Acellular Pertussis Vaccine Inhibits Bordetella pertussis Clearance from the Nasal Mucosa of Mice


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. B. pertussis Strains and Growth Conditions
2.2. Antigens
2.3. Composition of Vaccines Used in the Study
2.4. Ethics Statement
2.5. Mouse Immunization and Intranasal Infection
2.6. Analysis of Antibody Responses
2.7. Opsonophagocytic Uptake
2.8. Agglutination Titer Determination
2.9. Splenocyte Restimulation and Secreted Cytokine Determination
2.10. ACT Cytotoxicity Neutralization Assay
2.11. Statistical Analysis
3. Results
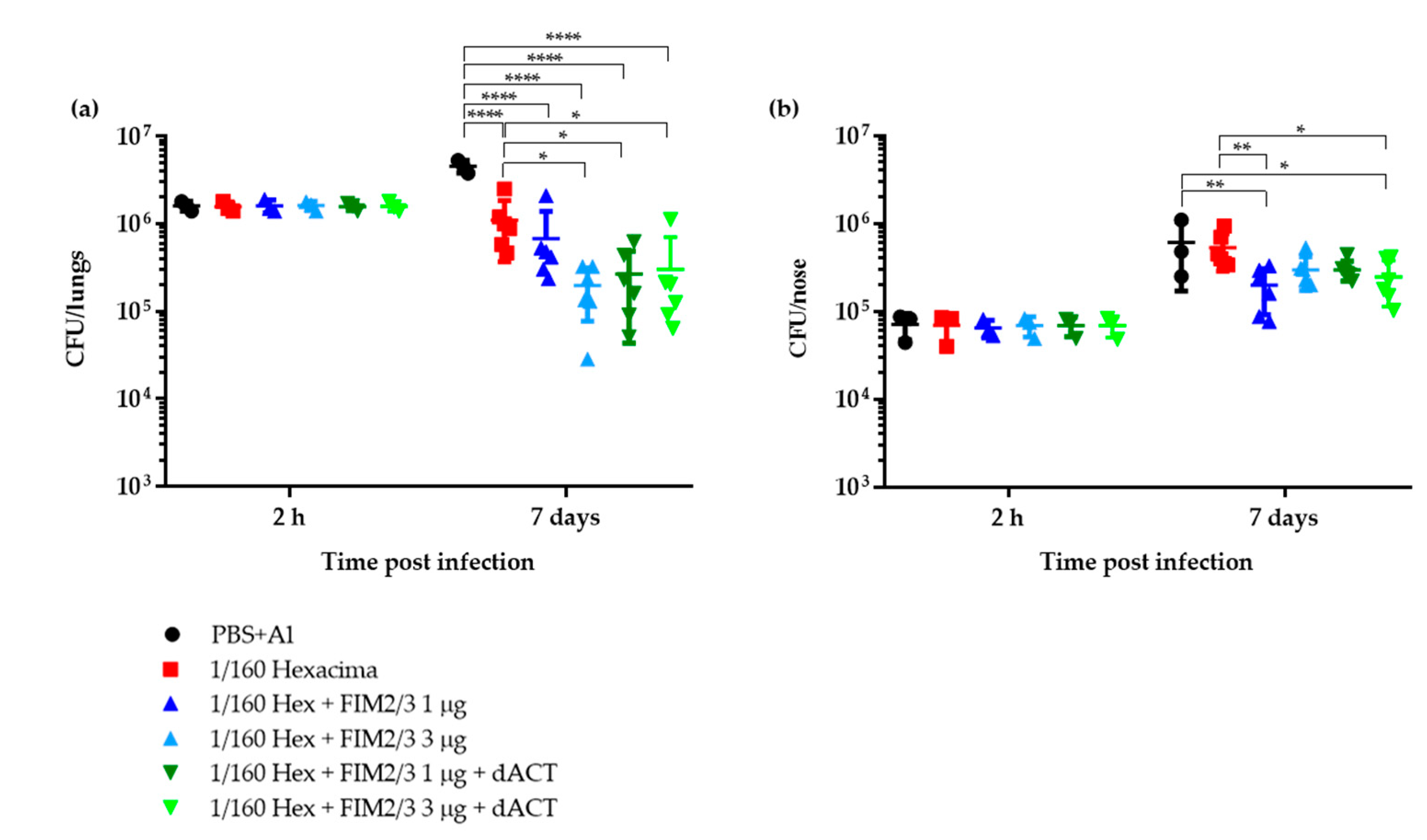
3.1. Limiting Dose of aP Supplemented with dACT and FIM2/3 Plus LOS Elicits Partial Protective Immunity against B. pertussis Infection of Mouse Lungs But No Protection of the Nasal Cavity
3.2. 1/20 Human Dose of dPT + FHA aP Vaccine Supplemented with PRN and FIM2/3 Induces Sterilizing Immunity in Mouse Lungs But Inhibits Elimination of B. pertussis Infection from the Nasal Cavity
3.3. 1/20 Human Dose of a dPT + FHA + PRN aP Vaccine Supplemented with FIM2/3 and dACT Induces Sterilizing Immunity in Mouse Lungs But Blocks Elimination of B. pertussis Infection from the Nasal Cavity
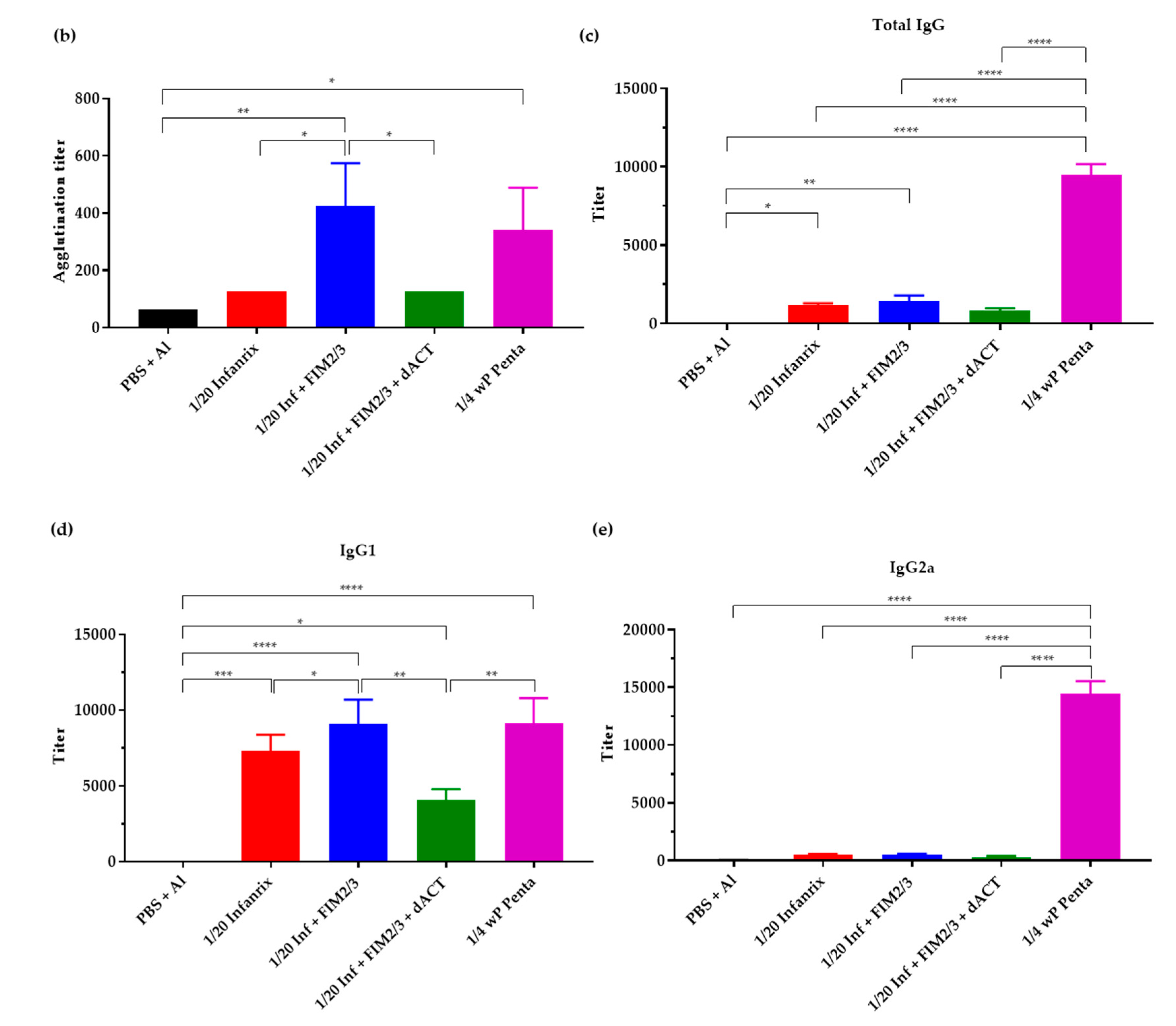
3.4. The Supplemented aP Vaccine Induces a Th2-Polarized Immune Response with IgG1 Isotype-Agglutinating Antibodies That Enhance Opsonophagocytic Uptake of B. pertussis Bacteria In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | adenylate cyclase toxin |
| AG | antigens |
| aP | acellular pertussis |
| CFUs | colony forming units |
| dACT | adenylate cyclase toxoid |
| D-MEM | Dulbecco’s Modified Eagle’s Medium |
| dPT | detoxified pertussis toxin |
| DTwP | diphtheria–tetanus–whole-cell pertussis vaccine |
| EU | endotoxin unit |
| FHA | filamentous hemagglutinin |
| FIM | fimbriae |
| HD | human dose |
| LOS | outer membrane lipooligosaccharide |
| LPS | lipopolysaccharide |
| MOI | multiplicity of infection |
| OPA | opsonophagocytic uptake assay |
| PBS | phosphate-buffered saline |
| PMA | phorbol myristate acetate |
| PRN | pertactin |
| PT | pertussis toxin |
| PRMI | Roswell Park Memorial Institute medium |
| RTX | repeats in toxin |
| TdaP | tetanus–diphtheria–acellular pertussis vaccine |
| TRM | tissue resident memory T cells |
| wP | whole-cell pertussis |
References
- Melvin, J.A.; Scheller, E.V.; Miller, J.F.; Cotter, P.A. Bordetella pertussis pathogenesis: Current and future challenges. Nat. Rev. Microbiol. 2014, 12, 274–288. [Google Scholar] [CrossRef]
- Cherry, J.D. The 112-Year Odyssey of Pertussis and Pertussis Vaccines-Mistakes Made and Implications for the Future. J. Pediatr. Infect. Dis. Soc. 2019, 8, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.H.T.; Duclos, P.; Nelson, E.A.S.; Hutubessy, R.C.W. An update of the global burden of pertussis in children younger than 5 years: A modelling study. Lancet Infect. Dis. 2017, 17, 974–980. [Google Scholar] [CrossRef]
