
Krüppel-like Factor 2 (KLF2) in Immune Cell Migration


Abstract
1. Introduction
2. KLF2—A Central Regulator of Quiescence and Activation
3. Requirements for Immune Cell Migration and Homing
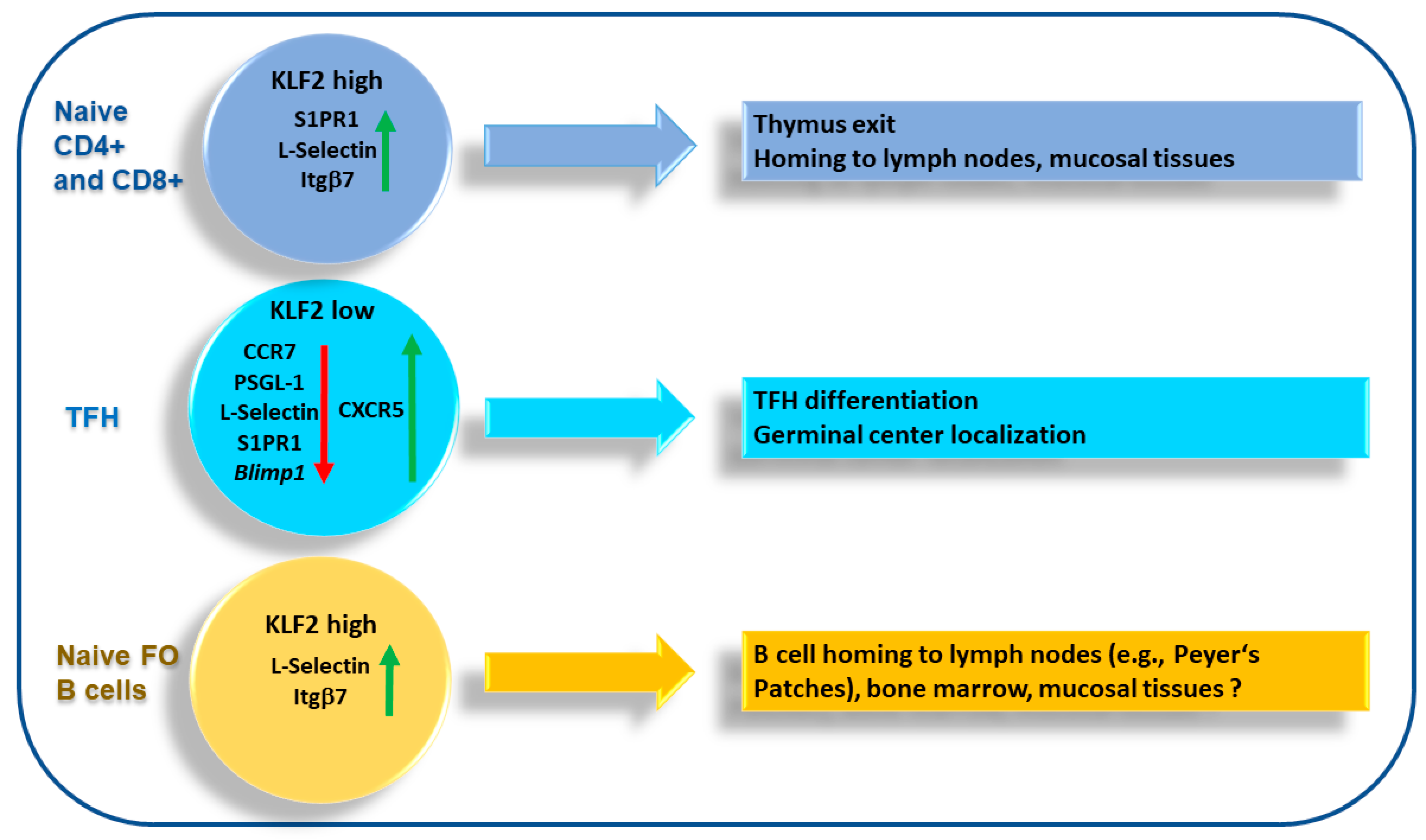
4. KLF2 and T Cell Migration
4.1. T Cell Activation
4.2. T Follicular Helper Cells
4.3. Regulatory T Cells
4.4. KLF2 and T Cells in Diseases
5. KLF2 and B Cell Migration
6. KLF2 and Myeloid Cell Migration
7. KLF2 and NK Cell Migration
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Disease | Involvement of KLF2 | KLF2-Related Effects | Cell Type | Refs. |
|---|---|---|---|---|
| HIV-1 infection | KLF2 downregulation -> S1PR1 and L-Selectin downregulation/CD69 upregulation | Defective CD4+ T cell migration | CD4+ T cells | [76,77] |
| Autoimmunity | KLF2 knockout (Foxp3-cre deleter) | Signs of psoriasis, signs of IBD | iTregs | [69] |
| Type1 Diabetes (Autoimmunity) | microRNA-92a regulating KLF2 -> dysregulated TFH precursor pool size | Autoimmunity against insulin producing beta-cells | TFH | [78] |
| Allergic Asthma | Notch knockout (CD4-cre deleter) -> downregulation of KLF2 -> downregulation of S1PR1 | Th2 cells are primed to leave the lymph node and invade the lung by KLF2-mediated upregulation of S1PR1; KLF2 downregulation by Notch-deficiency prevents Th2 lung infiltration | Th2 cells | [79] |
| Asthma | reduced KLF2 expression in blood neutrophils of Asthma patients -> upregulation of CXCR1 and CXCR2 | Enhanced neutrophil migration | Neutrophils | [97] |
| Arthritis | Hemizygous KLF2 mice | Increased pro-inflammatory factors, osteoclast differentiation | Monocytes, Osteoclasts | [88] |
| Marginal Zone B cell Lymphoma | KLF2 knockout (mb1-cre deleter, CD19-cre deleter) & KLF2 mutations in human SMZBL -> strongly enriched MZB pool size | Splenomegaly (mouse),Marginal Zone B cell Lymphoma (human) | B cells | [8,10,18,19,21] |
| Atherosclerosis | KLF2 knockout (LysM-cre deleter) on Ldlr-deficient background; hemizygous KLF2 on ApoE-deficient background | Increased adhesion to endothelial cells, Increased signs of atherosclerosis when combined with Ldlr-deficiency or ApoE-deficiency | Macrophages/Neutrophils | [93,94] |
| Atherosclerosis | KLF2 knockout (CD11c-cre deleter) -> CD86 and CD40 upregulation upon LPS activation | Enhanced T cell activation, Increased signs of atherosclerosis when combined with Ldlr-deficiency | Dendritic cells | [98] |
| Thrombosis | KLF2 knockout (LysM-cre deleter) -> decreased P-Selectin | Decreased rolling ability | Neutrophils | [96] |
| Muscle Injury | KLF2 knockout (LysM-cre deleter) -> CCR5 upregulation | Increased numbers of CCR5+ monocytes and of MerTK+ macrophages in injured muscles -> increased phagocytosis activity | Macrophages | [95] |
| Sepsis | KLF2 knockout (Lyz2-cre deleter) | Endotoxic shock | Myeloid cells | [90] |
References
- Preiss, A.; Rosenberg, U.B.; Kienlin, A.; Seifert, E.; Jackle, H. Molecular genetics of Kruppel, a gene required for segmentation of the Drosophila embryo. Nature 1985, 313, 27–32. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.B.; Yang, V.W. Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef]
