.J.; Rosen, H.; McHeyzer-Williams, M.G. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 2009, 10, 375–384. [Google Scholar] [CrossRef]
- Ise, W.; Inoue, T.; McLachlan, J.B.; Kometani, K.; Kubo, M.; Okada, T.; Kurosaki, T. Memory B cells contribute to rapid Bcl6 expression by memory follicular helper T cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11792–11797. [Google Scholar] [CrossRef] [PubMed]
- Warfel, J.M.; Edwards, K.M. Pertussis vaccines and the challenge of inducing durable immunity. Curr. Opin. Immunol. 2015, 35, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Auladell, M.; Jia, X.; Hensen, L.; Chua, B.; Fox, A.; Nguyen, T.H.O.; Doherty, P.C.; Kedzierska, K. Recalling the Future: Immunological Memory Toward Unpredictable Influenza Viruses. Front. Immunol. 2019, 10, 1400. [Google Scholar] [CrossRef] [PubMed]
- Huisman, W.; Martina, B.E.; Rimmelzwaan, G.F.; Gruters, R.A.; Osterhaus, A.D. Vaccine-induced enhancement of viral infections. Vaccine 2009, 27, 505–512. [Google Scholar] [CrossRef]
- Su, S.; Du, L.; Jiang, S. Learning from the past: Development of safe and effective COVID-19 vaccines. Nat. Rev. Microbiol. 2020, 16, 1–9. [Google Scholar] [CrossRef]
- Walker, J.C.; Smolders, M.A.; Gemen, E.F.; Antonius, T.A.; Leuvenink, J.; de Vries, E. Development of lymphocyte subpopulations in preterm infants. Scand. J. Immunol. 2011, 73, 53–58. [Google Scholar] [CrossRef]
- Kollmann, T.R.; Kampmann, B.; Mazmanian, S.K.; Marchant, A.; Levy, O. Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 2017, 46, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Vono, M.; Eberhardt, C.S.; Auderset, F.; Mastelic-Gavillet, B.; Lemeille, S.; Christensen, D.; Andersen, P.; Lambert, P.H.; Siegrist, C.A. Maternal Antibodies Inhibit Neonatal and Infant Responses to Vaccination by Shaping the Early-Life B Cell Repertoire within Germinal Centers. Cell Rep. 2019, 28, 1773–1784.e5. [Google Scholar] [CrossRef]
- Ygberg, S.; Nilsson, A. The developing immune system—From foetus to toddler. Acta Paediatr. 2012, 101, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Zandvoort, A.; Timens, W. The dual function of the splenic marginal zone: Essential for initiation of anti-TI-2 responses but also vital in the general first-line defense against blood-borne antigens. Clin. Exp. Immunol. 2002, 130, 4–11. [Google Scholar] [CrossRef]