- Klein, N.P.; Baine, Y.; Kolhe, D.; Baccarini, C.I.; Miller, J.M.; Van der Wielen, M. Five-year Antibody Persistence and Booster Response After 1 or 2 Doses of Meningococcal A, C, W and Y Tetanus Toxoid Conjugate Vaccine in Healthy Children. Pediatr. Infect. Dis. J. 2016, 35, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Koepke, R.; Eickhoff, J.C.; Ayele, R.A.; Petit, A.B.; Schauer, S.L.; Hopfensperger, D.J.; Conway, J.H.; Davis, J.P. Estimating the effectiveness of tetanus-diphtheria-acellular pertussis vaccine (Tdap) for preventing pertussis: Evidence of rapidly waning immunity and difference in effectiveness by Tdap brand. J. Infect. Dis. 2014, 210, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.D. Acellular vaccines and resurgence of pertussis. JAMA 2012, 308, 2149–2150. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.L.; Ware, R.S.; Grimwood, K.; Lambert, S.B. Number and order of whole cell pertussis vaccines in infancy and disease protection. JAMA 2012, 308, 454–456. [Google Scholar] [CrossRef]
- Tartof, S.Y.; Lewis, M.; Kenyon, C.; White, K.; Osborn, A.; Liko, J.; Zell, E.; Martin, S.; Messonnier, N.E.; Clark, T.A.; et al. Waning immunity to pertussis following 5 doses of DTaP. Pediatrics 2013, 131, e1047–e1052. [Google Scholar] [CrossRef]
- Witt, M.A.; Arias, L.; Katz, P.H.; Truong, E.T.; Witt, D.J. Reduced risk of pertussis among persons ever vaccinated with whole cell pertussis vaccine compared to recipients of acellular pertussis vaccines in a large US cohort. Clin. Infect. Dis. 2013, 56, 1248–1254. [Google Scholar] [CrossRef]
- Witt, M.A.; Katz, P.H.; Witt, D.J. Unexpectedly limited durability of immunity following acellular pertussis vaccination in preadolescents in a North American outbreak. Clin. Infect. Dis. 2012, 54, 1730–1735. [Google Scholar] [CrossRef]
- Barkoff, A.M.; Mertsola, J.; Pierard, D.; Dalby, T.; Hoegh, S.V.; Guillot, S.; Stefanelli, P.; van Gent, M.; Berbers, G.; Vestrheim, D.; et al. Pertactin-deficient Bordetella pertussis isolates: Evidence of increased circulation in Europe, 1998 to 2015. Eurosurveillance 2019, 24, 1700832. [Google Scholar] [CrossRef]
- Hellwig, S.M.; Rodriguez, M.E.; Berbers, G.A.; van de Winkel, J.G.; Mooi, F.R. Crucial role of antibodies to pertactin in Bordetella pertussis immunity. J. Infect. Dis. 2003, 188, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.W.; Pawloski, L.; Williams, M.; Weening, K.; DeBolt, C.; Qin, X.; Reynolds, L.; Kenyon, C.; Giambrone, G.; Kudish, K.; et al. Pertactin-negative Bordetella pertussis strains: Evidence for a possible selective advantage. Clin. Infect. Dis. 2015, 60, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Pawloski, L.C.; Queenan, A.M.; Cassiday, P.K.; Lynch, A.S.; Harrison, M.J.; Shang, W.; Williams, M.M.; Bowden, K.E.; Burgos-Rivera, B.; Qin, X.; et al. Prevalence and molecular characterization of pertactin-deficient Bordetella pertussis in the United States. Clin. Vaccine Immunol. 