
mRNA-Based Cancer Vaccines: A Therapeutic Strategy for the Treatment of Melanoma Patients

 ,
,  and
and
Abstract
:1. Introduction
2. Melanoma Antigens
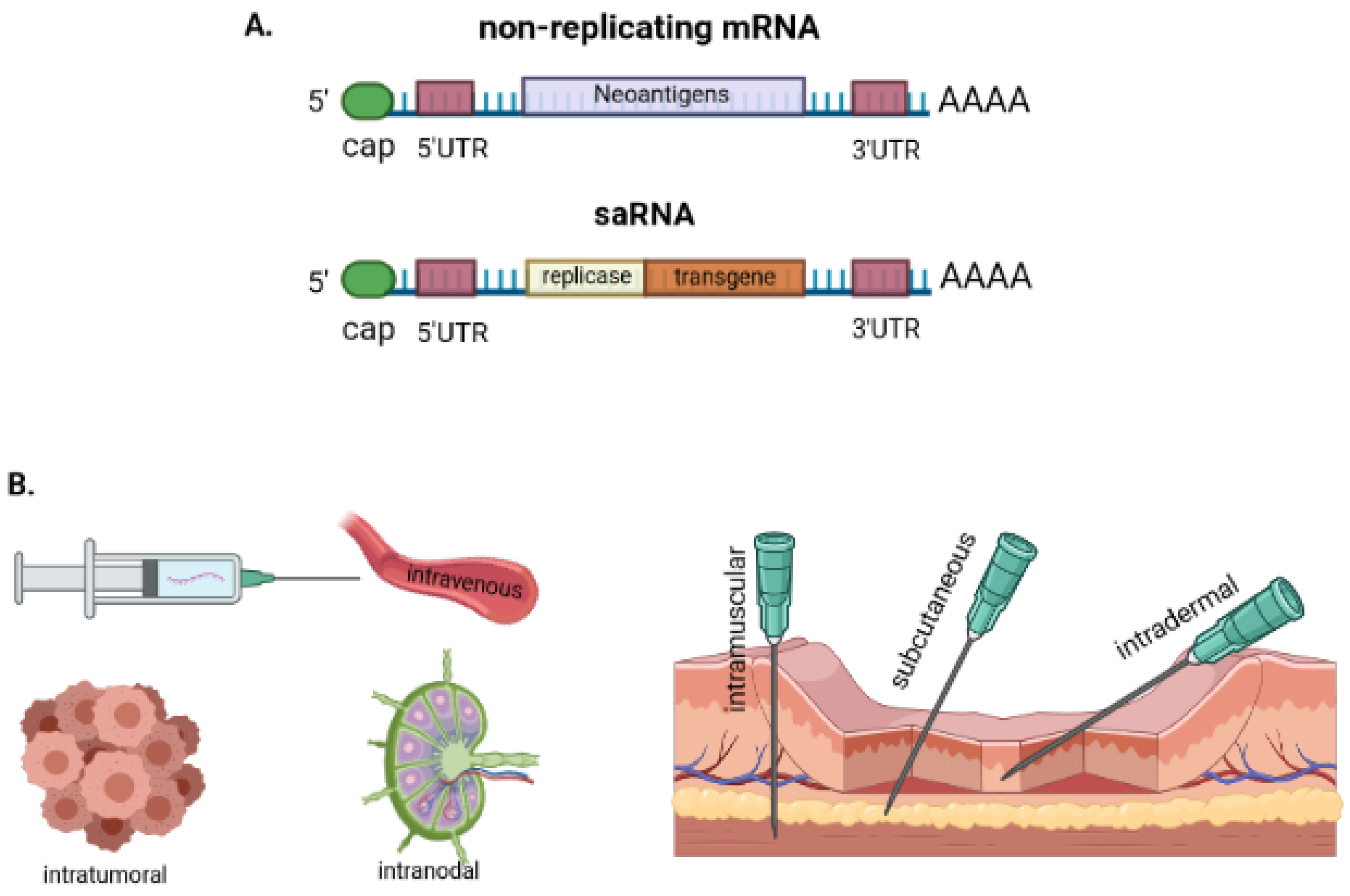
3. mRNA Vaccines: General Features
3.1. The Basis of mRNA Vaccines
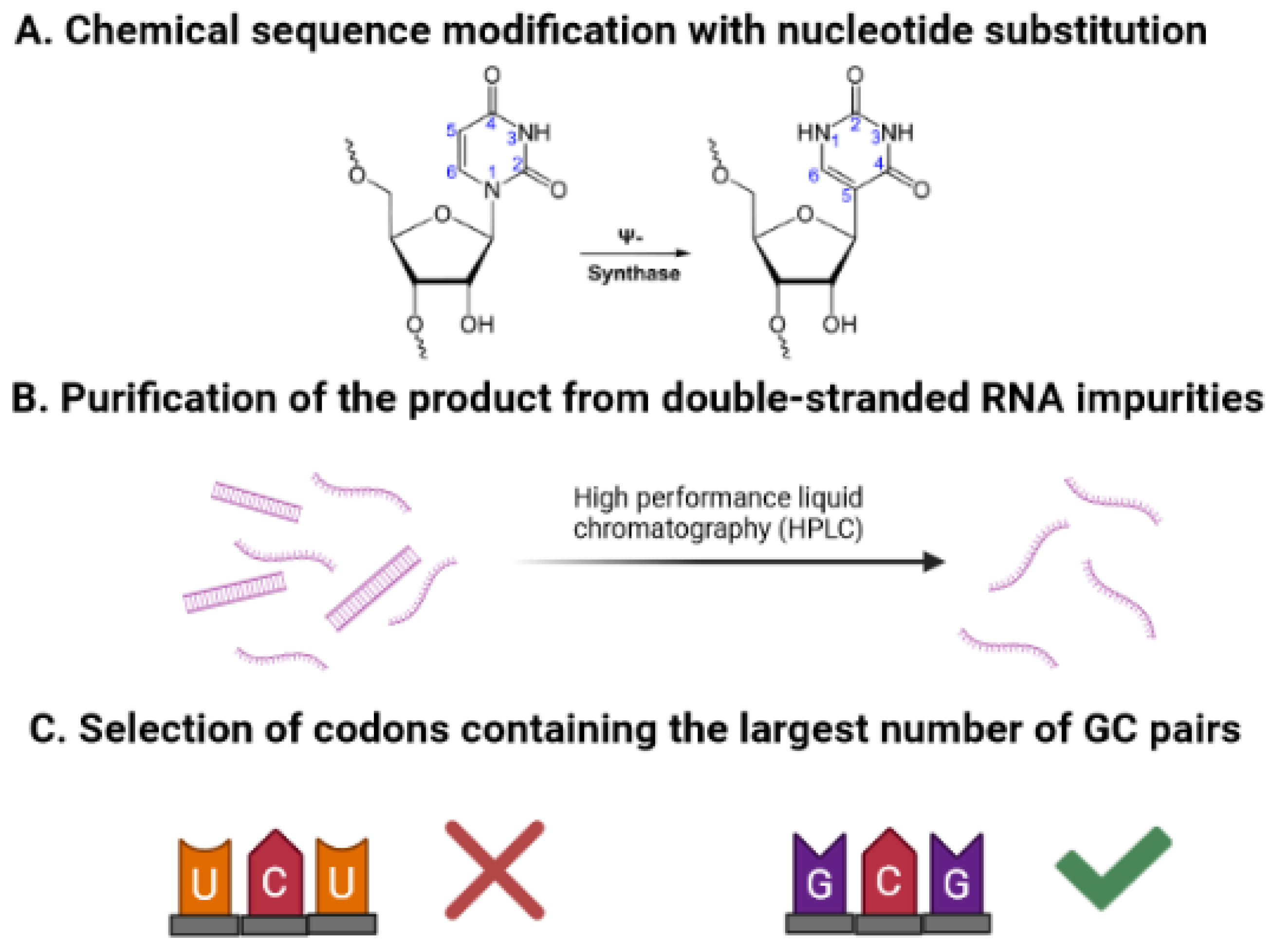
3.2. Vaccine Optimization by Improving mRNA Translation and Stability
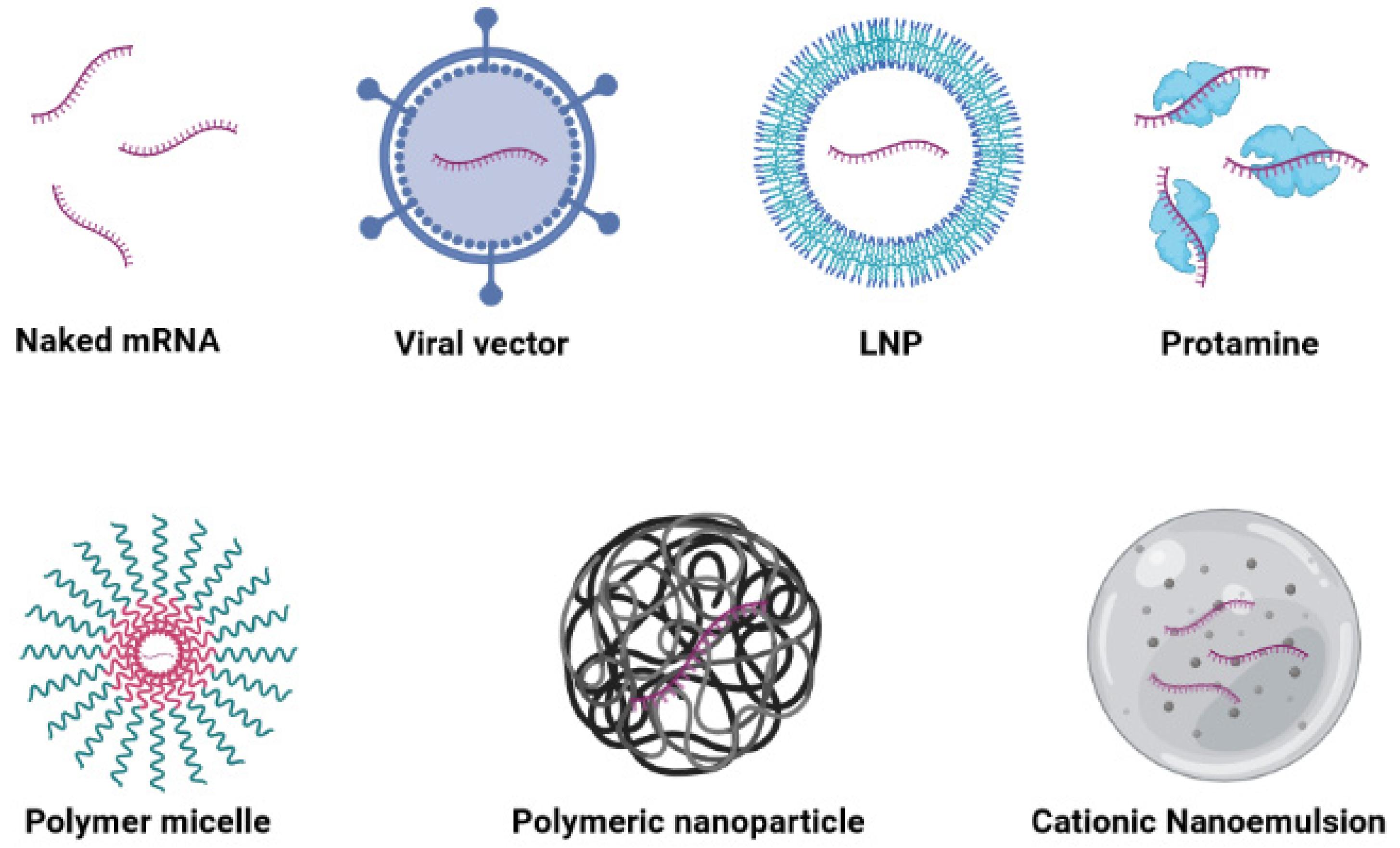
3.3. Various Carriers for mRNA Vaccine Delivery
3.3.1. Naked mRNA Vaccines
3.3.2. Viral Vectors
3.3.3. Lipid-Based Carriers
3.3.4. Polymer-Based Carriers
3.3.5. Hybrid Carriers
3.3.6. Peptide-Based Carriers
3.3.7. Dendritic-Cell-Based mRNA Vaccines
4. mRNA Vaccines in Combination with Checkpoint Blockade for the Treatment of Melanoma Cancer
5. Clinical Trials of In Vitro Transcription mRNA Vaccines in Melanoma
6. DC mRNA Vaccines in Melanoma
7. Therapeutic Considerations, Challenges, and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Statistics. Available online: http://cancerstatisticscenter.cancer.org/ (accessed on 3 July 2021).
