
Antibody-Drug Conjugates: Functional Principles and Applications in Oncology and Beyond

 , and
, and
Abstract
:1. Introduction
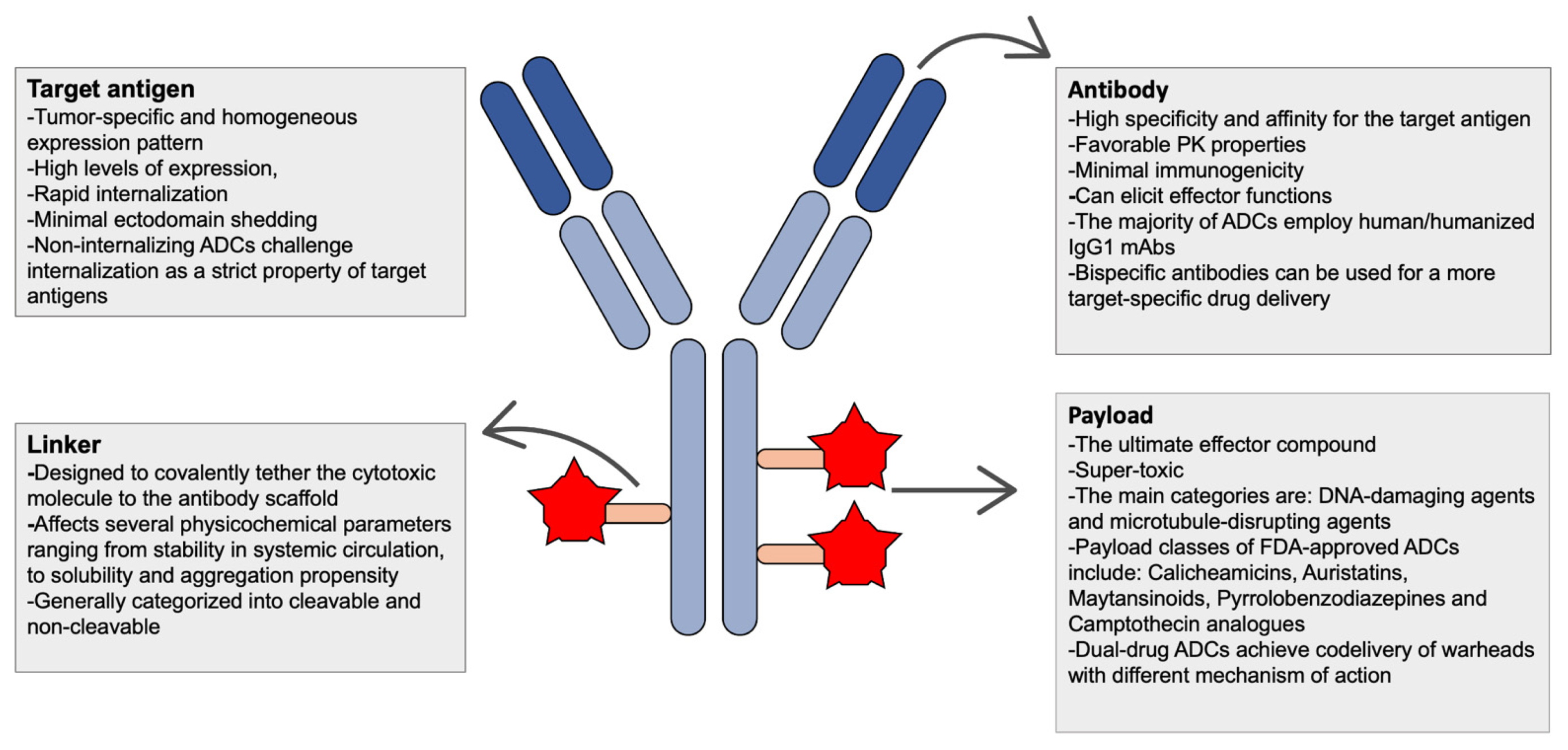
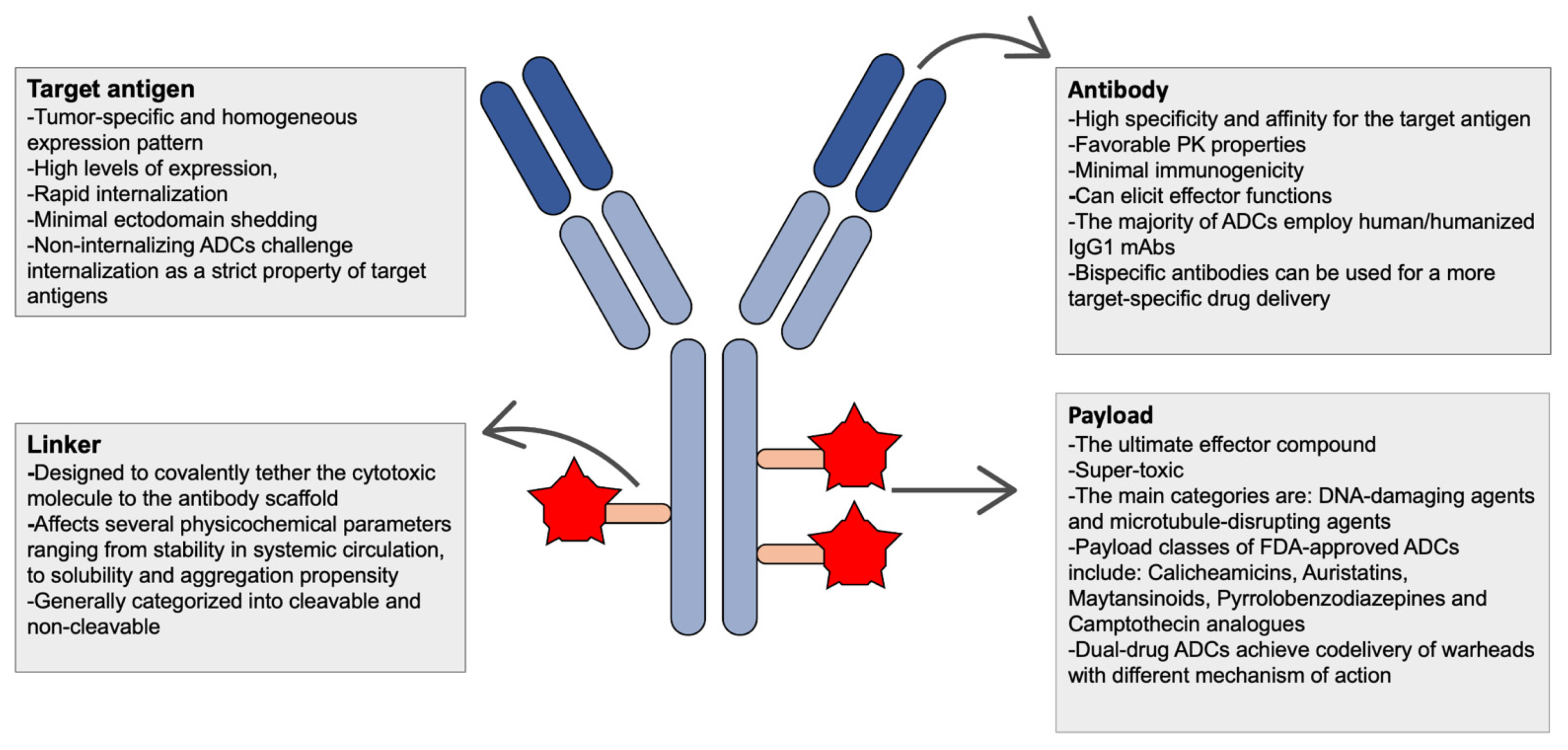
| Essential components of ADCS | ||
| ||
| Types | Considerations for Abs | Considerations for target antigen |
|
|
|
| ||
| Types | Considerations for linker | Considerations for conjugation |
|
|
|
| ||
| Types | Considerations for payload | Mechanisms of drug resistance |
|
|
|
2. Functional Principles and Essential Components of ADCs
2.1. Antigen Selection and Antibodies
2.2. Linkers and Conjugation Technologies
2.3. Payloads
2.3.1. Calicheamicins
2.3.2. Pyrrolobenzodiazepines (PBDs)
2.3.3. Auristatins
2.3.4. Maytansinoids
2.3.5. Camptothecin (CPT)
2.3.6. Dual-Drug ADCs
3. Bystander Killing and Resistance Phenomena
3.1. Bystander Killing Effect
3.2. Drug Resistance
3.2.1. Antigen-Related Resistance
3.2.2. Deficient Lysosomal Function
3.2.3. Upregulated Efflux Pumps
3.2.4. Survival/Apoptotic Signaling
3.2.5. Binding-Site Barrier (BSB) Phenomenon
4. Applications of ADCs for Oncological and Non-Oncological Conditions

{kind=link}
| ADC | Indication | Antibody | Linker | DAR | Testing Status | Initial Publication, Year | Reference |
|---|---|---|---|---|---|---|---|
| Anti-E Selectin dexamethasone (Dexa–AbhEsel) | Chronic models of inflammation | Murine anti-E-selectin mAb (H18/7) | Succinate linker | 2.3 | In vitro preclinical | J Immunol, 2002 | [134] |
| Anti-CD163 dexamethasone (Cymac-001) | Chronic models of inflammation | Murine anti-CD163 mAb (Ed-2) | Hemisuccinate linker | ~4 | In vivo preclinical | Mol Ther, 2012 | [125] |
| Anti-CD74 fluticasone propionate (Anti-CD74-flu449) | Autoimmune models | Human anti-CD74 mAb | Pyrophosphate acetal linker | ≥1.7 | In vivo preclinical | Bioconjug Chem, 2018 | [135] |
| Anti-CD70 budesonide | Chronic models of inflammation | Murine anti- CD70 mAb (Bu69) | CatPhos linker | 1.9 | In vitro preclinical | Bioconjug Chem, 2016 | [136] |
| Anti-CXCR4 dasatinib | Autoimmune and inflammatory models | Humanized anti-CXCR4 mAb (HLCX) | Tetra-poly-ethylene glycol linker | ~3 | In vitro preclinical | J Am Chem Soc, 2015 | [137] |
| Anti-CD11a PDE4 inhibitor | Chronic models of inflammation | Humanized anti-CD11 mAb | PEG4-Phe-Lys | ~2 | In vivo preclinical | Mol Ther, 2016 | [138] |
| Anti-CD11a LXR agonist | Atherosclerosis | Humanized anti-CD11 mAb | PEG4-Phe-Lys | 2 | In vitro preclinical | Bioconjug Chem, 2015 | [139] |
| Anti-CD71 siRNA | Muscular diseases | Murine ant-CD71 mAb | Maleimide linker | N/A | In vivo preclinical | J Control Release, 2016 | [140] |
| Anti-TNFRSF13c siRNA | Myasthenia gravis | Murine anti-TNFRSF13c mAb | Protamine linker | N/A | In vivo preclinical | Clin Immunol, 2017 | [141] |
| Anti-IL-7R MMAE (A7R-ADC-MMAE) | Steroid-resistant arthritis | Murine anti-IL-7R mAb | Val-Cit linker | N/A | In vivo preclinical | Sci Rep, 2017 | [142] |
| Anti-CD30 vedotin (ADCETRIS) | Systemic sclerosis | Chimeric anti-CD30 mAb (cAC10, SGN-30) | Val-Cit linker | ~4 | Phase II clinical trial (NCT03198689), (NCT03222492) | Ann Rheum Dis, 2021 | [4] |
| Anti-CD117 saporin (C117-ADC) | Conditioning for HSCT | Murine anti-CD117 mAb | N/A | N/A | In vivo preclinical | Nat Commun, 2019 | [143] |
| Anti-CD45 saporin (CD45-SAP) | Conditioning for HSCT | Murine anti-CD45 mAb | N/A | N/A | In vivo preclinical | Nat Biotechnol, 2016 | [144] |
| Anti-IL-6 alendronate | Rheumatoid arthritis | Humanized anti-IL-6 mAb (tocilizumab) | PDPH-PEG-NHS | N/A | In vivo preclinical | Bioconjug Chem, 2017 | [145] |
| Anti–C5aR1 C5 siRNA | Rheumatoid arthritis | Murine anti-C5aR1 mAb | Protamine linker | N/A | In vivo preclinical | J Immunol, 2015 | [146] |
| Anti-FRβ Pseudomonas exotoxin A (PE38) | Rheumatoid arthritis | Murine anti-FRβ mAb | N/A | N/A | In vivo preclinical | Arthritis Rheumatol, 2006 | [147] |
| Anti-TNFα glucocorticoid (ABBV-3373) | Rheumatoid arthritis | N/A | N/A | N/A | Phase II clinical trial (NCT03823391) | Ann Rheum Dis, 2021 | [5] |
| Anti-S. aureus antibiotic (DSTA4637S) | S. aureus bacteremia | Human anti-β-N- acetylglucosamine cell-wall teichoic acid (β-GlcNAc- WTA) mAb | MC-Val-Cit-PAB-OH | 2 | Phase I clinical trial (NCT03162250) | Nature, 2015 | [129] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCs | Antibody-drug conjugates |
| mAbs | Monoclonal antibodies |
| bsADCs | Bispecific ADCs |
| BSB | Binding-Site Barrier |
| PRLR | Prolactin receptor |
| GO | Gemtuzumab ozogamicin |
| BV | Brentuximab Vedotin |
| DAR | Drug-to-antibody ratio |
| T-DM1 | Τrastuzumab emtasine |
| AML | Acute myeloid leukemia |
| Val-Cit | Valine-citrulline |
| PABC | Para-amino-benzyloxycarbonyl |
| PBDs | Pyrrolobenzodiazepines |
| TRX | 1,2,4-trioxolane |
| uAA | Unnatural amino-acids |
| mTG | Microbial transglutaminase |
| SortA | Sortase A |
| FGE | Formylglycine-generating enzyme |
| V-ATPase | Vacuolar H+-ATPase |
| CPT | Camptothecin |
| AF-HPA | Auristatin F-hydroxypropylamide |
| MMAE | Monomethyl auristatin E |
| MMAF | Monomethyl auristatin F |
| TM-ADC | Trastuzumab–maytansinoid ADC |
| AAC | Antibody-antibiotic conjugate |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| MSSA | Methicillin-susceptible Staphylococcus aureus |
References
- Theocharopoulos, C.; Lialios, P.-P.; Gogas, H.; Ziogas, D.C. An overview of Antibody-Drug conjugates in oncological practice. Ther. Adv. Med. Oncol. 2020, 12, 1758835920962997. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody-Drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of Antibody-Drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Codina, A.; Nevskaya, T.; Pope, J. OP0172 brentuximab vedontin for skin involvement in refractory diffuse cutaneous systemic sclerosis, interim results of a phase IIb open-label trial. Ann. Rheum. Dis. 2021, 80, 103–104. [Google Scholar] [CrossRef]