- Anderson, K.P.; Kern, C.B.; Crable, S.C.; Lingrel, J.B. Isolation of a gene encoding a functional zinc finger protein homologous to erythroid Kruppel-like factor: Identification of a new multigene family. Mol. Cell Biol. 1995, 15, 5957–5965. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.T.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factors in lymphocyte biology. J. Immunol. 2012, 188, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.T.; Veselits, M.L.; Barton, K.P.; Lu, M.M.; Clendenin, C.; Leiden, J.M. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 1997, 11, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Marschall, J.S.; Wilhelm, T.; Schuh, W.; Huber, M. MEK/Erk-based negative feedback mechanism involved in control of Steel Factor-triggered production of Kruppel-like factor 2 in mast cells. Cell Signal. 2012, 24, 879–888. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, S. Role of kruppel-like transcription factors in adipogenesis. Dev. Biol. 2013, 373, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Clipson, A.; Wang, M.; de Leval, L.; Ashton-Key, M.; Wotherspoon, A.; Vassiliou, G.; Bolli, N.; Grove, C.; Moody, S.; Escudero-Ibarz, L.; et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia 2015, 29, 1177–1185. [Google Scholar] [CrossRef]
- Sweet, D.R.; Fan, L.; Hsieh, P.N.; Jain, M.K. Kruppel-Like Factors in Vascular Inflammation: Mechanistic Insights and Therapeutic Potential. Front. Cardiovasc. Med. 2018, 5, 6. [Google Scholar] [CrossRef]
- Spina, V.; Mensah, A.A.; Arribas, A.J. Biology of splenic and nodal marginal zone lymphomas. Ann. Lymphoma 2021, 5. [Google Scholar] [CrossRef]
- Kuo, C.T.; Veselits, M.L.; Leiden, J.M. LKLF: A transcriptional regulator of single-positive T cell quiescence and survival. Science 1997, 277, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.F.; Kuo, C.T.; Leiden, J.M. Transcription factor LKLF is sufficient to program T cell quiescence via a c-Myc-dependent pathway. Nat. Immunol. 2001, 2, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lingrel, J.B. KLF2 inhibits Jurkat T leukemia cell growth via upregulation of cyclin-dependent kinase inhibitor p21WAF1/CIP1. Oncogene 2004, 23, 8088–8096. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, M.; Hara, T.; Yoshita-Takahashi, M.; Kohda, T.; Tanaka, Y.; Nakamura, M. Promoter CpG methylation inhibits Kruppel-like factor 2 (KLF2)-Mediated repression of hTERT gene expression in human T-cells. Biochem. Biophys. Rep. 2021, 26, 100984. [Google Scholar] [CrossRef]
- Schuh, W.; Meister, S.; Herrmann, K.; Bradl, H.; Jack, H.M. Transcriptome analysis in primary B lymphoid precursors following induction of the pre-B cell receptor. Mol. Immunol. 2008, 45, 362–375. [Google Scholar] [CrossRef]
- Herglotz, J.; Unrau, L.; Hauschildt, F.; Fischer, M.; Kriebitzsch, N.; Alawi, M.; Indenbirken, D.; Spohn, M.; Muller, U.; Ziegler, M.; et al. Essential control of early B-cell development by Mef2 transcription factors. Blood 2016, 127, 572–581. [Google Scholar] [CrossRef]
- Glynne, R.; Ghandour, G.; Rayner, J.; Mack, D.H.; Goodnow, C.C. B-lymphocyte quiescence, tolerance and activation as viewed by global gene expression profiling on microarrays. Immunol. Rev. 2000, 176, 216–246. [Google Scholar] [CrossRef]
