The Age of Cyclic Dinucleotide Vaccine Adjuvants


Abstract
1. Introduction
2. Cyclic Dinucleotides (CDNs)—An Universal Adjuvant
2.1. CDN Adjuvants Induce Potent, Safe, and Balanced Vaccine Responses
2.2. CDN Adjuvants Induce Anti-Tumor Responses
2.3. CDN Adjuvants Induce Vaccine Responses on Mucosal Surface
2.4. CDN Adjuvants Induce Tolerogenic Responses
3. Delivering CDN Vaccine Adjuvants In Vivo
3.1. Encapsulated CDN Adjuvants for Infectious Diseases
3.2. Encapsulated CDN Adjuvants for Cancer Immunotherapy
4. Mode of Action of CDN Adjuvants
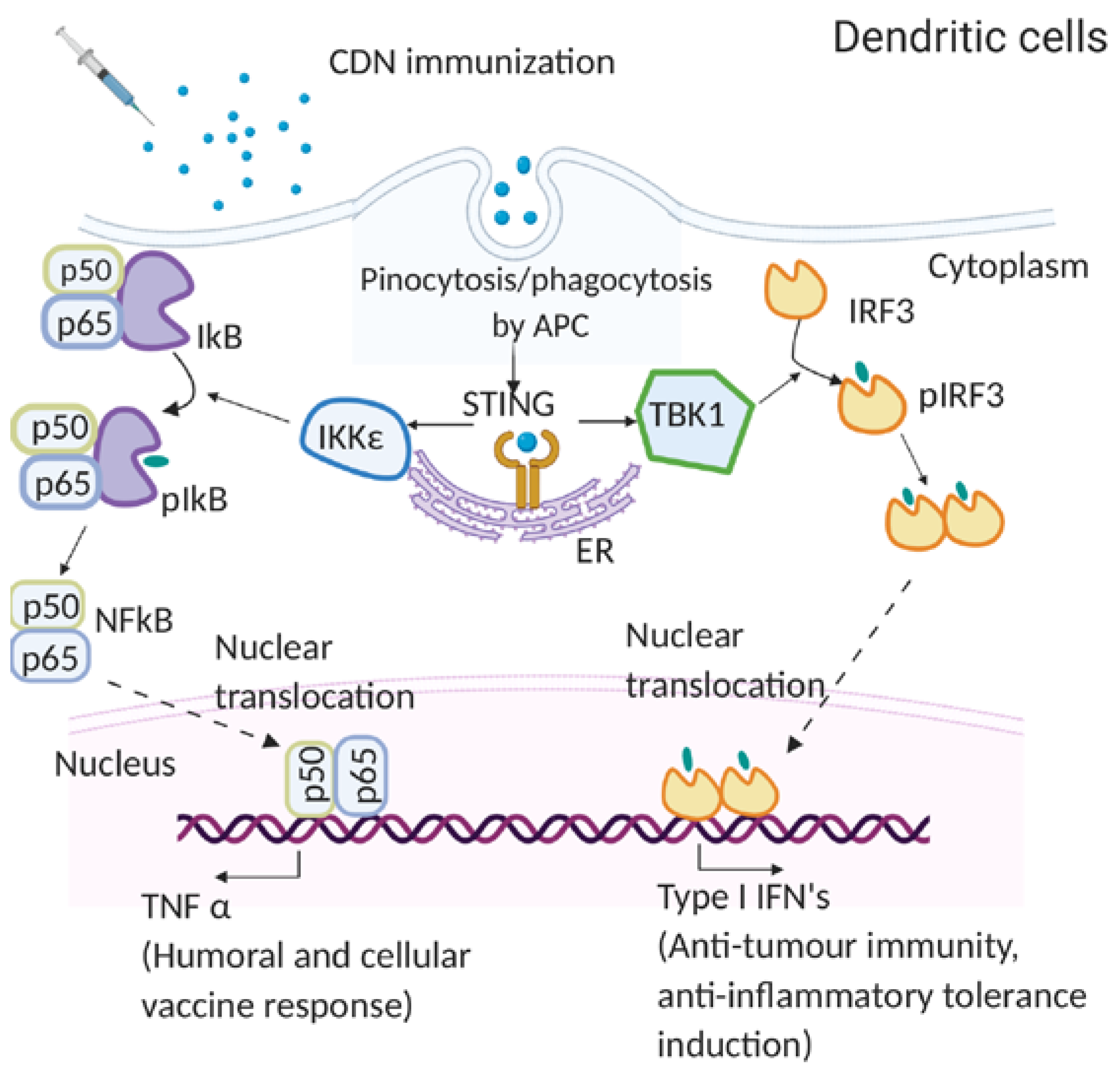
4.1. Molecular Mechanism of CDN Adjuvants
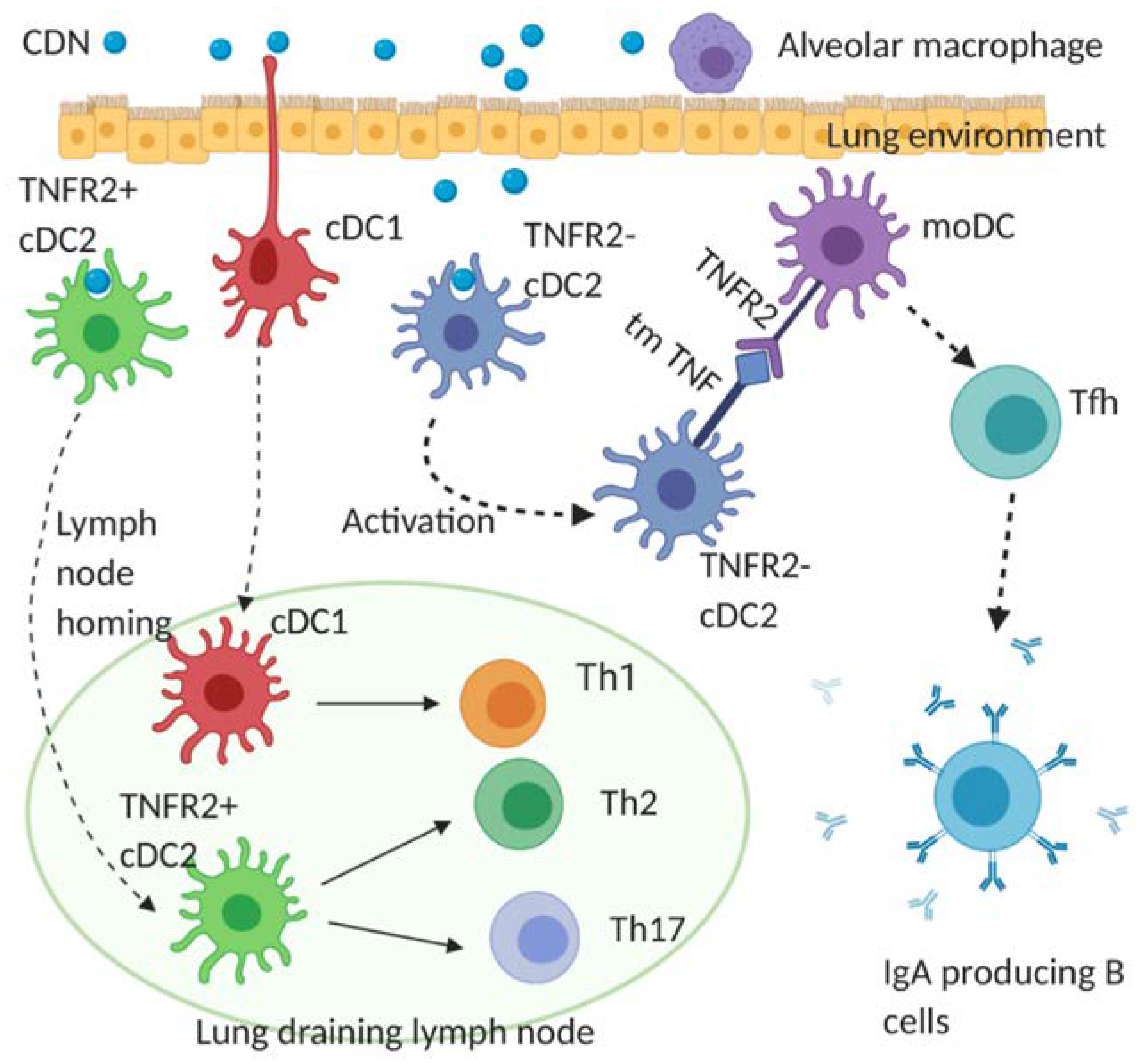
4.2. Cellular Mechanism of CDN Vaccine Adjuvants for Infectious Diseases
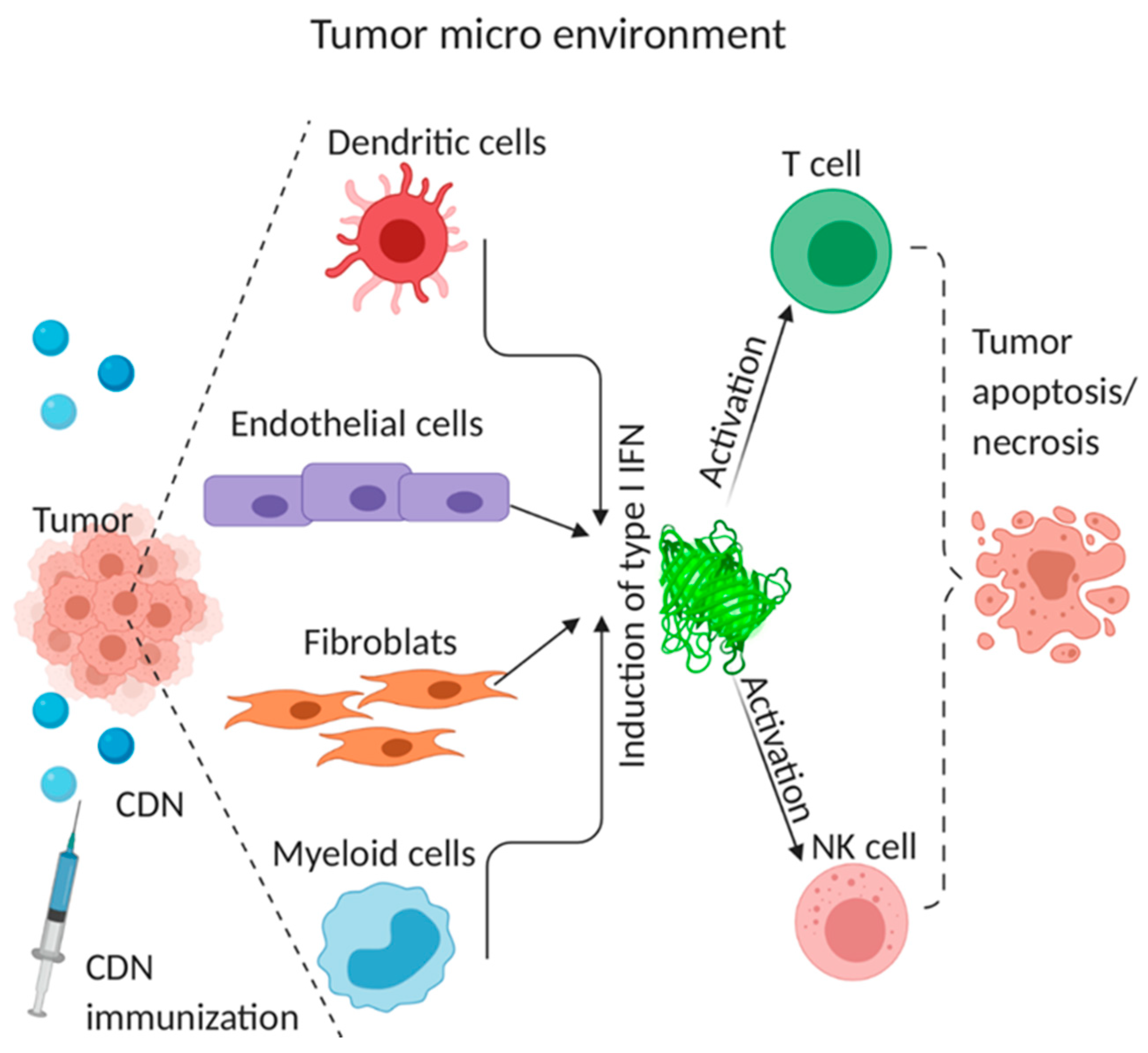
4.3. Cellular Mechanism of CDN Anti-Tumor Activity
4.4. Mechanism of CDN-Induced Immune Tolerance
5. Confounding Factors in CDN Adjuvanticity in Humans
5.1. The Heterogeneity of the Human STING Gene
5.2. The Impact of Age in CDN Adjuvanticity
6. Conclusions
7. Future of CDN Vaccine Adjuvants
Author Contributions
Funding
Conflicts of Interest
References