- Stoffel, B.; McPherson, M.; Hernandez, A.; Goess, C.; Mathieu, S.; Waegell, W.; Bryant, S.; Hobson, A.; Ruzek, M.; Pang, Y.; et al. POS0365 anti-TNF glucocorticoid receptor modulator antibody drug conjugate for the treatment of autoimmune diseases. Ann. Rheum. Dis. 2021, 80, 412–413. [Google Scholar] [CrossRef]
- Peck, M.; Rothenberg, M.E.; Deng, R.; Lewin-Koh, N.; She, G.; Kamath, A.V.; Carrasco-Triguero, M.; Saad, O.; Castro, A.; Teufel, L.; et al. A Phase 1, Randomized, Single-Ascending-Dose Study to Investigate the Safety, Tolerability, and Pharmacokinetics of DSTA4637S, an Anti-Staphylococcus aureus Thiomab Antibody-Antibiotic Conjugate, in Healthy Volunteers. Antimicrob. Agents Chemother. 2019, 63, e02588-18. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.; Hollingsworth, R. Selecting Optimal Antibody-Drug Conjugate Targets Using Indication-Dependent or Indication-Independent Approaches: Fundamentals, Drug Development, and Clinical Outcomes to Target Cancer; John Wiley & Sons, Inc.: New York, NY, USA, 2016; pp. 33–58. [Google Scholar]
- Ritchie, M.; Bloom, L.; Carven, G.; Sapra, P. Selecting an Optimal Antibody for Antibody- Drug Conjugate Therapy. In Antibody-Drug Conjugates; AAPS Advances in the Pharmaceutical Sciences Series; Springer: Cham, Switzerland, 2015; Volume 17, pp. 23–48. [Google Scholar] [CrossRef]
- Goldmacher, V.S.; Kovtun, Y.V. Antibody-Drug conjugates: Using monoclonal antibodies for delivery of cytotoxic payloads to cancer cells. Ther. Deliv. 2011, 2, 397–416. [Google Scholar] [CrossRef] [Green Version]
- Bander, N.H. Antibody-Drug Conjugate Target Selection: Critical Factors. In Antibody-Drug Conjugates; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1045, pp. 29–40. [Google Scholar] [CrossRef]
- Sharma, S.; Li, Z.; Bussing, D.; Shah, D.K. Evaluation of Quantitative Relationship Between Target Expression and Antibody-Drug Conjugate Exposure Inside Cancer Cells. Drug Metab. Dispos. 2020, 48, 368–377. [Google Scholar] [CrossRef]
- Bussing, D.; Sharma, S.; Li, Z.; Meyer, L.F.; Shah, D.K. Quantitative Evaluation of the Effect of Antigen Expression Level on Antibody-Drug Conjugate Exposure in Solid Tumor. AAPS J. 2021, 23, 1–11. [Google Scholar] [CrossRef]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; De Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-Drug Conjugates for the Treatment of Non–Hodgkin’s Lymphoma: Target and Linker-Drug Selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef] [Green Version]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Perrino, E.; Steiner, M.; Krall, N.; Bernardes, G.; Pretto, F.; Casi, G.; Neri, D. Curative Properties of Noninternalizing Antibody-Drug Conjugates Based on Maytansinoids. Cancer Res. 2014, 74, 2569–2578. [Google Scholar] [CrossRef] [Green Version]
- Gébleux, R.; Stringhini, M.; Casanova, R.; Soltermann, A.; Neri, D. Non-internalizing antibody-drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. Int. J. Cancer 2016, 140, 1670–1679. [Google Scholar] [CrossRef] [Green Version]
- Rossin, R.; Versteegen, R.M.; Wu, J.; Khasanov, A.; Wessels, H.J.; Steenbergen, E.J.; Hoeve, W.T.; Janssen, H.M.; Van Onzen, A.H.A.M.; Hudson, P.J.; et al. Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun. 2018, 9, 1484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pastan, I. High Shed Antigen Levels within Tumors: An Additional Barrier to Immunoconjugate Therapy. Clin. Cancer Res. 2008, 14, 7981–7986. [Google Scholar] [CrossRef] [Green Version]
- Bogen, J.P.; Hinz, S.C.; Grzeschik, J.; Ebenig, A.; Krah, S.; Zielonka, S.; Kolmar, H. Dual Function pH Responsive Bispecific Antibodies for Tumor Targeting and Antigen Depletion in Plasma. Front. Immunol. 2019, 10, 1892. [Google Scholar] [CrossRef] [Green Version]
- Pak, Y.; Zhang, Y.; Pastan, I.; Lee, B. Antigen Shedding May Improve Efficiencies for Delivery of Antibody-Based Anticancer Agents in Solid Tumors. Cancer Res. 2012, 72, 3143–3152. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). OncoImmunology 2017, 7, e1395127. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldmann, H. Human Monoclonal Antibodies: The Benefits of Humanization. In Human Monoclonal Antibodies; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1904, pp. 1–10. [Google Scholar] [CrossRef]
- De Taeye, S.W.; Rispens, T.; Vidarsson, G. The Ligands for Human IgG and Their Effector Functions. Antibodies 2019, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, R.; Hammond, S.A.; Oberst, M.; Wilkinson, R.W. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J. Immunother. Cancer 2014, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Song, Y.; Tian, W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 2020, 13, 45. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of Antibody-Drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Ackerman, M.E.; Pawlowski, D.; Wittrup, K.D. Effect of antigen turnover rate and expression level on antibody penetration into tumor spheroids. Mol. Cancer Ther. 2008, 7, 2233–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2016, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Comer, F.; Gao, C.; Coats, S. Bispecific and Biparatopic Antibody Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Cancer Drug Discovery and Development; Humana Press: Cham, Switzerland, 2018; pp. 267–280. [Google Scholar]
- Maruani, A. Bispecifics and Antibody-Drug conjugates: A positive synergy. Drug Discov. Today Technol. 2018, 30, 55–61. [Google Scholar] [CrossRef] [Green Version]
- De Goeij, B.E.; Vink, T.; Napel, H.T.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P. Efficient Payload Delivery by a Bispecific Antibody-Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef] [Green Version]
- Andreev, J.; Thambi, N.; Bay, A.E.P.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody-Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Bay, A.P.; Kalsy, A.; Tiwari, S.; Luan, B.; Kunz, A.; Chen, Z.; Zhang, L.; Potocky, T.; Nittoli, T.; Thurston, G.; et al. Abstract 233: Bispecific HER2 ADC: Making more potent HER2 ADC by improving target internalization. In Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; Volume 79. [Google Scholar]