- Siegrist, C.A.; Aspinall, R. B-cell responses to vaccination at the extremes of age. Nat. Rev. Immunol. 2009, 9, 185–194. [Google Scholar] [CrossRef]
- Mastelic, B.; Kamath, A.T.; Fontannaz, P.; Tougne, C.; Rochat, A.F.; Belnoue, E.; Combescure, C.; Auderset, F.; Lambert, P.H.; Tacchini-Cottier, F.; et al. Environmental and T cell-intrinsic factors limit the expansion of neonatal follicular T helper cells but may be circumvented by specific adjuvants. J. Immunol. 2012, 189, 5764–5772. [Google Scholar] [CrossRef]
- Morbach, H.; Eichhorn, E.M.; Liese, J.G.; Girschick, H.J. Reference values for B cell subpopulations from infancy to adulthood. Clin. Exp. Immunol. 2010, 162, 271–279. [Google Scholar] [CrossRef]
- Adkins, B.; Leclerc, C.; Marshall-Clarke, S. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 2004, 4, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Huygens, A.; Lecomte, S.; Tackoen, M.; Olislagers, V.; Delmarcelle, Y.; Burny, W.; Van Rysselberge, M.; Liesnard, C.; Larsen, M.; Appay, V.; et al. Functional Exhaustion Limits CD4+ and CD8+ T-Cell Responses to Congenital Cytomegalovirus Infection. J. Infect. Dis. 2015, 212, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Risdon, G.; Gaddy, J.; Horie, M.; Broxmeyer, H.E. Alloantigen priming induces a state of unresponsiveness in human umbilical cord blood T cells. Proc. Natl. Acad. Sci. USA 1995, 92, 2413–2417. [Google Scholar] [CrossRef]
- Morris, M.C.; Surendran, N. Neonatal Vaccination: Challenges and Intervention Strategies. Neonatology 2016, 109, 161–169. [Google Scholar] [CrossRef]
- Burt, T.D. Fetal regulatory T cells and peripheral immune tolerance in utero: Implications for development and disease. Am. J. Reprod. Immunol. 2013, 69, 346–358. [Google Scholar] [CrossRef]
- Boer, M.C.; Joosten, S.A.; Ottenhoff, T.H. Regulatory T-Cells at the Interface between Human Host and Pathogens in Infectious Diseases and Vaccination. Front. Immunol. 2015, 6, 217. [Google Scholar] [CrossRef] [PubMed]