- Marrack, P.; McKee, A.S.; Munks, M.W. Towards an understanding of the adjuvant action of aluminium. Nat. Rev. Immunol. 2009, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Harandi, A.M. Systems analysis of human vaccine adjuvants. Semin. Immunol. 2018, 39, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, G.; Rappuoli, R.; Didierlaurent, A.M. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin. Immunol. 2018, 39, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Ogunniyi, A.D.; Paton, J.C.; Kirby, A.C.; McCullers, J.A.; Cook, J.; Hyodo, M.; Hayakawa, Y.; Karaolis, D.K. C-di-gmp is an effective immunomodulator and vaccine adjuvant against pneumococcal infection. Vaccine 2008, 26, 4676–4685. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; KuoLee, R.; Tram, K.; Qiu, H.; Zhang, J.; Patel, G.B.; Chen, W. 3’,5’-cyclic diguanylic acid elicits mucosal immunity against bacterial infection. Biochem. Biophys. Res. Commun. 2009, 387, 581–584. [Google Scholar] [CrossRef]
- Gray, P.M.; Forrest, G.; Wisniewski, T.; Porter, G.; Freed, D.C.; DeMartino, J.A.; Zaller, D.M.; Guo, Z.; Leone, J.; Fu, T.M.; et al. Evidence for cyclic diguanylate as a vaccine adjuvant with novel immunostimulatory activities. Cell Immunol. 2012, 278, 113–119. [Google Scholar] [CrossRef]