2014, 21, 119–125. [Google Scholar] [CrossRef]
- Williams, M.M.; Sen, K.; Weigand, M.R.; Skoff, T.H.; Cunningham, V.A.; Halse, T.A.; Tondella, M.L. CDC Pertussis Working Group. Bordetella pertussis Strain Lacking Pertactin and Pertussis Toxin. Emerg. Infect. Dis. 2016, 22, 319–322. [Google Scholar] [CrossRef]
- Faulkner, A.E.; Skoff, T.H.; Tondella, M.L.; Cohn, A.; Clark, T.A.; Martin, S.W. Trends in Pertussis Diagnostic Testing in the United States, 1990 to 2012. Pediatr. Infect. Dis. J. 2016, 35, 39–44. [Google Scholar] [CrossRef]
- Marchant, C.D.; Loughlin, A.M.; Lett, S.M.; Todd, C.W.; Wetterlow, L.H.; Bicchieri, R.; Higham, S.; Etkind, P.; Silva, E.; Siber, G.R. Pertussis in Massachusetts, 1981-1991: Incidence, serologic diagnosis, and vaccine effectiveness. J. Infect. Dis. 1994, 169, 1297–1305. [Google Scholar] [CrossRef]
- Althouse, B.M.; Scarpino, S.V. Asymptomatic transmission and the resurgence of Bordetella pertussis. BMC Med. 2015, 13, 146. [Google Scholar] [CrossRef]
- Warfel, J.M.; Zimmerman, L.I.; Merkel, T.J. Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. Proc. Natl. Acad. Sci. USA 2014, 111, 787–792. [Google Scholar] [CrossRef]
- Mills, K.H.; Ross, P.J.; Allen, A.C.; Wilk, M.M. Do we need a new vaccine to control the re-emergence of pertussis? Trends Microbiol. 2014, 22, 49–52. [Google Scholar] [CrossRef]
- Redhead, K.; Watkins, J.; Barnard, A.; Mills, K.H. Effective immunization against Bordetella pertussis respiratory infection in mice is dependent on induction of cell-mediated immunity. Infect. Immun. 1993, 61, 3190–3198. [Google Scholar] [CrossRef]
- Allen, A.C.; Wilk, M.M.; Misiak, A.; Borkner, L.; Murphy, D.; Mills, K.H.G. Sustained protective immunity against Bordetella pertussis nasal colonization by intranasal immunization with a vaccine-adjuvant combination that induces IL-17-secreting TRM cells. Mucosal Immunol. 2018, 11, 1763–1776. [Google Scholar] [CrossRef]
- Misiak, A.; Wilk, M.M.; Raverdeau, M.; Mills, K.H. IL-17-Producing Innate and Pathogen-Specific Tissue Resident Memory gammadelta T Cells Expand in the Lungs of Bordetella pertussis-Infected Mice. J. Immunol. 2017, 198, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.J.; Sutton, C.E.; Higgins, S.; Allen, A.C.; Walsh, K.; Misiak, A.; Lavelle, E.C.; McLoughlin, R.M.; Mills, K.H. Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: Towards the rational design of an improved acellular pertussis vaccine. PLoS Pathog. 2013, 9, e1003264. [Google Scholar] [CrossRef]