- Hoek, K.L.; Gordy, L.E.; Collins, P.L.; Parekh, V.V.; Aune, T.M.; Joyce, S.; Thomas, J.W.; Van Kaer, L.; Sebzda, E. Follicular B cell trafficking within the spleen actively restricts humoral immune responses. Immunity 2010, 33, 254–265. [Google Scholar] [CrossRef]
- Winkelmann, R.; Sandrock, L.; Porstner, M.; Roth, E.; Mathews, M.; Hobeika, E.; Reth, M.; Kahn, M.L.; Schuh, W.; Jack, H.M. B cell homeostasis and plasma cell homing controlled by Kruppel-like factor 2. Proc. Natl. Acad. Sci. USA 2011, 108, 710–715. [Google Scholar] [CrossRef]
- Winkelmann, R.; Sandrock, L.; Kirberg, J.; Jack, H.M.; Schuh, W. KLF2--a negative regulator of pre-B cell clonal expansion and B cell activation. PLoS ONE 2014, 9, e97953. [Google Scholar] [CrossRef]
- Hart, G.T.; Wang, X.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc. Natl. Acad. Sci. USA 2011, 108, 716–721. [Google Scholar] [CrossRef]
- Hart, G.T.; Peery, S.L.; Hamilton, S.E.; Jameson, S.C. Cutting edge: Kruppel-like factor 2 is required for phenotypic maintenance but not development of B1 B cells. J. Immunol. 2012, 189, 3293–3297. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Cheah, M.T.; Franco, C.B.; Hosen, N.; Pin, C.L.; Sha, W.C.; Weissman, I.L. Transcriptional profiling of antigen-dependent murine B cell differentiation and memory formation. J. Immunol. 2007, 179, 6808–6819. [Google Scholar] [CrossRef]
- Das, H.; Kumar, A.; Lin, Z.; Patino, W.D.; Hwang, P.M.; Feinberg, M.W.; Majumder, P.K.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 6653–6658. [Google Scholar] [CrossRef]
- Salazar-Gonzalez, R.M.; Niess, J.H.; Zammit, D.J.; Ravindran, R.; Srinivasan, A.; Maxwell, J.R.; Stoklasek, T.; Yadav, R.; Williams, I.R.; Gu, X.; et al. CCR6-mediated dendritic cell activation of pathogen-specific T cells in Peyer’s patches. Immunity 2006, 24, 623–632. [Google Scholar] [CrossRef]
- Ahrendt, M.; Hammerschmidt, S.I.; Pabst, O.; Pabst, R.; Bode, U. Stromal cells confer lymph node-specific properties by shaping a unique microenvironment influencing local immune responses. J. Immunol. 2008, 181, 1898–1907. [Google Scholar] [CrossRef]
- Mora, J.R.; Bono, M.R.; Manjunath, N.; Weninger, W.; Cavanagh, L.L.; Rosemblatt, M.; Von Andrian, U.H. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 2003, 424, 88–93. [Google Scholar] [CrossRef]
- Hammerschmidt, S.I.; Ahrendt, M.; Bode, U.; Wahl, B.; Kremmer, E.; Forster, R.; Pabst, O. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J. Exp. Med. 2008, 205, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.D. Ligands for L-selectin: Homing, inflammation, and beyond. Annu. Rev. Immunol. 2004, 22, 129–156. [Google Scholar] [CrossRef] [PubMed]
- Girard, J.P.; Moussion, C.; Forster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.R.; Owens, T.W.; Naylor, M.J. Structural and mechanical functions of integrins. Biophys. Rev. 2014, 6, 203–213. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Habtezion, A.; Nguyen, L.P.; Hadeiba, H.; Butcher, E.C. Leukocyte Trafficking to the Small Intestine and Colon. Gastroenterology 2016, 150, 340–354. [Google Scholar] [CrossRef]