- Sautois, B.; Fillet, G.; Beguin, Y. Comparative cytokine production by in vitro stimulated mononucleated cells from cord blood and adult blood. Exp. Hematol 1997, 25, 103–108. [Google Scholar]
- Saso, A.; Kampmann, B. Vaccine responses in newborns. Semin. Immunopathol. 2017, 39, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Wood, N.; Siegrist, C.A. Neonatal immunization: Where do we stand? Curr. Opin. Infect. Dis. 2011, 24, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, K.A.; Liang, M.D.; Andresen, T.K.; Thomas, K.E.; Marty, A.M.; Cuesta, N.; Vogel, S.N.; Fenton, M.J. TLR2 and TLR4 serve distinct roles in the host immune response against Mycobacterium bovis BCG. J. Leukoc. Biol. 2003, 74, 277–286. [Google Scholar] [CrossRef]
- Marchant, A.; Goetghebuer, T.; Ota, M.O.; Wolfe, I.; Ceesay, S.J.; De Groote, D.; Corrah, T.; Bennett, S.; Wheeler, J.; Huygen, K.; et al. Newborns develop a Th1-type immune response to Mycobacterium bovis bacillus Calmette-Guerin vaccination. J. Immunol. 1999, 163, 2249–2255. [Google Scholar] [PubMed]
- Rousseau, M.C.; El-Zein, M.; Conus, F.; Legault, L.; Parent, M.E. Bacillus Calmette-Guerin (BCG) Vaccination in Infancy and Risk of Childhood Diabetes. Paediatr. Perinat. Epidemiol. 2016, 30, 141–148. [Google Scholar] [CrossRef]
- Thostesen, L.M.; Kjaergaard, J.; Pihl, G.T.; Birk, N.M.; Nissen, T.N.; Aaby, P.; Jensen, A.K.G.; Olesen, A.W.; Stensballe, L.G.; Jeppesen, D.L.; et al. Neonatal BCG vaccination and atopic dermatitis before 13 months of age: A randomized clinical trial. Allergy 2018, 73, 498–504. [Google Scholar] [CrossRef]
- Morra, M.E.; Kien, N.D.; Elmaraezy, A.; Abdelaziz, O.A.M.; Elsayed, A.L.; Halhouli, O.; Montasr, A.M.; Vu, T.L.; Ho, C.; Foly, A.S.; et al. Early vaccination protects against childhood leukemia: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 15986. [Google Scholar] [CrossRef]