- Solans, L.; Debrie, A.S.; Borkner, L.; Aguilo, N.; Thiriard, A.; Coutte, L.; Uranga, S.; Trottein, F.; Martin, C.; Mills, K.H.G.; et al. IL-17-dependent SIgA-mediated protection against nasal Bordetella pertussis infection by live attenuated BPZE1 vaccine. Mucosal Immunol. 2018, 11, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Warfel, J.M.; Edwards, K.M. Pertussis vaccines and the challenge of inducing durable immunity. Curr. Opin. Immunol. 2015, 35, 48–54. [Google Scholar] [CrossRef]
- Wilk, M.M.; Borkner, L.; Misiak, A.; Curham, L.; Allen, A.C.; Mills, K.H.G. Immunization with whole cell but not acellular pertussis vaccines primes CD4 TRM cells that sustain protective immunity against nasal colonization with Bordetella pertussis. Emerg. Microbes Infect. 2019, 8, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Wilk, M.M.; Misiak, A.; McManus, R.M.; Allen, A.C.; Lynch, M.A.; Mills, K.H.G. Lung CD4 Tissue-Resident Memory T Cells Mediate Adaptive Immunity Induced by Previous Infection of Mice with Bordetella pertussis. J. Immunol. 2017, 199, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, T.; Dillon, M.B.; da Silva Antunes, R.; Paul, S.; Peters, B.; Crotty, S.; Lindestam Arlehamn, C.S.; Sette, A. Th1 versus Th2 T cell polarization by whole-cell and acellular childhood pertussis vaccines persists upon re-immunization in adolescence and adulthood. Cell. Immunol. 2016, 304–305, 35–43. [Google Scholar] [CrossRef]
- da Silva Antunes, R.; Babor, M.; Carpenter, C.; Khalil, N.; Cortese, M.; Mentzer, A.J.; Seumois, G.; Petro, C.D.; Purcell, L.A.; Vijayanand, P.; et al. Th1/Th17 polarization persists following whole-cell pertussis vaccination despite repeated acellular boosters. J. Clin. Investig. 2018, 128, 3853–3865. [Google Scholar] [CrossRef]
- BMJ. VACCINATION against whooping-cough; relation between protection in children and results of laboratory tests; a report to the Whooping-cough Immunization Committee of the Medical Research Council and to the medical officers of health for Cardiff, Leeds, Leyton, Manchester, Middlesex, Oxford, Poole, Tottenham, Walthamstow, and Wembley. BMJ 1956, 2, 454–462. [Google Scholar]
- Paddock, C.D.; Sanden, G.N.; Cherry, J.D.; Gal, A.A.; Langston, C.; Tatti, K.M.; Wu, K.H.; Goldsmith, C.S.; Greer, P.W.; Montague, J.L.; et al. Pathology and pathogenesis of fatal Bordetella pertussis infection in infants. Clin. Infect. Dis. 2008, 47, 328–338. [Google Scholar] [CrossRef]
- Betsou, F.; Sebo, P.; Guiso, N. CyaC-mediated activation is important not only for toxic but also for protective activities of Bordetella pertussis adenylate cyclase-hemolysin. Infect. Immun. 1993, 61, 3583–3589. [Google Scholar] [CrossRef]