- Haddad, W.; Cooper, C.J.; Zhang, Z.; Brown, J.B.; Zhu, Y.; Issekutz, A.; Fuss, I.; Lee, H.O.; Kansas, G.S.; Barrett, T.A. P-selectin and P-selectin glycoprotein ligand 1 are major determinants for Th1 cell recruitment to nonlymphoid effector sites in the intestinal lamina propria. J. Exp. Med. 2003, 198, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Morteau, O.; Gerard, C.; Lu, B.; Ghiran, S.; Rits, M.; Fujiwara, Y.; Law, Y.; Distelhorst, K.; Nielsen, E.M.; Hill, E.D.; et al. An indispensable role for the chemokine receptor CCR10 in IgA antibody-secreting cell accumulation. J. Immunol. 2008, 181, 6309–6315. [Google Scholar] [CrossRef] [PubMed]
- Rott, L.S.; Briskin, M.J.; Butcher, E.C. Expression of alpha4beta7 and E-selectin ligand by circulating memory B cells: Implications for targeted trafficking to mucosal and systemic sites. J. Leukoc. Biol. 2000, 68, 807–814. [Google Scholar] [PubMed]
- Lindquist, R.L.; Niesner, R.A.; Hauser, A.E. In the Right Place, at the Right Time: Spatiotemporal Conditions Determining Plasma Cell Survival and Function. Front Immunol. 2019, 10, 788. [Google Scholar] [CrossRef] [PubMed]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Bixel, M.G.; Kusumbe, A.P.; Ramasamy, S.K.; Sivaraj, K.K.; Butz, S.; Vestweber, D.; Adams, R.H. Flow Dynamics and HSPC Homing in Bone Marrow Microvessels. Cell Rep. 2017, 18, 1804–1816. [Google Scholar] [CrossRef]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef]
- Gaylo-Moynihan, A.; Prizant, H.; Popovic, M.; Fernandes, N.R.J.; Anderson, C.S.; Chiou, K.K.; Bell, H.; Schrock, D.C.; Schumacher, J.; Capece, T.; et al. Programming of Distinct Chemokine-Dependent and -Independent Search Strategies for Th1 and Th2 Cells Optimizes Function at Inflamed Sites. Immunity 2019, 51, 298–309.e296. [Google Scholar] [CrossRef]
- Nanki, T.; Takada, K.; Komano, Y.; Morio, T.; Kanegane, H.; Nakajima, A.; Lipsky, P.E.; Miyasaka, N. Chemokine receptor expression and functional effects of chemokines on B cells: Implication in the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R149. [Google Scholar] [CrossRef]
- Thomas, S.Y.; Hou, R.; Boyson, J.E.; Means, T.K.; Hess, C.; Olson, D.P.; Strominger, J.L.; Brenner, M.B.; Gumperz, J.E.; Wilson, S.B.; et al. CD1d-restricted NKT cells express a chemokine receptor profile indicative of Th1-type inflammatory homing cells. J. Immunol. 2003, 171, 2571–2580. [Google Scholar] [CrossRef]
- Preston, G.C.; Feijoo-Carnero, C.; Schurch, N.; Cowling, V.H.; Cantrell, D.A. The impact of KLF2 modulation on the transcriptional program and function of CD8 T cells. PLoS ONE 2013, 8, e77537. [Google Scholar] [CrossRef]
- Fischer, A.; Zundler, S.; Atreya, R.; Rath, T.; Voskens, C.; Hirschmann, S.; Lopez-Posadas, R.; Watson, A.; Becker, C.; Schuler, G.; et al. Differential effects of alpha4beta7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut 2016, 65, 1642–1664. [Google Scholar] [CrossRef]