- Demirjian, A.; Levy, O. Safety and efficacy of neonatal vaccination. Eur. J. Immunol. 2009, 39, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, I.; Dieussaert, I.; Doherty, T.M.; Franck, V.; Hanssens, L.; Miller, J.; Bekkat-Berkani, R.; Kandeil, W.; Prado-Cohrs, D.; Vyse, A. Maternal immunization: Where are we now and how to move forward? Ann. Med. 2018, 50, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Klein Klouwenberg, P.; Bont, L. Neonatal and infantile immune responses to encapsulated bacteria and conjugate vaccines. Clin. Dev. Immunol. 2008, 2008, 628963. [Google Scholar] [CrossRef] [PubMed]
- Landers, C.D.; Chelvarajan, R.L.; Bondada, S. The role of B cells and accessory cells in the neonatal response to TI-2 antigens. Immunol. Res. 2005, 31, 25–36. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 2016, 17, 7–19. [Google Scholar] [CrossRef]
- Fuentes, E.; Fuentes, M.; Alarcon, M.; Palomo, I. Immune System Dysfunction in the Elderly. An. Acad. Bras. Cienc. 2017, 89, 285–299. [Google Scholar] [CrossRef]
- Pera, A.; Campos, C.; Lopez, N.; Hassouneh, F.; Alonso, C.; Tarazona, R.; Solana, R. Immunosenescence: Implications for response to infection and vaccination in older people. Maturitas 2015, 82, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.L.; Wu, Y.C.; Barnett, Y.; Duggan, O.; Vaughan, R.; Kondeatis, E.; Nilsson, B.O.; Wikby, A.; Kipling, D.; Dunn-Walters, D.K. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 2009, 8, 18–25. [Google Scholar] [CrossRef]
- Ciabattini, A.; Nardini, C.; Santoro, F.; Garagnani, P.; Franceschi, C.; Medaglini, D. Vaccination in the elderly: The challenge of immune changes with aging. Semin. Immunol. 2018, 40, 83–94. [Google Scholar] [CrossRef]
- Frasca, D.; Riley, R.L.; Blomberg, B.B. Aging murine B cells have decreased class switch induced by anti-CD40 or BAFF. Exp. Gerontol. 2007, 42, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Pritz, T.; Lair, J.; Ban, M.; Keller, M.; Weinberger, B.; Krismer, M.; Grubeck-Loebenstein, B. Plasma cell numbers decrease in bone marrow of old patients. Eur. J. Immunol. 2015, 45, 738–746. [Google Scholar] [CrossRef]
- Howard, W.A.; Gibson, K.L.; Dunn-Walters, D.K. Antibody quality in old age. Rejuvenation Res. 2006, 9, 117–125. [Google Scholar] [CrossRef]