- Bethell, R.; Eberlein, C.; Pollard, V.; Georgiou, T.; Orphanides, G.; Mooney, L.; Kendrew, J.; Daniels, T. Abstract 2887: Bispecific antibody drug conjugates targeting CD7 and CD33 for the treatment of acute myeloid leukemia. In Proceedings of the AACR Annual Meeting 2020, Philadelphia, PA, USA, 27–28 April and 22–24 June 2020; Volume 80. [Google Scholar] [CrossRef]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; DiMasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2019, 35, 948–949. [Google Scholar] [CrossRef] [Green Version]
- Pegram, M.D.; Hamilton, E.P.; Tan, A.R.; Storniolo, A.M.; Balic, K.; Rosenbaum, A.I.; Liang, M.; He, P.; Marshall, S.; Scheuber, A.; et al. First-in-Human, Phase 1 Dose-Escalation Study of Biparatopic Anti-HER2 Antibody-Drug Conjugate MEDI4276 in Patients with HER2+ Advanced Breast or Gastric Cancer. Mol. Cancer Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in Antibody-Drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody−Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjug. Chem. 2009, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Bryden, F.; Martin, C.; Letast, S.; Lles, E.; Viéitez-Villemin, I.; Rousseau, A.; Colas, C.; Brachet-Botineau, M.; Allard-Vannier, E.; Larbouret, C.; et al. Impact of cathepsin B-sensitive triggers and hydrophilic linkers on in vitro efficacy of novel site-specific Antibody-Drug conjugates. Org. Biomol. Chem. 2018, 16, 1882–1889. [Google Scholar] [CrossRef]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2016, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and Properties of β-Glucuronide Linkers for Monoclonal Antibody−Drug Conjugates. Bioconjug. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef]
- Conilh, L.; Fournet, G.; Fourmaux, E.; Murcia, A.; Matera, E.-L.; Joseph, B.; Dumontet, C.; Viricel, W. Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform. Pharmaceuticals 2021, 14, 247. [Google Scholar] [CrossRef]
- Nolting, B. Linker Technologies for Antibody-Drug Conjugates. In Antibody-Drug Conjugates; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1045, pp. 71–100. [Google Scholar] [CrossRef]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor Delivery and In Vivo Processing of Disulfide-Linked and Thioether-Linked Antibody−Maytansinoid Conjugates. Bioconjug. Chem. 2009, 21, 84–92. [Google Scholar] [CrossRef]
- Kern, J.C.; Cancilla, M.T.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.; Liang, L.; Tomazela, D.; et al. Discovery of Pyrophosphate Diesters as Tunable, Soluble, and Bioorthogonal Linkers for Site-Specific Antibody-Drug Conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef]
- Kolodych, S.; Michel, C.; Delacroix, S.; Koniev, O.; Ehkirch, A.; Eberova, J.; Cianférani, S.; Renoux, B.; Krezel, W.; Poinot, P.; et al. Development and evaluation of β-galactosidase-sensitive antibody-drug conjugates. Eur. J. Med. Chem. 2017, 142, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Walsh, S.J.; Ashman, N.; Isidro-Llobet, A.; Carroll, J.S.; Spring, D.R. A dual-enzyme cleavable linker for Antibody-Drug conjugates. Chem. Commun. 2021, 57, 3457–3460. [Google Scholar] [CrossRef]
- Kumar, A.; Kinneer, K.; Masterson, L.; Ezeadi, E.; Howard, P.; Wu, H.; Gao, C.; Dimasi, N. Synthesis of a heterotrifunctional linker for the site-specific preparation of antibody-drug conjugates with two distinct warheads. Bioorg. Med. Chem. Lett. 2018, 28, 3617–3621. [Google Scholar] [CrossRef] [PubMed]
- Spangler, B.; Kline, T.; Hanson, J.; Li, X.; Zhou, S.; Wells, J.A.; Sato, A.K.; Renslo, A.R. Toward a Ferrous Iron-Cleavable Linker for Antibody-Drug Conjugates. Mol. Pharm. 2018, 15, 2054–2059. [Google Scholar] [CrossRef]
- Merkul, E.; Sijbrandi, N.J.; Muns, J.A.; Aydin, I.; Adamzek, K.; Houthoff, H.-J.; Nijmeijer, B.; Van Dongen, G.A. First platinum(II)-based metal-organic linker technology (Lx®) for a plug-and-play development of antibody-drug conjugates (ADCs). Expert Opin. Drug Deliv. 2019, 16, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovgan, I.; Ehkirch, A.; Lehot, V.; Kuhn, I.; Koniev, O.; Kolodych, S.; Hentz, A.; Ripoll, M.; Ursuegui, S.; Nothisen, M.; et al. On the use of DNA as a linker in antibody-drug conjugates: Synthesis, stability and in vitro potency. Sci. Rep. 2020, 10, 7691–7699. [Google Scholar] [CrossRef]
- Li, J.; Xiao, D.; Xie, F.; Li, W.; Zhao, L.; Sun, W.; Yang, X.; Zhou, X. Novel antibody-drug conjugate with UV-controlled cleavage mechanism for cytotoxin release. Bioorg. Chem. 2020, 111, 104475. [Google Scholar] [CrossRef]
- Bird, M.; Nunes, J.; Frigerio, M. Bridged Cysteine Conjugations. In Antibody-Drug Conjugates; Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2078, pp. 113–129. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Tian, F.; Lu, Y.; Manibusan, A.; Sellers, A.; Tran, H.; Sun, Y.; Phuong, T.; Barnett, R.; Hehli, B.; Song, F.; et al. A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 1766–1771. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tran, C.; Uter, N.T.; Yin, G.; Rivers, P.J.; et al. Production of Site-Specific Antibody-Drug Conjugates Using Optimized Non-Natural Amino Acids in a Cell-Free Expression System. Bioconjug. Chem. 2014, 25, 351–361. [Google Scholar] [CrossRef]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grünberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-Specific and Stoichiometric Modification of Antibodies by Bacterial Transglutaminase. Angew. Chem. Int. Ed. 2010, 49, 9995–9997. [Google Scholar] [CrossRef]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Brégeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-Based Chemo-Enzymatic Conjugation Approach Yields Homogeneous Antibody-Drug Conjugates. Bioconjug. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef]