- Iijima, N.; Iwasaki, A. Tissue instruction for migration and retention of TRM cells. Trends Immunol. 2015, 36, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, T.A.; Plate, A.; Adam, M.; Wick, M.; Kastenbauer, S.; Schmidt, C.; Klein, M.; Pfister, H.W.; Koedel, U. The chemokine CXCL13 is a key regulator of B cell recruitment to the cerebrospinal fluid in acute Lyme neuroborreliosis. J. Neuroinflammation 2009, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Aira, L.E.; Debes, G.F. Skin-Homing Regulatory B Cells Required for Suppression of Cutaneous Inflammation. J. Investig. Dermatol. 2021, 141, 1995–2005.e1996. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Kabashima, K.; Haynes, N.M.; Xu, Y.; Nutt, S.L.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J. Exp. Med. 2006, 203, 2683–2690. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Dev, K.K. The structure and function of the S1P1 receptor. Trends Pharmacol. Sci. 2013, 34, 401–412. [Google Scholar] [CrossRef]
- Allende, M.L.; Dreier, J.L.; Mandala, S.; Proia, R.L. Expression of the sphingosine 1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J. Biol. Chem. 2004, 279, 15396–15401. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.M.; Endrizzi, B.T.; Wu, J.; Ding, X.; Weinreich, M.A.; Walsh, E.R.; Wani, M.A.; Lingrel, J.B.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 2006, 442, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Jaigirdar, S.A.; Benson, R.A.; Elmesmari, A.; Kurowska-Stolarska, M.S.; McInnes, I.B.; Garside, P.; MacLeod, M.K.L. Sphingosine-1-Phosphate Promotes the Persistence of Activated CD4 T Cells in Inflamed Sites. Front. Immunol. 2017, 8, 1627. [Google Scholar] [CrossRef]
- Zajac, A.J.; Harrington, L.E. Tissue-resident T cells lose their S1P1 exit visas. Cell Mol. Immunol. 2014, 11, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Raffler, N.A.; Ley, K. Transcriptional regulation of mouse L-selectin. Biochim. Biophys. Acta 2009, 1789, 146–152. [Google Scholar] [CrossRef][Green Version]
- Pabbisetty, S.K.; Rabacal, W.; Maseda, D.; Cendron, D.; Collins, P.L.; Hoek, K.L.; Parekh, V.V.; Aune, T.M.; Sebzda, E. KLF2 is a rate-limiting transcription factor that can be targeted to enhance regulatory T-cell production. Proc. Natl. Acad. Sci. USA 2014, 111, 9579–9584. [Google Scholar] [CrossRef]
- Takada, K.; Wang, X.; Hart, G.T.; Odumade, O.A.; Weinreich, M.A.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 is required for trafficking but not quiescence in postactivated T cells. J. Immunol. 2011, 186, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Alles, M.; Turchinovich, G.; Zhang, P.; Schuh, W.; Agenes, F.; Kirberg, J. Leukocyte beta7 integrin targeted by Kruppel-like factors. J. Immunol. 2014, 193, 1737–1746. [Google Scholar] [CrossRef]
- Sebzda, E.; Zou, Z.; Lee, J.S.; Wang, T.; Kahn, M.L. Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nat. Immunol. 2008, 3, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, M.A.; Takada, K.; Skon, C.; Reiner, S.L.; Jameson, S.C.; Hogquist, K.A. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity 2009, 31, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Skon, C.N.; Lee, Y.J.; Oh, S.; Taylor, J.J.; Malhotra, D.; Jenkins, M.K.; Rosenfeld, M.G.; Hogquist, K.A.; Jameson, S.C. The transcription factor KLF2 restrains CD4(+) T follicular helper cell differentiation. Immunity 2015, 42, 252–264. [Google Scholar] [CrossRef]
- Weber, J.P.; Fuhrmann, F.; Feist, R.K.; Lahmann, A.; Al Baz, M.S.; Gentz, L.J.; Vu Van, D.; Mages, H.W.; Haftmann, C.; Riedel, R.; et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J. Exp. Med. 2015, 212, 217–233. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2019, 10, 3100. [Google Scholar] [CrossRef]