- Ulmer, A.; Dietz, K.; Hodak, I.; Polzer, B.; Scheitler, S.; Yildiz, M.; Czyz, Z.; Lehnert, P.; Fehm, T.; Hafner, C.; et al. Quantitative Measurement of Melanoma Spread in Sentinel Lymph Nodes and Survival. PLoS Med. 2014, 11, e1001604. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, Z.; Yarahmadi, M.; Nariman-Saleh-Fam, Z.; Tarhriz, V.; Islami, M.; Aghdam, A.M.; Eyvazi, S. Immune therapy of melanoma: Overview of therapeutic vaccines. J. Cell. Physiol. 2019, 234, 14612–14621. [Google Scholar] [CrossRef] [PubMed]
- Reiman, J.M.; Kmieciak, M.; Manjili, M.H.; Knutson, K.L. Tumor immunoediting and immunosculpting pathways to cancer progression. Semin. Cancer Biol. 2007, 17, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H.; et al. U.S. FDA Approval Summary: Nivolumab for Treatment of Unresectable or Metastatic Melanoma Following Progression on Ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, A.; Hazarika, M.; Theoret, M.R.; Mishra-Kalyani, P.; Chen, H.; He, K.; Sridhara, R.; Subramaniam, S.; Pfuma, E.; Wang, Y.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Patients with Unresectable or Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 5661–5665. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.B. Suppression of T cell responses in the tumor microenvironment. Vaccine 2015, 33, 7393–7400. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Prendergast, G.C. Cancer Vaccines: A Brief Overview. Methods Mol. Biol. 2016, 1403, 755–761. [Google Scholar] [CrossRef]
- Alexandrov, L.; Initiative, A.P.C.G.; Nik-Zainal, S.; Wedge, D.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.; Bignell, G.R.; Bolli, N.; Borg, A.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; I Khushalani, N.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka, E.; et al. Nivolumab in Previously Untreated Melanoma withoutBRAFMutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hoecke, L.; Verbeke, R.; Dewitte, H.; Lentacker, I.; Vermaelen, K.; Breckpot, K.; Van Lint, S. mRNA in cancer immunotherapy: Beyond a source of antigen. Mol. Cancer 2021, 20, 48. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S. Well-Defined Melanoma Antigens as Progression Markers for Melanoma: Insights into Differential Expression and Host Response Based on Stage. Clin. Cancer Res. 2006, 12, 673–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitcovski, J.; Shahar, E.; Aizenshtein, E.; Gorodetsky, R. Melanoma antigens and related immunological markers. Crit. Rev. Oncol. 2017, 115, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, N.G. Value of melanocytic-associated immunohistochemical markers in the diagnosis of malignant melanoma: A review and update. Hum. Pathol. 2014, 45, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Progress in human tumour immunology and immunotherapy. Nature 2001, 411, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.; Browning, J.; MacGregor, D.; Davis, I.D.; Sturrock, S.; Jungbluth, A.A.; Cebon, J. Tumor Antigen Expression in Melanoma Varies According to Antigen and Stage. Clin. Cancer Res. 2006, 12, 764–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogal, S.; Carotti, M.; Giaretta, L.; Lanciai, F.; Nogara, L.; Bubacco, L.; Bergantino, E. Human Tyrosinase Produced in Insect Cells: A Landmark for the Screening of New Drugs Addressing its Activity. Mol. Biotechnol. 2015, 57, 45–57. [Google Scholar] [CrossRef]
- Hearing, V.J. Biogenesis of pigment granules: A sensitive way to regulate melanocyte function. J. Dermatol. Sci. 2005, 37, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Lu, J.; Celis, E. Identification of helper T-cell epitopes that encompass or lie proximal to cytotoxic T-cell epitopes in the gp100 melanoma tumor antigen. Cancer Res. 2001, 61, 7577–7584. [Google Scholar] [PubMed]
- Gjerstorff, M.F.; Kock, K.; Nielsen, O.; Ditzel, H.J. MAGE-A1, GAGE and NY-ESO-1 cancer/testis antigen expression during human gonadal development. Hum. Reprod. 2007, 22, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronwright, G.; Le Blanc, K.; Götherström, C.; Darcy, P.; Ehnman, M.; Brodin, B. Cancer/Testis Antigen Expression in Human Mesenchymal Stem Cells: Down-regulation of SSX Impairs Cell Migration and Matrix Metalloproteinase 2 Expression. Cancer Res. 2005, 65, 2207–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandler, N.; Mosch, B.; Pietzsch, J. Protein and non-protein biomarkers in melanoma: A critical update. Amino Acids 2012, 43, 2203–2230. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1–Reactive T-cell Receptor: Long-term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.B.; Haley, D.; Miller, W.; Floyd, K.; Wisner, K.P.; Sanjuan, N.; Maecker, H.T.; Romero, P.; Hu, H.-M.; Alvord, W.G.; et al. gp100209–2M Peptide Immunization of Human Lymphocyte Antigen-A2+ Stage I-III Melanoma Patients Induces Significant Increase in Antigen-Specific Effector and Long-Term Memory CD8+ T Cells. Clin. Cancer Res. 2004, 10, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Clin. Investig. 2015, 125, 3413–3421. [Google Scholar] [CrossRef]
- Cantwell-Dorris, E.R.; O’Leary, J.; Sheils, O. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Boespflug, A.; Caramel, J.; Dalle, S.; Thomas, L. Treatment of NRAS-mutated advanced or metastatic melanoma: Rationale, current trials and evidence to date. Ther. Adv. Med. Oncol. 2017, 9, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecek, R.J.; Storkus, W.J. Combination strategies to enhance the potency of monocyte-derived dendritic cell-based cancer vaccines. Immunotherapy 2016, 8, 1205–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Shuqiang, L.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nat. Cell Biol. 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Rubinsteyn, A.; Kodysh, J.; Hodes, I.; Mondet, S.; Aksoy, B.A.; Finnigan, J.P.; Bhardwaj, N.; Hammerbacher, J. Computational Pipeline for the PGV-001 Neoantigen Vaccine Trial. Front. Immunol. 2017, 8, 1807. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.; Wilmott, J.; Waddell, N.; Johansson, P.A.; Field, M.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Kramps, T.; Elbers, K. Introduction to RNA Vaccines. RNA Vaccines 2017, 1499, 1–11. [Google Scholar] [CrossRef]
- Desfarges, S.; Ciuffi, A. Viral Integration and Consequences on Host Gene Expression. In Viruses: Essential Agents of Life; Springer: Dordrecht, The Netherlands, 2012; pp. 147–175. [Google Scholar] [CrossRef] [Green Version]
- Iavarone, C.; O’hagan, D.; Yu, D.; Delahaye, N.; Ulmer, J. Mechanism of action of mRNA-based vaccines. Expert Rev. Vacc. 2017, 16, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Self-amplifying RNA viruses as RNA vaccines. Int. J. Mol. Sci. 2020, 21, 5130. [Google Scholar] [CrossRef]
- Deering, R.P.; Kommareddy, S.; Ulmer, J.B.; Brito, L.A.; Geall, A.J. Nucleic acid vaccines: Prospects for non-viral delivery of mRNA vaccines. Expert Opin. Drug Deliv. 2014, 11, 885–899. [Google Scholar] [CrossRef]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 2019, 28, 119–128. [Google Scholar] [CrossRef]