- Strop, P.; Liu, S.-H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.-T.; Ho, W.-H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anami, Y.; Xiong, W.; Gui, X.; Deng, M.; Zhang, C.C.; Zhang, N.; An, Z.; Tsuchikama, K. Enzymatic conjugation using branched linkers for constructing homogeneous Antibody-Drug conjugates with high potency. Org. Biomol. Chem. 2017, 15, 5635–5642. [Google Scholar] [CrossRef]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In Vitro and In Vivo Potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; De Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-Specific Antibody-Drug Conjugation through Glycoengineering. Bioconjug. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Shi, W.; Huang, W. Homogeneous Antibody-Drug Conjugates via Glycoengineering. In Bioconjugation; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 2033, pp. 221–238. [Google Scholar] [CrossRef]
- Maiese, W.M.; Lechevalier, M.P.; Lechevalier, H.A.; Korshalla, J.; Kuck, N.; Fantini, A.; Wildey, M.J.; Thomas, J.; Greenstein, M. Calicheamicins, a novel family of antitumor antibiotics. Taxonomy, fermentation and biological properties. J. Antibiot. 1989, 42, 558–563. [Google Scholar] [CrossRef]
- Ellestad, G.A. Structural and conformational features relevant to the anti-tumor activity of calicheamicin γ1I? 1I. Chirality 2011, 23, 660–671. [Google Scholar] [CrossRef]
- Vollmar, B.S.; Frantz, C.; Schutten, M.M.; Zhong, F.; del Rosario, G.; Go, M.A.T.; Yu, S.-F.; Leipold, D.D.; Kamath, A.V.; Ng, C.; et al. Calicheamicin Antibody-Drug Conjugates with Improved Properties. Mol. Cancer Ther. 2021, 20, 1112–1120. [Google Scholar] [CrossRef]
- Hartley, J.A.; Spanswick, V.J.; Brooks, N.; Clingen, P.H.; McHugh, P.J.; Hochhauser, D.; Pedley, R.B.; Kelland, L.R.; Alley, M.C.; Schultz, R.; et al. SJG-136 (NSC 694501), a Novel Rationally Designed DNA Minor Groove Interstrand Cross-Linking Agent with Potent and Broad Spectrum Antitumor Activity: Part 1: Cellular Pharmacology, In vitro and Initial In vivo Antitumor Activity. Cancer Res. 2004, 64, 6693–6699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Khojasteh, S.C.; Hop, C.E.; Erickson, H.K.; Polson, A.; Pillow, T.H.; Yu, S.-F.; Wang, H.; Dragovich, P.S.; Zhang, D. Antibody Drug Conjugates Differentiate Uptake and DNA Alkylation of Pyrrolobenzodiazepines in Tumors from Organs of Xenograft Mice. Drug Metab. Dispos. 2016, 44, 1958–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef]
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.; Sussman, D.; Prota, A.E. Structural Basis of Microtubule Destabilization by Potent Auristatin Anti-Mitotics. PLoS ONE 2016, 11, e0160890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, S.O.; Mendelsohn, B.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced Activity of Monomethylauristatin F through Monoclonal Antibody Delivery: Effects of Linker Technology on Efficacy and Toxicity. Bioconjug. Chem. 2005, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, A.V.; Bodyak, N.D.; Yin, M.; Thomas, J.D.; Clardy, S.M.; Conlon, P.R.; Stevenson, C.A.; Uttard, A.; Qin, L.; Gumerov, D.R.; et al. Dolaflexin: A Novel Antibody-Drug Conjugate Platform Featuring High Drug Loading and a Controlled Bystander Effect. Mol. Cancer Ther. 2021, 20, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Bodyak, N.D.; Mosher, R.; Yurkovetskiy, A.V.; Yin, M.; Bu, C.; Conlon, P.R.; Demady, D.R.; DeVit, M.J.; Gumerov, D.R.; Gurijala, V.R.; et al. The Dolaflexin-based Antibody-Drug Conjugate XMT-1536 Targets the Solid Tumor Lineage Antigen SLC34A2/NaPi2b. Mol. Cancer Ther. 2021, 20, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Desai, S.D.; Li, T.-K.; Mao, Y.; Sun, M.; Sim, S.-P. Mechanism of Action of Camptothecin. Ann. N. Y. Acad. Sci. 2000, 922, 1–10. [Google Scholar] [CrossRef]
- Levengood, M.R.; Zhang, X.; Hunter, J.H.; Emmerton, K.K.; Miyamoto, J.B.; Lewis, T.S.; Senter, P.D. Orthogonal Cysteine Protection Enables Homogeneous Multi-Drug Antibody-Drug Conjugates. Angew. Chem. Int. Ed. 2016, 56, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor Cells Chronically Treated with a Trastuzumab–Maytansinoid Antibody-Drug Conjugate Develop Varied Resistance Mechanisms but Respond to Alternate Treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levengood, M.R.; Zhang, X.; Emmerton, K.K.; Hunter, J.H.; Senter, P.D. Abstract 982: Development of homogeneous dual-drug ADCs: Application to the co-delivery of auristatin payloads with complementary antitumor activities. In Proceedings of the AACR Annual Meeting 2017, Washington, DC, USA, 1–5 April 2017; Volume 77. [Google Scholar] [CrossRef]
- Duvall, J.R.; Damelin, M.; Kozytska, M.V.; Nehilla, B.J.; Protopopova, M.; Conlon, P.R.; Qin, L.; Nazzaro, M.; Thomas, J.D.; Zhang, Q.; et al. Abstract 232: An antibody-drug conjugate carrying a microtubule inhibitor and a DNA alkylator exerts both mechanisms of action on tumor cells. In Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; Volume 79. [Google Scholar] [CrossRef]
- Nilchan, N.; Li, X.; Pedzisa, L.; Nanna, A.R.; Roush, W.R.; Rader, C. Dual-mechanistic antibody-drug conjugate via site-specific selenocysteine/cysteine conjugation. Antib. Ther. 2019, 2, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 2021, 12, 3528. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef] [Green Version]
- McCombs, J.R.; Owen, S.C. Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Sharma, S.; Shah, D.K. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J. Pharmacokinet. Pharmacodyn. 2016, 43, 567–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtun, Y.V.; Goldmacher, V.S. Cell killing by Antibody-Drug conjugates. Cancer Lett. 2007, 255, 232–240. [Google Scholar] [CrossRef]
- Burton, J.K.; Bottino, D.; Secomb, T.W. A Systems Pharmacology Model for Drug Delivery to Solid Tumors by Antibody-Drug Conjugates: Implications for Bystander Effects. AAPS J. 2019, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Seigel, G.M.; Guo, L.; Verma, A.; Wong, G.G.-L.; Chang, H.-P.; Shah, D.K. Evolution of the Systems Pharmacokinetics-Pharmacodynamics Model for Antibody-Drug Conjugates to Characterize Tumor Heterogeneity and In Vivo Bystander Effect. J. Pharmacol. Exp. Ther. 2020, 374, 184–199. [Google Scholar] [CrossRef] [Green Version]