- Pabbisetty, S.K.; Rabacal, W.; Volanakis, E.J.; Parekh, V.V.; Olivares-Villagomez, D.; Cendron, D.; Boyd, K.L.; Van Kaer, L.; Sebzda, E. Peripheral tolerance can be modified by altering KLF2-regulated Treg migration. Proc. Natl. Acad. Sci. USA 2016, 113, E4662–E4670. [Google Scholar] [CrossRef]
- Hewavisenti, R.V.; Ferguson, A.L.; Gasparini, G.; Ohashi, T.; Braun, A.; Watkins, T.S.; Miles, J.J.; Elliott, M.; Sierro, F.; Feng, C.G.; et al. Tissue-resident regulatory T cells accumulate at human barrier lymphoid organs. Immunol. Cell Biol. 2021, 99, 894–906. [Google Scholar] [CrossRef]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef]
- Ariotti, S.; Beltman, J.B.; Chodaczek, G.; Hoekstra, M.E.; van Beek, A.E.; Gomez-Eerland, R.; Ritsma, L.; van Rheenen, J.; Maree, A.F.; Zal, T.; et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc. Natl. Acad. Sci. USA 2012, 109, 19739–19744. [Google Scholar] [CrossRef]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef]
- Chien, Y.H.; Meyer, C.; Bonneville, M. gammadelta T cells: First line of defense and beyond. Annu. Rev. Immunol. 2014, 32, 121–155. [Google Scholar] [CrossRef]
- Odumade, O.A.; Weinreich, M.A.; Jameson, S.C.; Hogquist, K.A. Kruppel-like factor 2 regulates trafficking and homeostasis of gammadelta T cells. J. Immunol. 2010, 184, 6060–6066. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.W.; Jadlowsky, J.; Didigu, C.A.; Doms, R.W.; Riley, J.L. Kruppel-like factor 2 modulates CCR5 expression and susceptibility to HIV-1 infection. J. Immunol. 2012, 189, 3815–3821. [Google Scholar] [CrossRef] [PubMed]
- Trinite, B.; Chan, C.N.; Lee, C.S.; Mahajan, S.; Luo, Y.; Muesing, M.A.; Folkvord, J.M.; Pham, M.; Connick, E.; Levy, D.N. Suppression of Foxo1 activity and down-modulation of CD62L (L-selectin) in HIV-1 infected resting CD4 T cells. PLoS ONE 2014, 9, e110719. [Google Scholar] [CrossRef] [PubMed]
- Serr, I.; Furst, R.W.; Ott, V.B.; Scherm, M.G.; Nikolaev, A.; Gokmen, F.; Kalin, S.; Zillmer, S.; Bunk, M.; Weigmann, B.; et al. miRNA92a targets KLF2 and the phosphatase PTEN signaling to promote human T follicular helper precursors in T1D islet autoimmunity. Proc. Natl. Acad. Sci. USA 2016, 113, E6659–E6668. [Google Scholar] [CrossRef] [PubMed]
- Tindemans, I.; van Schoonhoven, A.; KleinJan, A.; de Bruijn, M.J.; Lukkes, M.; van Nimwegen, M.; van den Branden, A.; Bergen, I.M.; Corneth, O.B.; van IJcken, W.F.; et al. Notch signaling licenses allergic airway inflammation by promoting Th2 cell lymph node egress. J. Clin. Investig. 2020, 130, 3576–3591. [Google Scholar] [CrossRef] [PubMed]
- Riedel, R.; Addo, R.; Ferreira-Gomes, M.; Heinz, G.A.; Heinrich, F.; Kummer, J.; Greiff, V.; Schulz, D.; Klaeden, C.; Cornelis, R.; et al. Discrete populations of isotype-switched memory B lymphocytes are maintained in murine spleen and bone marrow. Nat. Commun. 2020, 11, 2570. [Google Scholar] [CrossRef]
- Zuccarino-Catania, G.V.; Sadanand, S.; Weisel, F.J.; Tomayko, M.M.; Meng, H.; Kleinstein, S.H.; Good-Jacobson, K.L.; Shlomchik, M.J. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol. 2014, 15, 631–637. [Google Scholar] [CrossRef]