- Hornung, V.; Barchet, W.; Schlee, M.; Hartmann, G. RNA recognition via TLR7 and TLR8. Toll-Like Receptors (TLRs) and Innate Immunity. Handb. Exp. Pharmacol. 2008, 83, 71–86. [Google Scholar]
- Nelson, J.; Sorensen, E.W.; Mintri, S.; Rabideau, A.E.; Zheng, W.; Besin, G.; Khatwani, N.; Su, S.V.; Miracco, E.J.; Issa, W.J.; et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020, 6, eaaz6893. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020, 40, 107534. [Google Scholar] [CrossRef] [PubMed]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.; Tollervey, D. mRNA stability in eukaryotes. Curr. Opin. Genet. Dev. 2000, 10, 193–198. [Google Scholar] [CrossRef]
- Wilson, T.M.A.; Treisman, R. Removal of poly(A) and consequent degradation of c-fos mRNA facilitated by 3′ AU-rich sequences. Nature 1988, 336, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, C.J.; Wormington, M.; Peltz, S.W. The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol. 2001, 2, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Kudla, G.; Lipinski, L.; Caffin, F.; Helwak, A.; Zylicz, M. High Guanine and Cytosine Content Increases mRNA Levels in Mammalian Cells. PLoS Biol. 2006, 4, e180. [Google Scholar] [CrossRef]
- Gallie, D.R. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991, 5, 2108–2116. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.; Paoletti, E.; Moss, B. Purification of mRNA guanylyltransferase and mRNA (guanine-7-) methyltransferase from vaccinia virions. J. Biol. Chem. 1975, 250, 9322–9329. [Google Scholar] [CrossRef]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl (3’-O-methyl) GpppG and 7-methyl (3’-deoxy) GpppG. Rna 2001, 7, 1486–1495. [Google Scholar] [PubMed]
- Weissman, D.; Karikó, K. mRNA: Fulfilling the Promise of Gene Therapy. Mol. Ther. 2015, 23, 1416–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Senti, G.; Kündig, T.M. Intralymphatic immunotherapy. World Allergy Organ. J. 2015, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Van Der Jeught, K.; Joe, P.T.; Bialkowski, L.; Heirman, C.; Daszkiewicz, L.; Liechtenstein, T.; Escors, D.; Thielemans, K.; Breckpot, K. Intratumoral administration of mRNA encoding a fusokine consisting of IFN-β and the ectodomain of the TGF-β receptor II potentiates antitumor immunity. Oncotarget 2014, 5, 10100–10113. [Google Scholar] [CrossRef] [Green Version]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Ziemniak, M.; Strenkowska, M.; Kowalska, J.; Jemielity, J. Potential therapeutic applications of RNA cap analogs. Futur. Med. Chem. 2013, 5, 1141–1172. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar] [PubMed]
- Kuhn, A.N.; Beißert, T.; Simon, P.; Vallazza, B.; Buck, J.; Davies, B.P.; Tureci, O.; Sahin, U. mRNA as a versatile tool for exogenous protein expression. Curr. Gene Ther. 2012, 12, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Sullivan, T.D. Half-lives of beta and gamma globin messenger RNAs and of protein synthetic capacity in cultured human reticulocytes. Blood 1985, 66, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.A.; Shyu, A.-B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Cannarozzi, G.; Schraudolph, N.N.; Faty, M.; von Rohr, P.; Friberg, M.T.; Roth, A.C.; Gonnet, P.; Gonnet, G.; Barral, Y. A Role for Codon Order in Translation Dynamics. Cell 2010, 141, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Kimchi-Sarfaty, C.; Oh, J.; Kim, I.-W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “Silent” Polymorphism in the MDR1 Gene Changes Substrate Specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Zhong, F.; Cao, W.; Chan, E.; Tay, P.N.; Cahya, F.F.; Zhang, H.; Lu, J. Deviation from major codons in the Toll-like receptor genes is associated with low Toll-like receptor expression. Immunology 2005, 114, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Keller, J.M.; Weissman, D. Increased Erythropoiesis in Mice Injected with Submicrogram Quantities of Pseudouridine-containing mRNA Encoding Erythropoietin. Mol. Ther. 2012, 20, 948–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gulck, E.R.A.; Ponsaerts, P.; Heyndrickx, L.; Vereecken, K.; Moerman, F.; De Roo, A.; Colebunders, R.; Bosch, G.V.D.; Van Bockstaele, D.R.; Van Tendeloo, V.F.I.; et al. Efficient stimulation of HIV-1-specific T cells using dendritic cells electroporated with mRNA encoding autologous HIV-1 Gag and Env proteins. Blood 2006, 107, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Diken, M.; Kreiter, S.; Selmi, A.; Britten, C.M.; Huber, C.; Tureci, O.; Sahin, U. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. 2011, 18, 702–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Sun, X. Recent advances in mRNA vaccine delivery. Nano Res. 2018, 11, 5338–5354. [Google Scholar] [CrossRef]
- Ringer, S. Regarding the Action of Hydrate of Soda, Hydrate of Ammonia, and Hydrate of Potash on the Ventricle of the Frog’s Heart. J. Physiol. 1882, 3, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.A. Sydney Ringer (1834–1910) and Alexis Hartmann (1898–1964). Anaesthesia 1981, 36, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Probst, J.; Weide, B.; Scheel, B.; Pichler, B.J.; Hoerr, I.; Rammensee, H.-G.; Pascolo, S. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. 2007, 14, 1175–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Luo, J.; Han, X.; Wei, Y.; Wei, X. mRNA vaccine: A potential therapeutic strategy. Mol. Cancer 2021, 20, 33. [Google Scholar] [CrossRef]
- Lorenz, C.; Fotin-Mleczek, M.; Roth, G.; Becker, C.; Dam, T.C.; Verdurmen, W.P.R.; Brock, R.; Probst, J.; Schlake, T. Protein expression from exogenous mRNA: Uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 2011, 8, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Selmi, A.; Vascotto, F.; Kautz-Neu, K.; Türeci, Ö.; Sahin, U.; Von Stebut, E.; Diken, M.; Kreiter, S. Uptake of synthetic naked RNA by skin-resident dendritic cells via macropinocytosis allows antigen expression and induction of T-cell responses in mice. Cancer Immunol. Immunother. 2016, 65, 1075–1083. [Google Scholar] [CrossRef]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and challenges in the delivery of mRNA-based vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.Y.; Mansfield, B.C. Recombinant AAV-directed gene therapy for type I glycogen storage diseases. Expert Opin. Biol. Ther. 2011, 11, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Ehrengruber, M.U.; Schlesinger, S.; Lundstrom, K. Alphaviruses: Semliki Forest virus and Sindbis virus vectors for gene transfer into neurons. Curr. Protoc. Neurosci. 2011, 57, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Rozovics, J.M.; Chase, A.J.; Cathcart, A.L.; Chou, W.; Gershon, P.D.; Palusa, S.; Wilusz, J.; Semler, B.L. Picornavirus Modification of a Host mRNA Decay Protein. mBio 2012, 3, e00431-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, J.W.; Morgan, M.; Galla, M.; Schambach, A. Viral and Synthetic RNA Vector Technologies and Applications. Mol. Ther. 2016, 24, 1513–1527. [Google Scholar] [CrossRef] [Green Version]