- Khera, E.; Cilliers, C.; Smith, M.D.; Ganno, M.L.; Lai, K.C.; Keating, T.A.; Kopp, A.; Nessler, I.; Abu-Yousif, A.O.; Thurber, G.M. Quantifying ADC bystander payload penetration with cellular resolution using pharmacodynamic mapping. Neoplasia 2020, 23, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Choi, J.U.; Cho, Y.S.; Kim, H.R.; Won, T.H.; Dimitrion, P.; Jeon, O.-C.; Kim, S.W.; Kim, I.-S.; Kim, S.Y.; et al. Self-Triggered Apoptosis Enzyme Prodrug Therapy (STAEPT): Enhancing Targeted Therapies via Recurrent Bystander Killing Effect by Exploiting Caspase-Cleavable Linker. Adv. Sci. 2018, 5, 1800368. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulou, A.; Zografos, E.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.-A.; Zagouri, F. Trastuzumab Deruxtecan (DS-8201a): The Latest Research and Advances in Breast Cancer. Clin. Breast Cancer 2020, 21, e212–e219. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 Antibody-Drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Collins, D.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired Resistance to Antibody-Drug Conjugates. Cancers 2019, 11, 394. [Google Scholar] [CrossRef] [Green Version]
- Hochberg, J.; Alexander, S. Resistance to Antibody-Drug Conjugate. In Resistance to Targeted Therapies in Lymphomas; Resistance to Targeted Anti-Cancer Therapeutics; Springer: Cham, Switzerland, 2019; pp. 57–69. [Google Scholar] [CrossRef]
- Luci, C.R.; García-Alonso, S.; Díaz-Rodriguez, E.; Nadal-Serrano, M.; Arribas, J.; Ocana, A.; Pandiella, A. Resistance to the Antibody-Drug Conjugate T-DM1 Is Based in a Reduction in Lysosomal Proteolytic Activity. Cancer Res. 2017, 77, 4639–4651. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, W.; Xu, Y.; Yang, Y.; Chen, X.; Quan, H.; Lou, L. Aberrant intracellular metabolism of T-DM1 confers T-DM1 resistance in human epidermal growth factor receptor 2-positive gastric cancer cells. Cancer Sci. 2017, 108, 1458–1468. [Google Scholar] [CrossRef]
- Trudeau, K.M.; Colby, A.H.; Zeng, J.; Las, G.; Feng, J.H.; Grinstaff, M.W.; Shirihai, O.S. Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity. J. Cell Biol. 2016, 214, 25–34. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocana, A.; Pandiella, A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.J.; Jacob, A.P.; Gurgel, J.L.; Tometsko, M.E.; Rock, B.M.; Patel, S.K.; Milburn, R.R.; Siu, S.; Ragan, S.P.; Rock, D.A.; et al. SLC46A3 Is Required to Transport Catabolites of Noncleavable Antibody Maytansine Conjugates from the Lysosome to the Cytoplasm. Cancer Res. 2015, 75, 5329–5340. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Herrera, A.F.; Hou, J.; Chen, L.; Wu, J.; Guo, Y.; Synold, T.W.; Ngo, V.; Puverel, S.; Mei, M.; et al. Inhibition of MDR1 Overcomes Resistance to Brentuximab Vedotin in Hodgkin Lymphoma. Clin. Cancer Res. 2019, 26, 1034–1044. [Google Scholar] [CrossRef]
- Rosen, D.B.; Harrington, K.H.; Cordeiro, J.A.; Leung, L.Y.; Putta, S.; Lacayo, N.; Laszlo, G.S.; Gudgeon, C.J.; Hogge, D.; Hawtin, R.E.; et al. AKT Signaling as a Novel Factor Associated with In Vitro Resistance of Human AML to Gemtuzumab Ozogamicin. PLoS ONE 2013, 8, e53518. [Google Scholar] [CrossRef] [PubMed]
- Maimaitili, Y.; Inase, A.; Miyata, Y.; Kitao, A.; Mizutani, Y.; Kakiuchi, S.; Shimono, Y.; Saito, Y.; Sonoki, T.; Minami, H.; et al. An mTORC1/2 kinase inhibitor enhances the cytotoxicity of gemtuzumab ozogamicin by activation of lysosomal function. Leuk. Res. 2018, 74, 68–74. [Google Scholar] [CrossRef]
- Li, G.; Guo, J.; Shen, B.-Q.; Yadav, D.B.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Phillips, G.D.L. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer Cells. Mol. Cancer Ther. 2018, 17, 1441–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, Q.; Gao, M.; Fu, L.; Li, Y.; Quan, H.; Lou, L. STAT3 activation confers trastuzumab-emtansine (T-DM1) resistance in HER2-positive breast cancer. Cancer Sci. 2018, 109, 3305–3315. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Raden, B.W.; Cronk, M.R.; Bernstein, I.D.; Appelbaum, F.R.; Banker, D.E.; Bello-Fernandez, C.; Stasakova, J.; Renner, A.; Carballido-Perrig, N.; et al. The peripheral benzodiazepine receptor ligand PK11195 overcomes different resistance mechanisms to sensitize AML cells to gemtuzumab ozogamicin. Blood 2004, 103, 4276–4284. [Google Scholar] [CrossRef]
- Saatci, Ö.; Borgoni, S.; Akbulut, Ö.; Durmuş, S.; Raza, U.; Eyüpoğlu, E.; Alkan, C.; Akyol, A.; Kutuk, O.; Wiemann, S.; et al. Targeting PLK1 overcomes T-DM1 resistance via CDK1-dependent phosphorylation and inactivation of Bcl-2/xL in HER2-positive breast cancer. Oncogene 2018, 37, 2251–2269. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Guo, L.; Verma, A.; Wong, G.G.-L.; Thurber, G.M.; Shah, D.K. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: A Quantitative Investigation. AAPS J. 2020, 22, 28. [Google Scholar] [CrossRef]
- Rudnick, S.I.; Adams, G.P. Affinity and Avidity in Antibody-Based Tumor Targeting. Cancer Biother. Radiopharm. 2009, 24, 155–161. [Google Scholar] [CrossRef]
- Khera, E.; Cilliers, C.; Bhatnagar, S.; Thurber, G.M. Computational transport analysis of antibody-drug conjugate bystander effects and payload tumoral distribution: Implications for therapy. Mol. Syst. Des. Eng. 2017, 3, 73–88. [Google Scholar] [CrossRef]
- Muchekehu, R.; Liu, D.; Horn, M.; Campbell, L.; Del Rosario, J.; Bacica, M.; Moskowitz, H.; Osothprarop, T.; Dirksen, A.; Doppalapudi, V.; et al. The Effect of Molecular Weight, PK, and Valency on Tumor Biodistribution and Efficacy of Antibody-Based Drugs. Transl. Oncol. 2013, 6, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar]
- Lazzerini, L.; Jöhrens, K.; Sehouli, J.; Cichon, G. Favorable therapeutic response after anti-Mesothelin Antibody-Drug conjugate treatment requires high expression of Mesothelin in tumor cells. Arch. Gynecol. Obstet. 2020, 302, 1255–1262. [Google Scholar] [CrossRef]