- Betsou, F.; Sebo, P.; Guiso, N. The C-terminal domain is essential for protective activity of the Bordetella pertussis adenylate cyclase-hemolysin. Infect. Immun. 1995, 63, 3309–3315. [Google Scholar] [CrossRef]
- Villarino Romero, R.; Bibova, I.; Cerny, O.; Vecerek, B.; Wald, T.; Benada, O.; Zavadilova, J.; Osicka, R.; Sebo, P. The Bordetella pertussis type III secretion system tip complex protein Bsp22 is not a protective antigen and fails to elicit serum antibody responses during infection of humans and mice. Infect. Immun. 2013, 81, 2761–2767. [Google Scholar] [CrossRef]
- Cheung, G.Y.; Xing, D.; Prior, S.; Corbel, M.J.; Parton, R.; Coote, J.G. Effect of different forms of adenylate cyclase toxin of Bordetella pertussis on protection afforded by an acellular pertussis vaccine in a murine model. Infect. Immun. 2006, 74, 6797–6805. [Google Scholar] [CrossRef]
- Boehm, D.T.; Hall, J.M.; Wong, T.Y.; DiVenere, A.M.; Sen-Kilic, E.; Bevere, J.R.; Bradford, S.D.; Blackwood, C.B.; Elkins, C.M.; DeRoos, K.A.; et al. Evaluation of Adenylate Cyclase Toxoid Antigen in Acellular Pertussis Vaccines by Using a Bordetella pertussis Challenge Model in Mice. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Ahmad, J.N.; Holubova, J.; Benada, O.; Kofronova, O.; Stehlik, L.; Vasakova, M.; Sebo, P. Bordetella Adenylate Cyclase Toxin Inhibits Monocyte-to-Macrophage Transition and Dedifferentiates Human Alveolar Macrophages into Monocyte-like Cells. mBio 2019, 10, e01743-19. [Google Scholar] [CrossRef]
- Cerny, O.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Sebo, P. cAMP Signaling of Adenylate Cyclase Toxin Blocks the Oxidative Burst of Neutrophils through Epac-Mediated Inhibition of Phospholipase C Activity. J. Immunol. 2017, 198, 1285–1296. [Google Scholar] [CrossRef]
- Sebo, P.; Osicka, R.; Masin, J. Adenylate cyclase toxin-hemolysin relevance for pertussis vaccines. Expert Rev. Vaccines 2014, 13, 1215–1227. [Google Scholar] [CrossRef]
- Stainer, D.W.; Scholte, M.J. A simple chemically defined medium for the production of phase I Bordetella pertussis. J. Gen. Microbiol. 1970, 63, 211–220. [Google Scholar] [CrossRef]
- Bindels, D.S.; Haarbosch, L.; van Weeren, L.; Postma, M.; Wiese, K.E.; Mastop, M.; Aumonier, S.; Gotthard, G.; Royant, A.; Hink, M.A.; et al. mScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 2017, 14, 53–56. [Google Scholar] [CrossRef]
- Kovach, M.E.; Phillips, R.W.; Elzer, P.H.; Roop, R.M., 2nd; Peterson, K.M. pBBR1MCS: A broad-host-range cloning vector. Biotechniques 1994, 16, 800–802. [Google Scholar]
- Alexander, F.; Matheson, M.; Fry, N.K.; Labram, B.; Gorringe, A.R. Antibody responses to individual Bordetella pertussis fimbrial antigen Fim2 or Fim3 following immunization with the five-component acellular pertussis vaccine or to pertussis disease. Clin. Vaccine Immunol. 2012, 19, 1776–1783. [Google Scholar] [CrossRef]