- Tezel, A.; Dokka, S.; Kelly, S.; Hardee, G.E.; Mitragotri, S. Topical Delivery of Anti-sense Oligonucleotides Using Low-Frequency Sonophoresis. Pharm. Res. 2004, 21, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy—An overview. J. Clin. Diagn. Res. JCDR 2015, 9, GE01. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Pichon, C. Lipid-based mRNA vaccine delivery systems. Expert Rev. Vaccines 2014, 14, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Kranz, L.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Grabbe, S.; Haas, H.; Diken, M.; Kranz, L.M.; Langguth, P.; Sahin, U. Translating nanoparticulate-personalized cancer vaccines into clinical applications: Case study with RNA-lipoplexes for the treatment of melanoma. Nanomedicine 2016, 11, 2723–2734. [Google Scholar] [CrossRef]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokugamage, M.; Gan, Z.; Zurla, C.; Levin, J.; Islam, F.; Kalathoor, S.; Sato, M.; Sago, C.D.; Santangelo, P.J.; Dahlman, J.E. Mild Innate Immune Activation Overrides Efficient Nanoparticle-Mediated RNA Delivery. Adv. Mater. 2019, 32, e1904905. [Google Scholar] [CrossRef] [PubMed]
- Ramishetti, S.; Hazan-Halevy, I.; Palakuri, R.; Chatterjee, S.; Naidu Gonna, S.; Dammes, N.; Freilich, I.; Kolik Shmuel, L.; Danino, D.; Peer, D. A combinatorial library of lipid nanoparticles for RNA delivery to leukocytes. Adv. Mater. 2020, 32, 1906128. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.; Pelc, R.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2016, 17, 1326–1335. [Google Scholar] [CrossRef]
- Gary, D.J.; Lee, H.; Sharma, R.; Lee, J.-S.; Kim, Y.; Cui, Z.Y.; Jia, D.; Bowman, V.D.; Chipman, P.R.; Wan, L.; et al. Influence of Nano-Carrier Architecture on In Vitro siRNA Delivery Performance and In Vivo Biodistribution: Polyplexes vs Micelleplexes. ACS Nano 2011, 5, 3493–3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussif, O.; Lezoualc’H, F.; Zanta, M.A.; Djavaheri-Mergny, M.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and In Vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lungwitz, U.; Breunig, M.; Blunk, T.; Göpferich, A. Polyethylenimine-based non-viral gene delivery systems. Eur. J. Pharm. Biopharm. 2005, 60, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Howard, K.A.; Rahbek, U.L.; Liu, X.; Damgaard, C.; Glud, S.Z.; Andersen, M.; Hovgaard, M.B.; Schmitz, A.; Nyengaard, J.R.; Besenbacher, F.; et al. RNA Interference in Vitro and in Vivo Using a Novel Chitosan/siRNA Nanoparticle System. Mol. Ther. 2006, 14, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. A Cross-Discip. J. Res. Sci. Gene Transf. Its Clin. Appl. 2005, 7, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Démoulins, T.; Milona, P.; Englezou, P.C.; Ebensen, T.; Schulze, K.; Suter, R.; Pichon, C.; Midoux, P.; Guzmán, C.A.; Ruggli, N.; et al. Polyethylenimine-based polyplex delivery of self-replicating RNA vaccines. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Üzgün, S.; Nica, G.; Pfeifer, C.; Bosinco, M.; Michaelis, K.; Lutz, J.-F.; Schneider, M.; Rosenecker, J.; Rudolph, C. PEGylation Improves Nanoparticle Formation and Transfection Efficiency of Messenger RNA. Pharm. Res. 2011, 28, 2223–2232. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Orr, B.G.; Holl, M.M.B. Role of Cell Membrane–Vector Interactions in Successful Gene Delivery. Acc. Chem. Res. 2016, 49, 1486–1493. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Jiang, Y.; Gaudin, A.; Zhang, J.; Agarwal, T.; Song, E.; Kauffman, A.C.; Tietjen, G.T.; Wang, Y.; Jiang, Z.; Cheng, C.J.; et al. A “top-down” approach to actuate poly(amine-co-ester) terpolymers for potent and safe mRNA delivery. Biomaterials 2018, 176, 122–130. [Google Scholar] [CrossRef]
- Stefan, J.; Kus, K.; Wisniewska, A.; Lorkowska-Zawicka, B.; Kaminski, K.; Szczubialka, K.; Nowakowska, M.; Korbut, R. The antiatherogenic effect of new biocompatible cationically modified polysaccharides: Chitosan and pullulan—The comparison study. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2019, 69, 995–1007. [Google Scholar]
- Yang, X.-Z.; Dou, S.; Sun, T.-M.; Mao, C.; Wang, H.; Wang, J. Systemic delivery of siRNA with cationic lipid assisted PEG-PLA nanoparticles for cancer therapy. J. Control. Release 2011, 156, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Magalhães, M.; Veiga, F.; Figueiras, A. Poloxamers, poloxamines and polymeric micelles: Definition, structure and therapeutic applications in cancer. J. Polym. Res. 2018, 25, 31. [Google Scholar] [CrossRef]
- Jhaveri, A.; Torchilin, V.P. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front. Pharmacol. 2014, 5, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Li, M.; Zhang, Z.; Gong, T.; Sun, X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA. Drug Deliv. 2015, 23, 2596–2607. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffrès, P.-A.; Midoux, P. Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Persano, S.; Guevara, M.L.; Li, Z.; Mai, J.; Ferrari, M.; Pompa, P.P.; Shen, H. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 2017, 125, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaee, M.; Oskuee, R.K.; Nassirli, H.; Malaekeh-Nikouei, B. Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J. Control. Release 2016, 236, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Rosenecker, J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 2017, 24, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.; Cun, D.; Remaut, K.; Bunker, M.; Zhang, J.; Martin-Bertelsen, B.; Yaghmur, A.; Braeckmans, K.; Nielsen, H.M.; Foged, C. Mechanistic profiling of the siRNA delivery dynamics of lipid–polymer hybrid nanoparticles. J. Control. Release 2015, 201, 22–31. [Google Scholar] [CrossRef]
- Mockey, M.; Bourseau, E.; Chandrashekhar, V.; Chaudhuri, A.; Lafosse, S.; Le Cam, E.; Quesniaux, V.F.J.; Ryffel, B.; Pichon, C.; Midoux, P. mRNA-based cancer vaccine: Prevention of B16 melanoma progression and metastasis by systemic injection of MART1 mRNA histidylated lipopolyplexes. Cancer Gene Ther. 2007, 14, 802–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Ott, G.; Barchfeld, G.L.; Chernoff, D.; Radhakrishnan, R.; van Hoogevest, P.; Van Nest, G. MF59 Design and Evaluation of a Safe and Potent Adjuvant for Human Vaccines. Vaccine Des. 1995, 6, 277–296. [Google Scholar] [CrossRef]
- Lovelyn, C.; Attama, A.A. Current state of nanoemulsions in drug delivery. J. Biomater. Nanobiotechnol. 2011, 2, 626. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, J.; Neundorf, I. Peptide Vectors for the Nonviral Delivery of Nucleic Acids. Acc. Chem. Res. 2012, 45, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Man, R.C.; Liao, Q.; Kung, K.L.; Chow, M.Y.; Lam, J.K. Effective mRNA pulmonary delivery by dry powder formulation of PEGylated synthetic KL4 peptide. J. Control. Release 2019, 314, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Hoerr, I.; Obst, R.; Rammensee, H.G.; Jung, G. In Vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef]