- Cilliers, C.; Guo, H.; Liao, J.; Christodolu, N.; Thurber, G.M. Multiscale Modeling of Antibody-Drug Conjugates: Connecting Tissue and Cellular Distribution to Whole Animal Pharmacokinetics and Potential Implications for Efficacy. AAPS J. 2016, 18, 1117–1130. [Google Scholar] [CrossRef] [Green Version]
- Rosellini, M.; Santoni, M.; Mollica, V.; Rizzo, A.; Cimadamore, A.; Scarpelli, M.; Storti, N.; Battelli, N.; Montironi, R.; Massari, F. Treating Prostate Cancer by Antibody-Drug Conjugates. Int. J. Mol. Sci. 2021, 22, 1551. [Google Scholar] [CrossRef] [PubMed]
- Koganemaru, S.; Shitara, K. Antibody-Drug conjugates to treat gastric cancer. Expert Opin. Biol. Ther. 2020, 21, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Agoston, A.T.; Guo, P.; Moses, M.A. A Rationally Designed ICAM1 Antibody Drug Conjugate for Pancreatic Cancer. Adv. Sci. 2020, 7, 2002852. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Lennernäs, H. Antibody-Drug Conjugates and Targeted Treatment Strategies for Hepatocellular Carcinoma: A Drug-Delivery Perspective. Molecules 2020, 25, 2861. [Google Scholar] [CrossRef]
- Graversen, J.H.; Svendsen, P.; Dagnæs-Hansen, F.; Dal, J.; Anton, G.; Etzerodt, A.; Petersen, M.D.; Christensen, P.A.; Møller, H.J.; Moestrup, S.K. Targeting the Hemoglobin Scavenger receptor CD163 in Macrophages Highly Increases the Anti-inflammatory Potency of Dexamethasone. Mol. Ther. 2012, 20, 1550–1558. [Google Scholar] [CrossRef] [Green Version]
- Møller, L.N.O.; Knudsen, A.R.; Andersen, K.J.; Nyengaard, J.R.; Hamilton-Dutoit, S.; Møller, E.M.O.; Svendsen, P.; Møller, H.J.; Moestrup, S.K.; Graversen, J.H.; et al. Anti-CD163-dexamethasone protects against apoptosis after ischemia/reperfusion injuries in the rat liver. Ann. Med. Surg. 2015, 4, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svendsen, P.; Graversen, J.H.; Etzerodt, A.; Hager, H.; Røge, R.; Grønbæk, H.; Christensen, E.I.; Møller, H.J.; Vilstrup, H.; Moestrup, S.K. Antibody-Directed Glucocorticoid Targeting to CD163 in M2-type Macrophages Attenuates Fructose-Induced Liver Inflammatory Changes. Mol. Ther. Methods Clin. Dev. 2017, 4, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Buttgereit, F.; Aelion, J.; Rojkovich, B.; Zubrzycka-Sienkiewicz, A.; Radstake, T.; Chen, S.; Arikan, D.; Kupper, H.; Amital, H. OP0115 efficacy and safety of ABBV-3373, a novel anti-tnf glucocorticoid receptor modulator antibody drug conjugate, in patients with moderate to severe rheumatoid arthritis despite methotrexate therapy: A phase 2a proof of concept study. Ann. Rheum. Dis. 2021, 80, 64. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.H.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Cai, H.; Yip, V.; Lee, M.V.; Wong, S.; Saad, O.; Ma, S.; Ljumanovic, N.; Khojasteh, S.C.; Kamath, A.V.; Shen, B.-Q. Characterization of Tissue Distribution, Catabolism, and Elimination of an Anti-Staphylococcus aureus THIOMAB Antibody-Antibiotic Conjugate in Rats. Drug Metab. Dispos. 2020, 48, 1161–1168. [Google Scholar] [CrossRef]
- Zhou, C.; Lehar, S.; Gutierrez, J.; Rosenberger, C.M.; Ljumanovic, N.; Dinoso, J.; Koppada, N.; Hong, K.; Baruch, A.; Carrasco-Triguero, M.; et al. Pharmacokinetics and pharmacodynamics of DSTA4637A: A novel THIOMAB™ antibody antibiotic conjugate against Staphylococcus aureus in mice. mAbs 2016, 8, 1612–1619. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Cai, H.; Baruch, A.; Lewin-Koh, N.; Yang, M.; Guo, F.; Xu, D.; Deng, R.; Hazenbos, W.; Kamath, A.V. Sustained activity of novel THIOMAB antibody-antibiotic conjugate against Staphylococcus aureus in a mouse model: Longitudinal pharmacodynamic assessment by bioluminescence imaging. PLoS ONE 2019, 14, e0224096. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Pan, B.; O’Connor, O.A. Brentuximab vedotin. Clin. Cancer Res. 2013, 19, 22–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everts, M.; Kok, R.J.; Asgeirsdottir, S.A.; Melgert, B.N.; Moolenaar, T.J.; Koning, G.A.; van Luyn, M.J.; Meijer, D.K.; Molema, G. Selective intracellular delivery of dexamethasone into activated endothelial cells using an E-selectin-directed immunoconjugate. J. Immunol. 2002, 168, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandish, P.E.; Palmieri, A.; Antonenko, S.; Beaumont, M.; Benso, L.; Cancilla, M.; Cheng, M.; Fayadat-Dilman, L.; Feng, G.; Figueroa, I.; et al. Development of Anti-CD74 Antibody-Drug Conjugates to Target Glucocorticoids to Immune Cells. Bioconjug. Chem. 2018, 29, 2357–2369. [Google Scholar] [CrossRef]
- Kern, J.C.; Dooney, D.; Zhang, R.; Liang, L.; Brandish, P.E.; Cheng, M.; Feng, G.; Beck, A.; Bresson, D.; Firdos, J.; et al. Novel Phosphate Modified Cathepsin B Linkers: Improving Aqueous Solubility and Enhancing Payload Scope of ADCs. Bioconjug. Chem. 2016, 27, 2081–2088. [Google Scholar] [CrossRef]
- Wang, R.E.; Liu, T.; Wang, Y.; Cao, Y.; Du, J.; Luo, X.; Deshmukh, V.; Kim, C.H.; Lawson, B.R.; Tremblay, M.S.; et al. An immunosuppressive antibody-drug conjugate. J. Am. Chem. Soc. 2015, 137, 3229–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Pearson, A.D.; Lim, R.K.; Rodgers, D.T.; Li, S.; Parker, H.B.; Weglarz, M.; Hampton, E.N.; Bollong, M.J.; Shen, J.; et al. Targeted Delivery of an Anti-inflammatory PDE4 Inhibitor to Immune Cells via an Antibody-drug Conjugate. Mol. Ther. 2016, 24, 2078–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, R.K.; Yu, S.; Cheng, B.; Li, S.; Kim, N.J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G.; et al. Targeted Delivery of LXR Agonist Using a Site-Specific Antibody-Drug Conjugate. Bioconjug. Chem. 2015, 26, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ibtehaj, N.; Huda, R. High-dose BAFF receptor specific mAb-siRNA conjugate generates Fas-expressing B cells in lymph nodes and high-affinity serum autoantibody in a myasthenia mouse model. Clin. Immunol. 2017, 176, 122–130. [Google Scholar] [CrossRef]
- Yasunaga, M.; Manabe, S.; Matsumura, Y. Immunoregulation by IL-7R-targeting antibody-drug conjugates: Overcoming steroid-resistance in cancer and autoimmune disease. Sci. Rep. 2017, 7, 10735. [Google Scholar] [CrossRef] [Green Version]