- Frasca, D.; Landin, A.M.; Lechner, S.C.; Ryan, J.G.; Schwartz, R.; Riley, R.L.; Blomberg, B.B. Aging down-regulates the transcription factor E2A, activation-induced cytidine deaminase, and Ig class switch in human B cells. J. Immunol. 2008, 180, 5283–5290. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, B.B.; Frasca, D. Age effects on mouse and human B cells. Immunol. Res. 2013, 57, 354–360. [Google Scholar] [CrossRef]
- Frasca, D.; Van der Put, E.; Riley, R.L.; Blomberg, B.B. Reduced Ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J. Immunol. 2004, 172, 2155–2162. [Google Scholar] [CrossRef]
- Colonna-Romano, G.; Bulati, M.; Aquino, A.; Pellicano, M.; Vitello, S.; Lio, D.; Candore, G.; Caruso, C. A double-negative (IgD-CD27-) B cell population is increased in the peripheral blood of elderly people. Mech. Ageing Dev. 2009, 130, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yang, K.; Ozen, Z.; Peng, W.; Marinova, E.; Kelsoe, G.; Zheng, B. Enhanced differentiation of splenic plasma cells but diminished long-lived high-affinity bone marrow plasma cells in aged mice. J. Immunol. 2003, 170, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- den Braber, I.; Mugwagwa, T.; Vrisekoop, N.; Westera, L.; Mogling, R.; de Boer, A.B.; Willems, N.; Schrijver, E.H.; Spierenburg, G.; Gaiser, K.; et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 2012, 36, 288–297. [Google Scholar] [CrossRef]
- Malek, T.R.; Bayer, A.L. Tolerance, not immunity, crucially depends on IL-2. Nat. Rev. Immunol. 2004, 4, 665–674. [Google Scholar] [CrossRef]
- Ahmed, M.; Lanzer, K.G.; Yager, E.J.; Adams, P.S.; Johnson, L.L.; Blackman, M.A. Clonal expansions and loss of receptor diversity in the naive CD8 T cell repertoire of aged mice. J. Immunol. 2009, 182, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Fang, F.; Cavanagh, M.M.; Qi, Q.; Weyand, C.M. Naive T cell maintenance and function in human aging. J. Immunol. 2015, 194, 4073–4080. [Google Scholar] [CrossRef] [PubMed]
- Renkema, K.R.; Li, G.; Wu, A.; Smithey, M.J.; Nikolich-Zugich, J. Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J. Immunol. 2014, 192, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef]