- Ebensen, T.; Libanova, R.; Schulze, K.; Yevsa, T.; Morr, M.; Guzman, C.A. Bis-(3’,5’)-cyclic dimeric adenosine monophosphate: Strong th1/th2/th17 promoting mucosal adjuvant. Vaccine 2011, 29, 5210–5220. [Google Scholar] [CrossRef]
- Madhun, A.S.; Haaheim, L.R.; Nostbakken, J.K.; Ebensen, T.; Chichester, J.; Yusibov, V.; Guzman, C.A.; Cox, R.J. Intranasal c-di-gmp-adjuvanted plant-derived h5 influenza vaccine induces multifunctional th1 cd4+ cells and strong mucosal and systemic antibody responses in mice. Vaccine 2011, 29, 4973–4982. [Google Scholar] [CrossRef]
- Ebensen, T.; Schulze, K.; Riese, P.; Link, C.; Morr, M.; Guzman, C.A. The bacterial second messenger cyclic digmp exhibits potent adjuvant properties. Vaccine 2007, 25, 1464–1469. [Google Scholar] [CrossRef]
- Wang, J.; Li, P.; Yu, Y.; Fu, Y.; Jiang, H.; Lu, M.; Sun, Z.; Jiang, S.; Lu, L.; Wu, M.X. Pulmonary surfactant-biomimetic nanoparticles potentiate heterosubtypic influenza immunity. Science 2020, 367, eaau0810. [Google Scholar] [CrossRef]