- Czechowicz, A.; Palchaudhuri, R.; Scheck, A.; Hu, Y.; Hoggatt, J.; Saez, B.; Pang, W.W.; Mansour, M.K.; Tate, T.A.; Chan, Y.Y.; et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat. Commun. 2019, 10, 617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palchaudhuri, R.; Saez, B.; Hoggatt, J.; Schajnovitz, A.; Sykes, D.B.; Tate, T.A.; Czechowicz, A.; Kfoury, Y.; Ruchika, F.; Rossi, D.J.; et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat. Biotechnol. 2016, 34, 738–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Bhang, S.H.; Lee, J.H.; Kim, H.; Hahn, S.K. Tocilizumab-Alendronate Conjugate for Treatment of Rheumatoid Arthritis. Bioconjug. Chem. 2017, 28, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Scheinman, R.I.; Holers, V.M.; Banda, N.K. A New Approach for the Treatment of Arthritis in Mice with a Novel Conjugate of an Anti-C5aR1 Antibody and C5 Small Interfering RNA. J. Immunol. 2015, 194, 5446–5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, T.; Tanaka, M.; Tsuneyoshi, Y.; Matsushita, K.; Sunahara, N.; Matsuda, T.; Yoshida, H.; Komiya, S.; Onda, M.; Matsuyama, T. In vitro and in vivo efficacy of a recombinant immunotoxin against folate receptor beta on the activation and proliferation of rheumatoid arthritis synovial cells. Arthritis Rheum. 2006, 54, 3126–3134. [Google Scholar] [CrossRef] [PubMed]
| ADC | Manufacturer | Year of Initial FDA Approval | Indications | Target | Antibody | Payload | Linker | DAR | Common Adverse Events (>10%) |
|---|---|---|---|---|---|---|---|---|---|
| Gemtuzumab Ozogamicin (Mylotarg®, CMA-676) | Pfizer/Wyeth | 2000, withdrawn 2010, re-approved 2017 | Newly diagnosed (de novo) CD33+ AML in adults (as a monotherapy or combined with chemotherapy) and pediatric patients 1 month and older (combined with chemotherapy) and relapsed/refractory CD33+ AML in adults and pediatric patients ≥ 2 years of age | CD33 | Humanized IgG4 | Calicheamicin derivative | Acid-labile hydrazone linker | ~2–3 | Infection, hemorrhage, thrombocytopenia, hypophosphatemia, hypokalemia, hyponatremia, nausea, vomiting, elevated ALP, elevated aminotransferase, fatigue, febrile neutropenia, constipation, abdominal pain, pyrexia, mucositis |
| Brentuximab Vedotin (Adcetris®, SGN-35) | Seattle Genetics, Millennium/ Takeda | 2011 | Previously untreated Stage III/IV cHL (combined with chemotherapy), cHL at high risk of relapse or progression as post-auto-HSCT consolidation, cHL after failure of auto-HSCT or after failure of ≥ 2 prior chemotherapy regimens, previously untreated sALCL or other CD30+ peripheral T-cell lymphomas (combined with chemotherapy), relapsed sALCL, relapsed peripheral cutaneous ALCL or CD30+ MF | CD30 | Chimeric IgG1 | MMAE | Protease-cleavable dipeptide (Val-Cit) linker | ~4 | Neutropenia, peripheral sensory neuropathy, fatigue, upper respiratory tract infection, nausea, diarrhea, anemia, thrombocytopenia, pyrexia, rash, abdominal pain, vomiting, arthralgia, myalgia, pruritus, peripheral motor neuropathy, headache, constipation, dizziness, lymphadenopathy, dyspnea, back pain, anxiety |
| Ado-trastuzumab emtansine (T-DM1, Kadcyla®) | Genentech, Roche | 2013 | Unresectable locally advanced or metastatic HER2+ breast cancer, previously treated with trastuzumab and a taxane, adjuvant treatment for HER2+ early breast cancer with residual invasive disease after neoadjuvant taxane and trastuzumab | HER2/ERB2 | Humanized IgG1 | DM1 | Thioether (non-cleavable) linker | 3.5 | Nausea, constipation, diarrhea, vomiting, abdominal pain, dry mouth, stomatitis, headache, peripheral neuropathy, dizziness, epistaxis, cough, dyspnea, fatigue, musculoskeletal pain, arthralgia, myalgia, pyrexia, thrombocytopenia, anemia, increased aminotransferases, insomnia, rash, hypokalemia |
| Inotuzumab ozogamicin (Besponsa®, CMC-544) | Pfizer/Wyeth | 2017 | Relapsed or refractory CD22+ B-cell precursor ALL in adults | CD22 | Humanized IgG4 | Calicheamicin derivative | Acid-labile hydrazone linker | ~4 | Thrombocytopenia, neutropenia, infection, anemia, leukopenia, nausea, fatigue, hemorrhage, pyrexia, elevated transaminases, febrile neutropenia, elevated gamma-glutamyltransferase, lymphopenia, headache, abdominal pain, diarrhea, constipation, vomiting, stomatitis, elevated ALP |
| Polatuzumab vedotin-piiq (Polivy®, DCDS4501A, RG7596) | Genentech, Roche | 2019 | Relapsed or refractory diffuse large B-cell lymphoma (combined with bendamustine and rituximab) in adult patients after ≥ 2 prior therapies | CD79b | Humanized IgG1 | MMAE | Protease-cleavable dipeptide (Val-Cit) linker | 3.5 | Neutropenia, thrombocytopenia, anemia, leukopenia, lymphopenia, febrile neutropenia, peripheral neuropathy, dizziness, diarrhea, vomiting, infusion-related reactions, pyrexia, decreased appetite, fatigue, pneumonia, upper respiratory tract infection, decreased weight, hypokalemia, hypoalbuminemia, hypocalcemia |
| Enfortumab vedotin (Padcev®, AGS-22M6E, AGS-22CE) | Astellas/ Seattle Genetics | 2019 | Locally advanced or metastatic urothelial cancer in adult patients who had received prior treatment with a PD-1/L1 inhibitor and platinum-based chemotherapy in neoadjuvant/adjuvant setting | Nectin 4 | Fully human IgG1 | MMAE | Protease-cleavable dipeptide (Val-Cit) linker | ~3.8 | Peripheral neuropathy, dysgeusia, fatigue, decreased appetite, rash, alopecia, dry skin, pruritus, dry eye, nausea, vomiting, constipation |
| Fam-trastuzumab deruxtecan-nxki (Enhertu®, DS-8201a, T-DXd) | AstraZeneca/Daiichi Sankyo | 2019 | Unresectable or metastatic HER2+ breast cancer in adult patients who have previously received ≥ 2 HER2 blockade regimens in the metastatic setting, locally advanced or metastatic HER2+ gastric or gastroesophageal adenocarcinoma after trastuzumab-based treatment | HER2/ERB2 | Humanized IgG1 | DXd (exatecan derivative) | Protease-cleavable tetrapeptide (Gly-Gly-Phe-Gly) linker | 7–8 | Nausea, vomiting, constipation, diarrhea, abdominal pain, stomatitis, dyspepsia, fatigue, alopecia, rash, decreased appetite, hypokalemia, anemia, neutropenia, leukopenia, thrombocytopenia, cough, dyspnea, epistaxis, headache, dizziness, upper respiratory tract infection, dry eye |