- Naylor, K.; Li, G.; Vallejo, A.N.; Lee, W.W.; Koetz, K.; Bryl, E.; Witkowski, J.; Fulbright, J.; Weyand, C.M.; Goronzy, J.J. The influence of age on T cell generation and TCR diversity. J. Immunol. 2005, 174, 7446–7452. [Google Scholar] [CrossRef] [PubMed]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L. Human T cell immunosenescence and inflammation in aging. J. Leukoc. Biol. 2017, 102, 977–988. [Google Scholar] [CrossRef]
- Goodwin, K.; Viboud, C.; Simonsen, L. Antibody response to influenza vaccination in the elderly: A quantitative review. Vaccine 2006, 24, 1159–1169. [Google Scholar] [CrossRef]
- Young, B.; Zhao, X.; Cook, A.R.; Parry, C.M.; Wilder-Smith, A.; MC, I.C. Do antibody responses to the influenza vaccine persist year-round in the elderly? A systematic review and meta-analysis. Vaccine 2017, 35, 212–221. [Google Scholar] [CrossRef]
- Lefebvre, J.S.; Masters, A.R.; Hopkins, J.W.; Haynes, L. Age-related impairment of humoral response to influenza is associated with changes in antigen specific T follicular helper cell responses. Sci. Rep. 2016, 6, 25051. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Self, W.H.; Wunderink, R.G.; Fakhran, S.; Balk, R.; Bramley, A.M.; Reed, C.; Grijalva, C.G.; Anderson, E.J.; Courtney, D.M.; et al. Community-Acquired Pneumonia Requiring Hospitalization among U.S. Adults. N. Engl. J. Med. 2015, 373, 415–427. [Google Scholar] [CrossRef]
- Schmader, K.E.; Johnson, G.R.; Saddier, P.; Ciarleglio, M.; Wang, W.W.; Zhang, J.H.; Chan, I.S.; Yeh, S.S.; Levin, M.J.; Harbecke, R.M.; et al. Effect of a zoster vaccine on herpes zoster-related interference with functional status and health-related quality-of-life measures in older adults. J. Am. Geriatr. Soc. 2010, 58, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.F.; Harpaz, R.; Luo, Y.; Hales, C.M.; Sy, L.S.; Tartof, S.Y.; Bialek, S.; Hechter, R.C.; Jacobsen, S.J. Declining Effectiveness of Herpes Zoster Vaccine in Adults Aged ≥60 Years. J. Infect. Dis. 2016, 213, 1872–1875. [Google Scholar] [CrossRef]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



 ,
, 



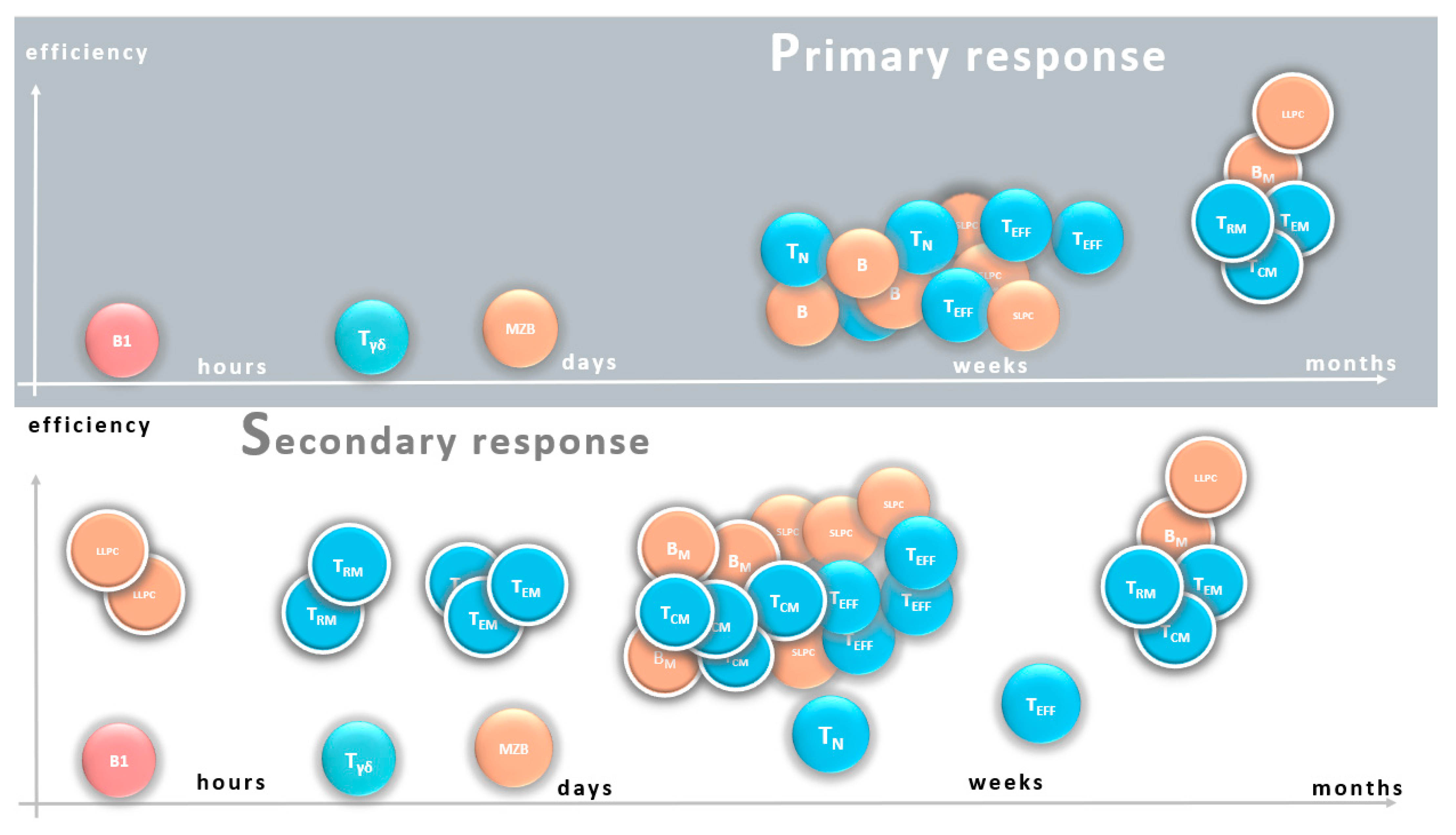{kind=link}
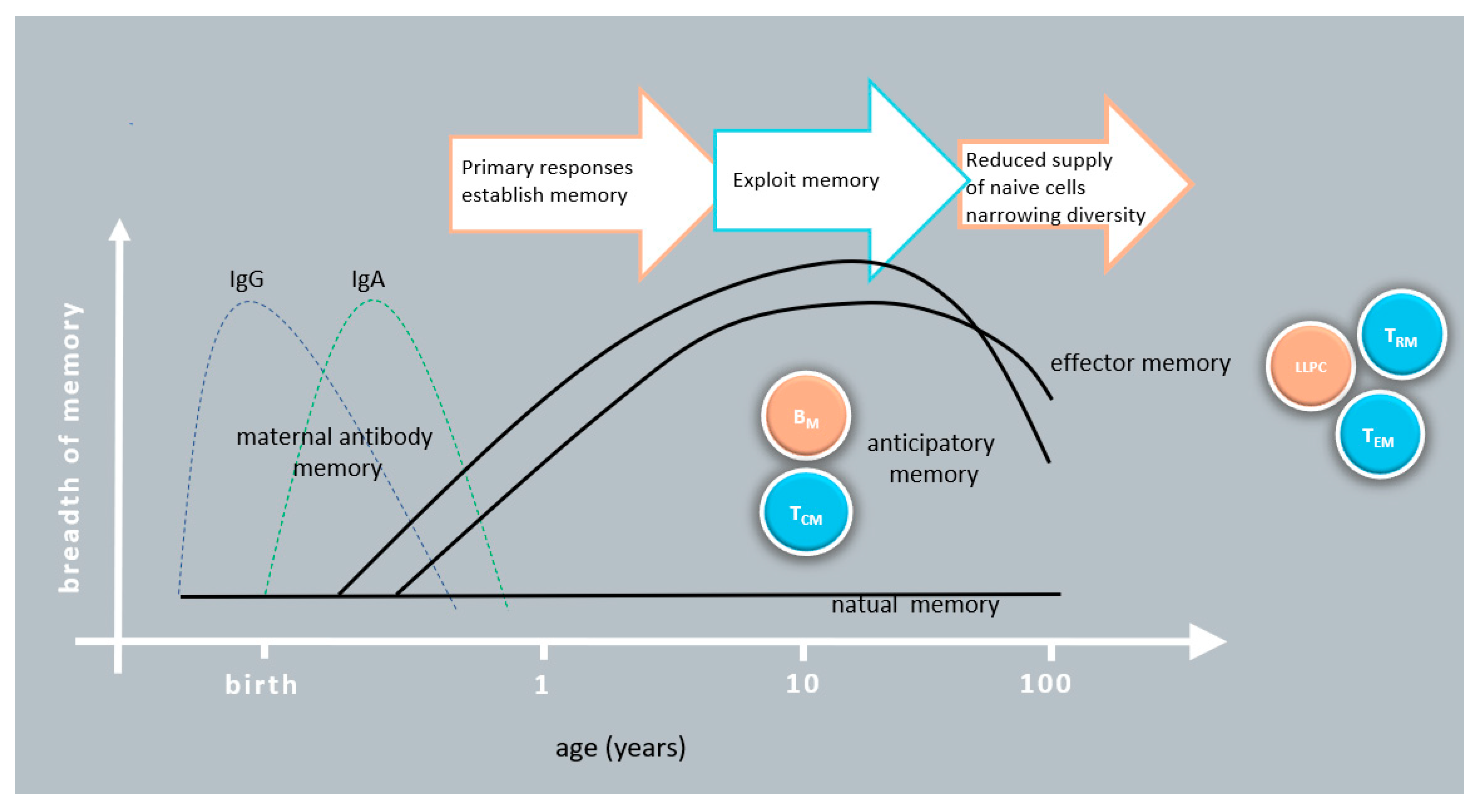{kind=link}