- Kallen, K.-J.; Heidenreich, R.; Schnee, M.; Petsch, B.; Schlake, T.; Thess, A.; Baumhof, P.; Scheel, B.; Koch, S.D.; Fotin-Mleczek, M. A novel, disruptive vaccination technology: Self-adjuvanted RNActive® vaccines. Hum. Vaccines Immunother. 2013, 9, 2263–2276. [Google Scholar] [CrossRef] [Green Version]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.-J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Kallen, K.-J.; Theß, A. A development that may evolve into a revolution in medicine: mRNA as the basis for novel, nucleotide-based vaccines and drugs. Ther. Adv. Vaccines 2014, 2, 10–31. [Google Scholar] [CrossRef] [Green Version]
- Bell, G.D.; Yang, Y.; Leung, E.; Krissansen, G.W. mRNA transfection by a Xentry-protamine cell-penetrating peptide is enhanced by TLR antagonist E6446. PLoS ONE 2018, 13, e0201464. [Google Scholar] [CrossRef] [Green Version]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based mRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Health Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Baru, M.; Nahum, O.; Jaaro, H.; Sha’Anani, J.; Nur, I. Lysosome-disrupting Peptide Increases the Efficiency of In-VivoGene Transfer by Liposome-encapsulated DNA. J. Drug Target. 1998, 6, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Wyman, T.B.; Nicol, F.; Zelphati, O.; Scaria, P.V.; Plank, C.; Szoka, F.C. Design, Synthesis, and Characterization of a Cationic Peptide That Binds to Nucleic Acids and Permeabilizes Bilayers. Biochemistry 1997, 36, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Lou, B.; De Koker, S.; Lau, C.Y.; Hennink, W.E.; Mastrobattista, E. mRNA Polyplexes with Post-Conjugated GALA Peptides Efficiently Target, Transfect, and Activate Antigen Presenting Cells. Bioconjugate Chem. 2018, 30, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigenpresentation Andt Cellstimulation Bydendriticcells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Sozio, F.; Barbazza, I.; Salvi, V.; Tiberio, L.; Laffranchi, M.; Gismondi, A.; Bosisio, D.; Schioppa, T.; Sozzani, S. Functional Role of Dendritic Cell Subsets in Cancer Progression and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 3930. [Google Scholar] [CrossRef] [PubMed]
- Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, J.A.A. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers 2020, 12, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabado, R.L.; Meseck, M.; Bhardwaj, N. Dendritic cell vaccines. In Vaccine Design: Methods and Protocols: Volume 1: Vaccines for Human Diseases; Thomas, S., Ed.; Springer: New York, NY, USA, 2016; pp. 763–777. [Google Scholar]
- Ahmed, R.; Sayegh, N.; Graciotti, M.; Kandalaft, L.E. Electroporation as a method of choice to generate genetically modified dendritic cell cancer vaccines. Curr. Opin. Biotechnol. 2020, 65, 142–155. [Google Scholar] [CrossRef]
- Melhem, N.M.; Gleason, S.M.; Liu, X.D.; Boyes, S.B. High-Level Antigen Expression and Sustained Antigen Presentation in Dendritic Cells Nucleofected with Wild-Type Viral mRNA but Not DNA. Clin. Vaccine Immunol. 2008, 15, 1337–1344. [Google Scholar] [CrossRef] [Green Version]
- De Temmerman, M.-L.; Dewitte, H.; Vandenbroucke, R.; Lucas, B.; Libert, C.; Demeester, J.; De Smedt, S.; Lentacker, I.; Rejman, J. mRNA-Lipoplex loaded microbubble contrast agents for ultrasound-assisted transfection of dendritic cells. Biomaterials 2011, 32, 9128–9135. [Google Scholar] [CrossRef] [PubMed]
- Aarntzen, E.H.J.G.; Schreibelt, G.; Bol, K.F.; Lesterhuis, W.J.; Croockewit, A.J.; De Wilt, J.H.W.; Van Rossum, M.M.; Blokx, W.; Jacobs, H.; Boer, T.D.-D.; et al. Vaccination with mRNA-Electroporated Dendritic Cells Induces Robust Tumor Antigen-Specific CD4+ and CD8+ T Cells Responses in Stage III and IV Melanoma Patients. Clin. Cancer Res. 2012, 18, 5460–5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markov, O.O.; Mironova, N.; Maslov, M.; Petukhov, I.A.; Morozova, N.G.; Vlassov, V.; Zenkova, M.A. Novel cationic liposomes provide highly efficient delivery of DNA and RNA into dendritic cell progenitors and their immature offsets. J. Control. Release 2012, 160, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical Evaluation of TriMix and Antigen mRNA-Based Antitumor Therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef] [Green Version]
- Johansen, P.; Häffner, A.C.; Koch, F.; Zepter, K.; Erdmann, I.; Maloy, K.; Simard, J.J.; Storni, T.; Senti, G.; Bot, A.; et al. Direct intralymphatic injection of peptide vaccines enhances immunogenicity. Eur. J. Immunol. 2005, 35, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Bonehill, A.; Van Nuffel, A.; Corthals, J.; Tuyaerts, S.; Heirman, C.; François, V.; Colau, D.; Van Der Bruggen, P.; Neyns, B.; Thielemans, K. Single-Step Antigen Loading and Activation of Dendritic Cells by mRNA Electroporation for the Purpose of Therapeutic Vaccination in Melanoma Patients. Clin. Cancer Res. 2009, 15, 3366–3375. [Google Scholar] [CrossRef] [Green Version]
- Le Moignic, A.; Malard, V.; Benvegnu, T.; Lemiègre, L.; Berchel, M.; Jaffrès, P.-A.; Baillou, C.; Delost, M.; Macedo, R.; Rochefort, J.; et al. Preclinical evaluation of mRNA trimannosylated lipopolyplexes as therapeutic cancer vaccines targeting dendritic cells. J. Control. Release 2018, 278, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Van Der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) Binds Both CD28 and CTLA-4 with a Low Affinity and Very Fast Kinetics. J. Exp. Med. 1997, 185, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Dyck, L.; Mills, K.H. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Van Elsas, A.; Hurwitz, A.A.; Allison, J.P. Combination Immunotherapy of B16 Melanoma Using Anti–Cytotoxic T Lymphocyte–Associated Antigen 4 (Ctla-4) and Granulocyte/Macrophage Colony-Stimulating Factor (Gm-Csf)-Producing Vaccines Induces Rejection of Subcutaneous and Metastatic Tumors Accompanied by Autoimmune Depigmentation. J. Exp. Med. 1999, 190, 355–366. [Google Scholar] [CrossRef]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual Blockade of PD-1 and CTLA-4 Combined with Tumor Vaccine Effectively Restores T-Cell Rejection Function in Tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic PD-1 Pathway Blockade Augments with Other Modalities of Immunotherapy T-Cell Function to Prevent Immune Decline in Ovarian Cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Liu, C.; Xu, C.; Lou, Y.; Chen, J.; Yang, Y.; Yagita, H.; Overwijk, W.W.; Lizée, G.; Radvanyi, L.; et al. PD-1 Blockade Enhances T-cell Migration to Tumors by Elevating IFN-γ Inducible Chemokines. Cancer Res. 2012, 72, 5209–5218. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016, 39, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Ai, M.; Curran, M.A. Immune checkpoint combinations from mouse to man. Cancer Immunol. Immunother. 2015, 64, 885–892. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, Y.; Kruse, V.; Corthals, J.; Schats, K.; Van Dam, P.-J.; Seremet, T.; Heirman, C.; Brochez, L.; Kockx, M.; Thielemans, K.; et al. A randomized controlled phase II clinical trial on mRNA electroporated autologous monocyte-derived dendritic cells (TriMixDC-MEL) as adjuvant treatment for stage III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol. Immunother. 2020, 69, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Clinical Translation of Nanomedicine and Biomaterials for Cancer Immunotherapy: Progress and Perspectives. Adv. Ther. 2020, 3. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med. 2019, 11, eaat9143. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct Gene Transfer into Mouse Muscle In Vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Baldin, A.; Isayev, O.; Werner, J.; Zamyatnin, A.; Bazhin, A. Cancer Vaccines: Antigen Selection Strategy. Vaccines 2021, 9, 85. [Google Scholar] [CrossRef]