| Sacituzumab govitecan-hziy (Trodelvy® IMMU-132, HRS7-SN38) | Immunomedics | 2020 | Unresectable locally advanced or metastatic triple negative (HR-/HER2-) breast cancer after ≥2 prior systemic therapies, locally advanced or metastatic urothelial carcinoma after platinum-based chemotherapy and either a PD-1 or PD-L1 inhibitor | Trop-2 | Humanized IgG1 | SN-38 | HydrolysableCL2A linker | 7.6 | Nausea, diarrhea, neutropenia, fatigue, anemia, vomiting, constipation, alopecia, rash, headache, respiratory tract infection, decreased appetite, urinary tract infection, hyperglycemia, arthralgia, dyspnea, dizziness, neuropathy, back pain, edema, thrombocytopenia, hypomagnesemia, hypokalemia, hypophosphatemia, pruritus, mucositis |
| Belantamab mafadotin-blm (Blenrep®, GSK2857916) | GlaxoSmithKline | 2020 | Relapsed or refractory multiple myeloma in adult patients who have received ≥ 4 therapies, including an anti-CD38 mAb, a proteasome inhibitor and an immunomodulatory agent | BCMA | Afucosylated Humanized IgG1 | MMAF | Maleimidocaproyl (mc) linker | ~4 | Keratopathy, decreased visual acuity, blurred vision, dry eyes, nausea, diarrhea, constipation, blurred vision, pyrexia, infusion-related reactions, arthralgia, back pain, upper respiratory tract infections, decreased appetite, fatigue |
| Loncastuximab tesirine-lpyl (Zynlonta®, ADCT-402) | ADC Therapeutics | 2021 | Relapsed or refractory large B-cell lymphoma in adult patients after ≥ 2 lines of systemic therapy | CD19 | Humanized IgG1 | PDB dimer SCX (SG3199) | Protease-cleavable valine-alanine linker | 2.8 | Fatigue, edema, rash, pruritus, nausea, diarrea, abdominal pain, vomiting, constipation, musculoskeletal pain, decreased appetite, dyspnea, pleural effusion, upper respiratory tract infection |
| ADC Name | FDA Status | Target/Payload | NCT Number | Current Trial Status | Indication | Assigned Interventions |
|---|---|---|---|---|---|---|
| Tisotumab vedotin (TF-011-MMAE) | Priority review granted in April 2021 | Tissue factor/MMAE | NCT04697628 (innovaTV 301) | 3 | Second or Third-line Recurrent or Metastatic Cervical cancer | Tisotumab vedotin 2.0 mg/kg IV Q3W Topotecan 1 or 1.25 mg/m2 IV on D1-5 Q3W Vinorelbine 30 mg/m2 IV on D1 and 8 Q3W Gemcitabine 1000 mg/m2 IV on D1 and 8 Q3W Irinotecan 100 or 125 mg/m2 IV weekly for 28 days, Q6W Pemetrexed 500 mg/m2 IV on D1 Q3W |
| Trastuzumab duocarmazine (SYD985) | Fast Track designation granted in January 2018 | HER2/seco-DUBA | NCT03262935 (TULIP) | 3 | HER2+ unresectable locally advanced or metastatic breast cancer | Trastuzumab duocarmazine RP2D 1.2 mg/kg IV Q3W Lapatinib + capecitabine or Trastuzumab + capecitabine or Trastuzumab + vinorelbine Trastuzumab + eribulin |
| Mirvetuximab soravtansine (IMGN853) | Fast Track designation granted in June 2018. Accelerated approval pathway includes pivotal trial SORAYA and confirmatory trial MIRASOL | Folate receptor α/DM4 | NCT04296890 (SORAYA) | 3 | Platinum-resistant advanced high-grade epithelial ovarian, primary peritoneal, or fallopian tube cancer, with high Folate receptor α expression | Mirvetuximab soravtasine 6 mg/kg IV Q3W |
| NCT04209855 (MIRASOL) | 3 | Platinum-resistant advanced high-grade epithelial ovarian, primary peritoneal or fallopian tube cancer with high Folate receptor α expression | Mirvetuximab soravtasine 6 mg/kg IV Q3W Paclitaxel 80 mg/m^2 QW within a 4-week cycle or Pegylated liposomal doxorubicin 40 mg/m^2 Q4W or Topotexan 4 mg/m^2 IV either on D1, 8, 15 Q4W or 1.25 mg/m^2 on D1-5 Q3W | |||
| Upifitamab rilsodotin (XMT-1536) | Fast Track designation granted in August 2020 | NaPi2b/DolaLock (auristatin F- hydroxypropylamide payload molecules) | NCT03319628 (UPLIFT; Pivotal Cohort) | 1b/2 | Platinum-resistant ovarian cancer and non-small cell lung cancer, adenocarcinoma subtype | Upifitamab rilsodotin RP2D IV Q4W |
| Disitamab vedotin (RC48) | Breakthrough Therapy designation granted in September 2020 | HER2/MMAE | NCT04879329 | 2 | HER2+ locally advanced or metastatic urothelial carcinoma in second-line treatment of patients pre-treated with platinum-containing chemotherapy | Disitamab vedotin 2.0 mg/kg IV once every 2 weeks (maximum dose 200 mg) |
| IMGN632 | Breakthrough Therapy designation granted in October 2020 | CD123/DNA mono-alkylating payload of the indolinobenzodiazepine pseudodimer (IGN) class. | NCT03386513 | 1/2 | Relapsed or refractory or Untreated blastic plasmacytoid dendritic cell neoplasm (BPDCN) | IMGN632 IV |
| ARX788 | Fast Track designation granted in January 2021 | HER2/MMAF | NCT04829604 (ACE-Breast03) | 2 | HER2+ metastatic breast cancer, resistant or refractory to T-DM1, and/or T-DXd, and/or tucatinib-containing regimens | ARX788 IV Q4W |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theocharopoulos, C.; Lialios, P.-P.; Samarkos, M.; Gogas, H.; Ziogas, D.C. Antibody-Drug Conjugates: Functional Principles and Applications in Oncology and Beyond. Vaccines 2021, 9, 1111. https://doi.org/10.3390/vaccines9101111
Theocharopoulos C, Lialios P-P, Samarkos M, Gogas H, Ziogas DC. Antibody-Drug Conjugates: Functional Principles and Applications in Oncology and Beyond. Vaccines. 2021; 9(10):1111. https://doi.org/10.3390/vaccines9101111
Chicago/Turabian StyleTheocharopoulos, Charalampos, Panagiotis-Petros Lialios, Michael Samarkos, Helen Gogas, and Dimitrios C. Ziogas. 2021. "Antibody-Drug Conjugates: Functional Principles and Applications in Oncology and Beyond" Vaccines 9, no. 10: 1111. https://doi.org/10.3390/vaccines9101111
APA StyleTheocharopoulos, C., Lialios, P.-P., Samarkos, M., Gogas, H., & Ziogas, D. C. (2021). Antibody-Drug Conjugates: Functional Principles and Applications in Oncology and Beyond. Vaccines, 9(10), 1111. https://doi.org/10.3390/vaccines9101111