- Pereira, J.P.; Kelly, L.M.; Cyster, J.G. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int. Immunol. 2010, 22, 413–419. [Google Scholar] [CrossRef] [PubMed]
- McHeyzer-Williams, M.; Okitsu, S.; Wang, N.; McHeyzer-Williams, L. Molecular programming of B cell memory. Nat. Rev. Immunol. 2011, 12, 24–34. [Google Scholar] [CrossRef]
- Groenendijk, B.C.; Van der Heiden, K.; Hierck, B.P.; Poelmann, R.E. The role of shear stress on ET-1, KLF2, and NOS-3 expression in the developing cardiovascular system of chicken embryos in a venous ligation model. Physiology 2007, 22, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Cinamon, G.; Zachariah, M.A.; Lam, O.M.; Foss, F.W., Jr.; Cyster, J.G. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008, 9, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Das, H. KLF2 in Regulation of NF-kappaB-Mediated Immune Cell Function and Inflammation. Int. J. Mol. Sci. 2017, 18, 2383. [Google Scholar] [CrossRef]
- Komano, Y.; Nanki, T.; Hayashida, K.; Taniguchi, K.; Miyasaka, N. Identification of a human peripheral blood monocyte subset that differentiates into osteoclasts. Arthritis Res. Ther. 2006, 8, R152. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Lu, J.; Joseph, M.; Aggarwal, R.; Kanji, S.; McMichael, B.K.; Lee, B.S.; Agarwal, S.; Ray-Chaudhury, A.; Iwenofu, O.H.; et al. Kruppel-like factor 2 (KLF2) regulates monocyte differentiation and functions in mBSA and IL-1beta-induced arthritis. Curr. Mol. Med. 2012, 12, 113–125. [Google Scholar] [CrossRef]
- Manoharan, P.; Basford, J.E.; Pilcher-Roberts, R.; Neumann, J.; Hui, D.Y.; Lingrel, J.B. Reduced levels of microRNAs miR-124a and miR-150 are associated with increased proinflammatory mediator expression in Kruppel-like factor 2 (KLF2)-deficient macrophages. J. Biol. Chem. 2014, 289, 31638–31646. [Google Scholar] [CrossRef]
- Mahabeleshwar, G.H.; Kawanami, D.; Sharma, N.; Takami, Y.; Zhou, G.; Shi, H.; Nayak, L.; Jeyaraj, D.; Grealy, R.; White, M.; et al. The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock. Immunity 2011, 34, 715–728. [Google Scholar] [CrossRef]
- Tuomisto, T.T.; Lumivuori, H.; Kansanen, E.; Hakkinen, S.K.; Turunen, M.P.; van Thienen, J.V.; Horrevoets, A.J.; Levonen, A.L.; Yla-Herttuala, S. Simvastatin has an anti-inflammatory effect on macrophages via upregulation of an atheroprotective transcription factor, Kruppel-like factor 2. Cardiovasc. Res. 2008, 78, 175–184. [Google Scholar] [CrossRef]
- Roberts, A.W.; Lee, B.L.; Deguine, J.; John, S.; Shlomchik, M.J.; Barton, G.M. Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity 2017, 47, 913–927.e916. [Google Scholar] [CrossRef]
- Lingrel, J.B.; Pilcher-Roberts, R.; Basford, J.E.; Manoharan, P.; Neumann, J.; Konaniah, E.S.; Srinivasan, R.; Bogdanov, V.Y.; Hui, D.Y. Myeloid-specific Kruppel-like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circ. Res. 2012, 110, 1294–1302. [Google Scholar] [CrossRef]
- Atkins, G.B.; Wang, Y.; Mahabeleshwar, G.H.; Shi, H.; Gao, H.; Kawanami, D.; Natesan, V.; Lin, Z.; Simon, D.I.; Jain, M.K. Hemizygous deficiency of Kruppel-like factor 2 augments experimental atherosclerosis. Circ. Res. 2008, 103, 690–693. [Google Scholar] [CrossRef]