- Liu, C.-C.; Yang, H.; Zhang, R.; Zhao, J.-J.; Hao, D.-J. Tumour-associated antigens and their anti-cancer applications. Eur. J. Cancer Care 2016, 26, e12446. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Goedegebuure, S.; Gillanders, W. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.; Pawelec, G.; Hoerr, I.; Rammensee, H.-G.; Garbe, C. Direct Injection of Protamine-protected mRNA: Results of a Phase 1/2 Vaccination Trial in Metastatic Melanoma Patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.G.; Jacobs, T.; Lamarca, A.; Hubner, R.A.; Valle, J.W.; Amir, E. Impact of high tumor mutational burden in solid tumors and challenges for biomarker application. Cancer Treat. Rev. 2020, 89, 102084. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Patel, M.R.; Cho, D.C.; Clarke, J.M.; Gutierrez, M.; Zaks, T.Z.; Frederick, J.; Hopson, K.; Mody, K.; Binanti-Berube, A.; et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J. Clin. Oncol. 2019, 37, 2523. [Google Scholar] [CrossRef]
- Hadden, J.W. Immunostimulants. Trends Pharmacol. Sci. 1993, 14, 169–174. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Bonehill, A.; Tuyaerts, S.; Van Nuffel, A.; Heirman, C.; Bos, T.J.; Fostier, K.; Neyns, B.; Thielemans, K. Enhancing the T-cell Stimulatory Capacity of Human Dendritic Cells by Co-electroporation With CD40L, CD70 and Constitutively Active TLR4 Encoding mRNA. Mol. Ther. 2008, 16, 1170–1180. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Van Nuffel, A.; Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Neyns, B. Long-term clinical outcome of melanoma patients treated with messenger RNA-electroporated dendritic cell therapy following complete resection of metastases. Cancer Immunol. Immunother. 2014, 64, 381–388. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients with Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Arance Fernandez, A.M.; Baurain, J.-F.; Vulsteke, C.; Rutten, A.; Soria, A.; Carrasco, J.; Neyns, B.; De Keersmaecker, B.; Van Assche, T.; Lindmark, B. A phase I study (E011-MEL) of a TriMix-based mRNA immunotherapy (ECI-006) in resected melanoma patients: Analysis of safety and immunogenicity. J. Clin. Oncol. 2019, 37, 15. [Google Scholar] [CrossRef]
- Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic cells pulsed with RNA are potent antigen-presenting Cells In Vitro and In Vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Lesterhuis, W.J.; De Vries, I.J.M.; Schreibelt, G.; Schuurhuis, D.H.; Aarntzen, E.H.; De Boer, A.; Scharenborg, N.M.; Van De Rakt, M.; Hesselink, E.J.; Figdor, C.; et al. Immunogenicity of dendritic cells pulsed with CEA peptide or transfected with CEA mRNA for vaccination of colorectal cancer patients. Anticancer. Res. 2010, 30, 5091–5097. [Google Scholar] [PubMed]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tønnessen, P.; Suso, E.M.I.; Sæbøe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kongsted, P.; Borch, T.H.; Ellebaek, E.; Iversen, T.Z.; Andersen, R.; Met, Ö.; Hansen, M.; Lindberg, H.; Sengeløv, L.; Svane, I.M. Dendritic cell vaccination in combination with docetaxel for patients with metastatic castration-resistant prostate cancer: A randomized phase II study. Cytotherapy 2017, 19, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Van Tendeloo, V.F.; Van de Velde, A.; Van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyte, J.A.; Mu, L.; Aamdal, S.; Kvalheim, G.; Dueland, S.; Hauser, M.A.; Gullestad, H.P.; Ryder, T.; Lislerud, K.; Hammerstad, H.; et al. Phase I/II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther. 2006, 13, 905–918. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.A.; Kvalheim, G.; Lislerud, K.; Straten, P.T.; Dueland, S.; Aamdal, S.; Gaudernack, G. T cell responses in melanoma patients after vaccination with tumor-mRNA transfected dendritic cells. Cancer Immunol. Immunother. 2006, 56, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Caballero, O.L.; Yung, W.A.; Weinstein, J.N.; Riggins, G.J.; Strausberg, R.L.; Zhao, Q. Tumor Subtype-Specific Cancer–Testis Antigens as Potential Biomarkers and Immunotherapeutic Targets for Cancers. Cancer Immunol. Res. 2014, 2, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, A.J.G.; Caballero, O.L.; Jungbluth, A.; Chen, Y.-T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Sang, M.; Lian, Y.; Zhou, X.; Shan, B. MAGE-A family: Attractive targets for cancer immunotherapy. Vaccine 2011, 29, 8496–8500. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Van Nuffel, A.; Corthals, J.; Heirman, C.; Tuyaerts, S.; Benteyn, D.; De Coninck, A.; Van Riet, I.; Verfaillie, G.; Vandeloo, J.; et al. Therapeutic Vaccination with an Autologous mRNA Electroporated Dendritic Cell Vaccine in Patients with Advanced Melanoma. J. Immunother. 2011, 34, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2015, 4, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nuffel, A.; Benteyn, D.; Wilgenhof, S.; Pierret, L.; Corthals, J.; Heirman, C.; van der Bruggen, P.; Coulie, P.G.; Neyns, B.; Thielemans, K.; et al. Dendritic Cells Loaded with mRNA Encoding Full-length Tumor Antigens Prime CD4+ and CD8+ T Cells in Melanoma Patients. Mol. Ther. 2012, 20, 1063–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudewijns, S.; Bloemendal, M.; De Haas, N.; Westdorp, H.; Bol, K.F.; Schreibelt, G.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Gorris, M.A.J.; Croockewit, A.; et al. Autologous monocyte-derived DC vaccination combined with cisplatin in stage III and IV melanoma patients: A prospective, randomized phase 2 trial. Cancer Immunol. Immunother. 2020, 69, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, I.J.M.; Krooshoop, D.J.E.B.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.S.; Van Muijen, G.N.P.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.G.; et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003, 63, 12–17. [Google Scholar]
- Morse, M.A.; Coleman, R.E.; Akabani, G.; Niehaus, N.; Coleman, D.; Lyerly, H. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999, 59, 56–58. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Encoding Content | Trial ID | Start Date | Phase | Enrollment Status | Brand | Target Antigens | Formulation | Route | Combination | Study Results | Sponsor |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MAAs | NCT 00204607 | Jun 2004 | I/II | Completed | NA | Melan-A, MAGE-A1, MAGE-A3, Survivin, GP100, Tyrosinase | Protamine-protected mRNA | i.d. | GM-CSF | An increase of vaccine-specific T cells was observed in two of four immunologically evaluable patients. One of seven patients with measurable diseases showed a CR. | University Hospital Tuebingen |