- Stanek, O.; Masin, J.; Osicka, R.; Jurnecka, D.; Osickova, A.; Sebo, P. Rapid Purification of Endotoxin-Free RTX Toxins. Toxins 2019, 11, 336. [Google Scholar] [CrossRef]
- Villarino Romero, R.; Hasan, S.; Fae, K.; Holubova, J.; Geurtsen, J.; Schwarzer, M.; Wiertsema, S.; Osicka, R.; Poolman, J.; Sebo, P. Bordetella pertussis filamentous hemagglutinin itself does not trigger anti-inflammatory interleukin-10 production by human dendritic cells. Int. J. Med. Microbiol. 2016, 306, 38–47. [Google Scholar] [CrossRef]
- van der Ark, A.; van Straaten-van de Kappelle, I.; Akkermans, A.; Hendriksen, C.; van de Donk, H. Development of pertussis serological potency test. Serological assessment of antibody response induced by whole cell vaccine as an alternative to mouse protection in an intracerebral challenge model. Biologicals 1994, 22, 233–242. [Google Scholar] [CrossRef]
- Fabbrini, M.; Sammicheli, C.; Margarit, I.; Maione, D.; Grandi, G.; Giuliani, M.M.; Mori, E.; Nuti, S. A new flow-cytometry-based opsonophagocytosis assay for the rapid measurement of functional antibody levels against Group B Streptococcus. J. Immunol. Methods 2012, 378, 11–19. [Google Scholar] [CrossRef]
- Godfroid, F.; Denoel, P.; de Grave, D.; Schuerman, L.; Poolman, J. Diphtheria-tetanus-pertussis (DTP) combination vaccines and evaluation of pertussis immune responses. Int. J. Med. Microbiol. 2004, 294, 269–276. [Google Scholar] [CrossRef]
- Guiso, N.; Capiau, C.; Carletti, G.; Poolman, J.; Hauser, P. Intranasal murine model of Bordetella pertussis infection. I. Prediction of protection in human infants by acellular vaccines. Vaccine 1999, 17, 2366–2376. [Google Scholar] [CrossRef]
- Mills, K.H.; Barnard, A.; Watkins, J.; Redhead, K. Cell-mediated immunity to Bordetella pertussis: Role of Th1 cells in bacterial clearance in a murine respiratory infection model. Infect. Immun. 1993, 61, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Queenan, A.M.; Dowling, D.J.; Cheng, W.K.; Fae, K.; Fernandez, J.; Flynn, P.J.; Joshi, S.; Brightman, S.E.; Ramirez, J.; Serroyen, J.; et al. Increasing FIM2/3 antigen-content improves efficacy of Bordetella pertussis vaccines in mice in vivo without altering vaccine-induced human reactogenicity biomarkers in vitro. Vaccine 2019, 37, 80–89. [Google Scholar] [CrossRef]
- Zaghouani, H.; Hoeman, C.M.; Adkins, B. Neonatal immunity: Faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol. 2009, 30, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Conover, M.S.; Sloan, G.P.; Love, C.F.; Sukumar, N.; Deora, R. The Bps polysaccharide of Bordetella pertussis promotes colonization and biofilm formation in the nose by functioning as an adhesin. Mol. Microbiol. 2010, 77, 1439–1455. [Google Scholar] [CrossRef]