- Manoharan, P.; Song, T.; Radzyukevich, T.L.; Sadayappan, S.; Lingrel, J.B.; Heiny, J.A. KLF2 in Myeloid Lineage Cells Regulates the Innate Immune Response during Skeletal Muscle Injury and Regeneration. iScience 2019, 17, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.V.; Lapping, S.; Maiseyeu, A.; Schmaier, A.; Jain, M. Neutrophil KLF2 Regulates Arterial and Venous Thrombosis. Blood 2018, 132 (Suppl. 1), 75. [Google Scholar] [CrossRef]
- Zhu, L.M.; Zeng, D.; Lei, X.C.; Huang, J.; Deng, Y.F.; Ji, Y.B.; Liu, J.; Dai, F.F.; Li, Y.Z.; Shi, D.D.; et al. KLF2 regulates neutrophil migration by modulating CXCR1 and CXCR2 in asthma. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165920. [Google Scholar] [CrossRef] [PubMed]
- Alberts-Grill, N.; Engelbertsen, D.; Bu, D.; Foks, A.; Grabie, N.; Herter, J.M.; Kuperwaser, F.; Chen, T.; Destefano, G.; Jarolim, P.; et al. Dendritic Cell KLF2 Expression Regulates T Cell Activation and Proatherogenic Immune Responses. J. Immunol. 2016, 197, 4651–4662. [Google Scholar] [CrossRef]
- Deng, Y.; Kerdiles, Y.; Chu, J.; Yuan, S.; Wang, Y.; Chen, X.; Mao, H.; Zhang, L.; Zhang, J.; Hughes, T.; et al. Transcription factor Foxo1 is a negative regulator of natural killer cell maturation and function. Immunity 2015, 42, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Fabre, S.; Carrette, F.; Chen, J.; Lang, V.; Semichon, M.; Denoyelle, C.; Lazar, V.; Cagnard, N.; Dubart-Kupperschmitt, A.; Mangeney, M.; et al. FOXO1 regulates L-Selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J. Immunol. 2008, 181, 2980–2989. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Limon, J.J.; Blanc, C.; Peng, S.L.; Fruman, D.A. Foxo1 regulates marginal zone B-cell development. Eur. J. Immunol. 2010, 40, 1890–1896. [Google Scholar] [CrossRef]
- Crinier, A.; Milpied, P.; Escaliere, B.; Piperoglou, C.; Galluso, J.; Balsamo, A.; Spinelli, L.; Cervera-Marzal, I.; Ebbo, M.; Girard-Madoux, M.; et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 2018, 49, 971–986.e975. [Google Scholar] [CrossRef]
- Rabacal, W.; Pabbisetty, S.K.; Hoek, K.L.; Cendron, D.; Guo, Y.; Maseda, D.; Sebzda, E. Transcription factor KLF2 regulates homeostatic NK cell proliferation and survival. Proc. Natl. Acad. Sci. USA 2016, 113, 5370–5375. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, N.; Kekalainen, E.; Chen, P.; Lourda, M.; Wilson, J.N.; Scharenberg, M.; Bergman, P.; Al-Ameri, M.; Hard, J.; Mold, J.E.; et al. Unique transcriptional and protein-expression signature in human lung tissue-resident NK cells. Nat. Commun. 2019, 10, 3841. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wittner, J.; Schuh, W. Krüppel-like Factor 2 (KLF2) in Immune Cell Migration. Vaccines 2021, 9, 1171. https://doi.org/10.3390/vaccines9101171
Wittner J, Schuh W. Krüppel-like Factor 2 (KLF2) in Immune Cell Migration. Vaccines. 2021; 9(10):1171. https://doi.org/10.3390/vaccines9101171
Chicago/Turabian StyleWittner, Jens, and Wolfgang Schuh. 2021. "Krüppel-like Factor 2 (KLF2) in Immune Cell Migration" Vaccines 9, no. 10: 1171. https://doi.org/10.3390/vaccines9101171
APA StyleWittner, J., & Schuh, W. (2021). Krüppel-like Factor 2 (KLF2) in Immune Cell Migration. Vaccines, 9(10), 1171. https://doi.org/10.3390/vaccines9101171