| NCT 00204516 | Apr 2007 | I/II | Completed | NA | Melan-A, MAGE-A1, MAGE-A3, Survivin, GP100, Tyrosinase | Naked mRNA | i.d. | GM-CSF | University Hospital Tuebingen | ||
| NCT 01684241 | Jun 2012 | I | Completed | NY-ESO-1, tyrosinase | Naked mRNA | i.n. | None | BioNTech | |||
| NCT 02410733 | Mar 2015 | I | Active, not recruiting | FixVac (BNT111) | NY-ESO-1, MAGE-A3, TPTE, tyrosinase | RNA-LPX | i.v. | Anti-PD1 | More than 75% showed immune responses against at least one MAA in 50 patients. In the FixVac monotherapy group (n = 25), three patients experienced a PR and seven had SD, while in the FixVac/anti-PD1 combination group, 6 out of 17 patients developed a PR. | BioNTech | |
| Neoantigens | NCT 02035956 | Oct 2013 | I | Completed | IVACMUTANOME | Neopeptides | Naked mRNA | i.n. | mRNA encoding NY-ESO-1, tyrosinase | One-third of pre-existing weak responses against neo-epitopes were augmented while two-thirds were de novo responses. 13 patients in total. Eight patients without measurable lesions at the start of the trial developed robust immune responses against neo-epitopes and achieved recurrence-free for the whole follow-up period. | BioNTech |
| NCT 03289962 | Dec 2017 | I | Recruiting | RO7198457 | Neopeptides | LNP | i.n. | Atezolizumab | BioNTech GenenTech | ||
| NCT 03815058 | Jan 2019 | II | Recruiting | RO7198458 | Neopeptides | LNP | i.v. | Pembrolizumab | BioNTech GenenTech | ||
| NCT 03480152 | Nov 2019 | I/II | Terminated | mRNA-4650 | Neopeptides | LNP | i.m. | None | Moderna | ||
| NCT 03897881 | Jul 2019 | II | Recruiting | mRNA-4157 | Neopeptides | LNP | i.v. | Pembrolizumab | No ≥ grade III AEs occurred. Of the 13 patients on monotherapy, 12 patients remain disease-free. In the combination group (n = 20), one CR, two PR, and five SD were observed for at least five administration cycles. | Moderna Merck | |
| Immunostimulants | NCT 03394937 | Jun 2017 | I | Recruiting | ECI-006 | CD70, CD40L, caTLR4 | Naked mRNA | i.n. | mRNA encoding tyrosinase, gp100, MAGE-A3, MAGE-C2, PRAME | No AEs Grade 3 or higher were reported. Vaccine-induced immune responses were detected in 4/10 and 3/9 patients treated with the low and high dose, respectively. | eTheRNA immunotherapies |
| Year | Trial ID | Phase | Antigen | Formulation | Route | Combination | Grade ≥3 Adverse Events | Study Results | Refs |
|---|---|---|---|---|---|---|---|---|---|
| 2006 | NA | I/II | Autologous tumor-mRNA | Electroporation | i.d. and i.n. | None | None | A vaccine-specific immune response was demonstrated in 9/19 patients evaluated by T-cell assays and in 8/18 patients evaluated by DTH reaction. The response rates do not suggest an advantage in applying i.n. vaccination compared with i.d. vaccination. | [190] |
| 2007 | NA | I/II | Autologous tumor-mRNA | Electroporation | i.d. and i.n. | None | None | The immunological data indicated sustained T cell responses and suggested an enhancing effect of booster vaccinations. | [191] |
| 2011 | NA | NA | MAGE-A3, MAGE-C2, tyrosinase, gp100 | Electroporation with TriMix-mRNA | i.d. | IFN-α-2b | None | Vaccinal antigen-specific DIL were found in 0/6 patients tested at vaccine initiation and in 12/21 (57.1%) assessed after the fourth vaccine. During TriMixDC/IFN-a-2b combination therapy, one PR and five SD were observed in 17 patients with evaluable disease at baseline. | [195] |
| 2012 | NA | NA | MAGE-A3, MAGE-C2, tyrosinase, gp100 | Electroporation with TriMix-mRNA | i.v. and i.d. | None | NA | Ex vivo-generated mRNA-modified DCs can induce effector CD8+ and CD4+ T cells from the naive T-cell repertoire of melanoma patients. | [197] |
| 2013 | NCT 01066390 | Ib | MAGE-A3, MAGE-C2, tyrosinase, gp100 | Electroporation with TriMix-mRNA | i.v. | None | None | In a total of 15 patients, two patients achieved a CR and two patients a PR. All objective responders achieved a PFS. Antigen-specific SKILs were documented in 6 of 12 patients, and antigen-specific CD8+ T-cells were detected in the blood of four of five patients. | [181] |
| 2015 | NA | NA | MAGE-A1, MAGE-A3, MAGE-C2, MelanA/MART-1, tyrosinase, gp100 | Electroporation with TriMix-mRNA | i.d. | IFN-α-2b | None | The median relapse-free survival is 22 months (95 % CI 12–32 months), the 2-year and 4-year survival rates are 93% and 70%, respectively. | [182] |
| 2016 | NCT 01302496 | II | MAGE-A3, MAGE-C2, tyrosinase, gp100 | Electroporation with TriMix-mRNA | i.v. and i.d. | Ipilimumab | 17 | The 6-month disease control rate was 51% (95% CI, 36% to 67%), and the overall tumor response rate was 38%, seven CR and one PR are ongoing after a median follow-up time of 36 months | [183] |
| 2020 | NCT 01676779 | II | MAGEA3, MAGE-C2, tyrosinase, gp100 | Electroporation with TriMix-mRNA | i.v. and i.d. | None | None | 71% of patients in the study arm were free of disease compared with 35% in the control arm after one year. The median time to non-salvageable recurrence was superior in the study arm. | [198] |
| 2020 | NCT 02285413 | II | tyrosinase, gp100 | Electroporation | i.v. and i.d. | Cisplatin | 1 | Antigen-specific CD8+ T cells were found in 44% versus 67%, and functional T cell responses in 28% versus 19% in patients receiving DC vaccination with and without cisplatin, respectively. A significantly better OS is observed in stage III patients treated with combination therapy compared with DC monotherapy. | [166] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bidram, M.; Zhao, Y.; Shebardina, N.G.; Baldin, A.V.; Bazhin, A.V.; Ganjalikhany, M.R.; Zamyatnin, A.A., Jr.; Ganjalikhani-hakemi, M. mRNA-Based Cancer Vaccines: A Therapeutic Strategy for the Treatment of Melanoma Patients. Vaccines 2021, 9, 1060. https://doi.org/10.3390/vaccines9101060
Bidram M, Zhao Y, Shebardina NG, Baldin AV, Bazhin AV, Ganjalikhany MR, Zamyatnin AA Jr., Ganjalikhani-hakemi M. mRNA-Based Cancer Vaccines: A Therapeutic Strategy for the Treatment of Melanoma Patients. Vaccines. 2021; 9(10):1060. https://doi.org/10.3390/vaccines9101060
Chicago/Turabian StyleBidram, Maryam, Yue Zhao, Natalia G. Shebardina, Alexey V. Baldin, Alexandr V. Bazhin, Mohamad Reza Ganjalikhany, Andrey A. Zamyatnin, Jr., and Mazdak Ganjalikhani-hakemi. 2021. "mRNA-Based Cancer Vaccines: A Therapeutic Strategy for the Treatment of Melanoma Patients" Vaccines 9, no. 10: 1060. https://doi.org/10.3390/vaccines9101060
APA StyleBidram, M., Zhao, Y., Shebardina, N. G., Baldin, A. V., Bazhin, A. V., Ganjalikhany, M. R., Zamyatnin, A. A., Jr., & Ganjalikhani-hakemi, M. (2021). mRNA-Based Cancer Vaccines: A Therapeutic Strategy for the Treatment of Melanoma Patients. Vaccines, 9(10), 1060. https://doi.org/10.3390/vaccines9101060