- Weyrich, L.S.; Feaga, H.A.; Park, J.; Muse, S.J.; Safi, C.Y.; Rolin, O.Y.; Young, S.E.; Harvill, E.T. Resident microbiota affect Bordetella pertussis infectious dose and host specificity. J. Infect. Dis. 2014, 209, 913–921. [Google Scholar] [CrossRef][Green Version]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar] [CrossRef]
- Kamanova, J.; Kofronova, O.; Masin, J.; Genth, H.; Vojtova, J.; Linhartova, I.; Benada, O.; Just, I.; Sebo, P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 2008, 181, 5587–5597. [Google Scholar] [CrossRef] [PubMed]
- Mobberley-Schuman, P.S.; Connelly, B.; Weiss, A.A. Phagocytosis of Bordetella pertussis incubated with convalescent serum. J. Infect. Dis. 2003, 187, 1646–1653. [Google Scholar] [CrossRef]
- Weingart, C.L.; Mobberley-Schuman, P.S.; Hewlett, E.L.; Gray, M.C.; Weiss, A.A. Neutralizing antibodies to adenylate cyclase toxin promote phagocytosis of Bordetella pertussis by human neutrophils. Infect. Immun. 2000, 68, 7152–7155. [Google Scholar] [CrossRef]
- Ibsen, P.H. The effect of formaldehyde, hydrogen peroxide and genetic detoxification of pertussis toxin on epitope recognition by murine monoclonal antibodies. Vaccine 1996, 14, 359–368. [Google Scholar] [CrossRef]
- Auderset, F.; Ballester, M.; Mastelic-Gavillet, B.; Fontannaz, P.; Chabaud-Riou, M.; Reveneau, N.; Garinot, M.; Mistretta, N.; Liu, Y.; Lambert, P.H.; et al. Reactivating Immunity Primed by Acellular Pertussis Vaccines in the Absence of Circulating Antibodies: Enhanced Bacterial Control by TLR9 Rather Than TLR4 Agonist-Including Formulation. Front. Immunol. 2019, 10, 1520. [Google Scholar] [CrossRef] [PubMed]
- Ausar, S.F.; Zhu, S.; Duprez, J.; Cohen, M.; Bertrand, T.; Steier, V.; Wilson, D.J.; Li, S.; Sheung, A.; Brookes, R.H.; et al. Genetically detoxified pertussis toxin displays near identical structure to its wild-type and exhibits robust immunogenicity. Commun. Biol. 2020, 3, 427. [Google Scholar] [CrossRef]
- Blanchard Rohner, G.; Chatzis, O.; Chinwangso, P.; Rohr, M.; Grillet, S.; Salomon, C.; Lemaitre, B.; Boonrak, P.; Lawpoolsri, S.; Clutterbuck, E.; et al. Boosting Teenagers With Acellular Pertussis Vaccines Containing Recombinant or Chemically Inactivated Pertussis Toxin: A Randomized Clinical Trial. Clin. Infect. Dis. 2019, 68, 1213–1222. [Google Scholar] [CrossRef]
- Pitisuttithum, P.; Chokephaibulkit, K.; Sirivichayakul, C.; Sricharoenchai, S.; Dhitavat, J.; Pitisuthitham, A.; Phongsamart, W.; Boonnak, K.; Lapphra, K.; Sabmee, Y.; et al. Antibody persistence after vaccination of adolescents with monovalent and combined acellular pertussis vaccines containing genetically inactivated pertussis toxin: A phase 2/3 randomised, controlled, non-inferiority trial. Lancet Infect. Dis. 2018, 18, 1260–1268. [Google Scholar] [CrossRef]
- Sricharoenchai, S.; Sirivichayakul, C.; Chokephaibulkit, K.; Pitisuttithum, P.; Dhitavat, J.; Pitisuthitham, A.; Phongsamart, W.; Boonnak, K.; Lapphra, K.; Sabmee, Y.; et al. A genetically inactivated two-component acellular pertussis vaccine, alone or combined with tetanus and reduced-dose diphtheria vaccines, in adolescents: A phase 2/3, randomised controlled non-inferiority trial. Lancet Infect. Dis. 2018, 18, 58–67. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine | Brand Name | dPT (µg) | FHA (µg) | PRN (µg) | FIM2/3 (µg) | LOS (EU) | dACT (µg) | Al3+ (µg) |
|---|---|---|---|---|---|---|---|---|
| 1/160 aP Hex | Hexacima® | 0.156 | 0.156 | - | - | - | 137 | |
| 1/160 aP Hex + FIM2/3 (1 µg) | 0.156 | 0.156 | - | 1 | 6000 | - | ||
| 1/160 aP Hex + FIM2/3 (3 µg) | 0.156 | 0.156 | - | 3 | 18,000 | - | ||
| 1/160 aP Hex + FIM2/3 (1 µg) + dACT | 0.156 | 0.156 | - | 1 | 6000 | 30 | ||
| 1/160 aP Hex + FIM2/3 (3 µg) + dACT | 0.156 | 0.156 | - | 3 | 18,000 | 30 | ||
| 1/20 aP Hex | Hexacima® | 1.25 | 1.25 | - | - | - | ||
| 1/20 aP Hex + FIM2/3 + PRN | 1.25 | 1.25 | 0.2 | 5 | 30,000 | - | 153 | |
| 1/20 aP Hex + FIM2/3 + PRN + dACT | 1.25 | 1.25 | 0.2 | 5 | 30,000 | 30 | ||
| 1/20 aP Inf | Infanrix® | 1.25 | 1.25 | 0.4 | - | - | ||
| 1/20 aP Inf + FIM2/3 | 1.25 | 1.25 | 0.4 | 5 | 30,000 | - | 131 | |
| 1/20 aP Inf + FIM2/3 + dACT | 1.25 | 1.25 | 0.4 | 5 | 30,000 | 30 | ||
| ¼ wP Penta | Pentavac® SD | nd | nd | nd | nd | nd | nd | ≤312 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holubová, J.; Staněk, O.; Brázdilová, L.; Mašín, J.; Bumba, L.; Gorringe, A.R.; Alexander, F.; Šebo, P. Acellular Pertussis Vaccine Inhibits Bordetella pertussis Clearance from the Nasal Mucosa of Mice. Vaccines 2020, 8, 695. https://doi.org/10.3390/vaccines8040695
Holubová J, Staněk O, Brázdilová L, Mašín J, Bumba L, Gorringe AR, Alexander F, Šebo P. Acellular Pertussis Vaccine Inhibits Bordetella pertussis Clearance from the Nasal Mucosa of Mice. Vaccines. 2020; 8(4):695. https://doi.org/10.3390/vaccines8040695
Chicago/Turabian StyleHolubová, Jana, Ondřej Staněk, Ludmila Brázdilová, Jiří Mašín, Ladislav Bumba, Andrew R. Gorringe, Frances Alexander, and Peter Šebo. 2020. "Acellular Pertussis Vaccine Inhibits Bordetella pertussis Clearance from the Nasal Mucosa of Mice" Vaccines 8, no. 4: 695. https://doi.org/10.3390/vaccines8040695
APA StyleHolubová, J., Staněk, O., Brázdilová, L., Mašín, J., Bumba, L., Gorringe, A. R., Alexander, F., & Šebo, P. (2020). Acellular Pertussis Vaccine Inhibits Bordetella pertussis Clearance from the Nasal Mucosa of Mice. Vaccines, 8(4), 695. https://doi.org/10.3390/vaccines8040695