- Van Dis, E.; Sogi, K.M.; Rae, C.S.; Sivick, K.E.; Surh, N.H.; Leong, M.L.; Kanne, D.B.; Metchette, K.; Leong, J.J.; Bruml, J.R.; et al. Sting-activating adjuvants elicit a th17 immune response and protect against mycobacterium tuberculosis infection. Cell Rep. 2018, 23, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.L.; Jee, J.; Kim, E.; Steiner, H.E.; Cormet-Boyaka, E.; Boyaka, P.N. Sublingual targeting of sting with 3’3’-cgamp promotes systemic and mucosal immunity against anthrax toxins. Vaccine 2017, 35, 2511–2519. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karaolis, D.K.; Newstead, M.W.; Zeng, X.; Hyodo, M.; Hayakawa, Y.; Bhan, U.; Liang, H.; Standiford, T.J. Cyclic di-gmp stimulates protective innate immunity in bacterial pneumonia. Infect. Immun. 2007, 75, 4942–4950. [Google Scholar] [CrossRef]
- Zhao, L.; KuoLee, R.; Harris, G.; Tram, K.; Yan, H.; Chen, W. C-di-gmp protects against intranasal acinetobacter baumannii infection in mice by chemokine induction and enhanced neutrophil recruitment. Int. Immunopharmacol. 2011, 11, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.L.; Narita, K.; Hyodo, M.; Hayakawa, Y.; Nakane, A.; Karaolis, D.K. C-di-gmp as a vaccine adjuvant enhances protection against systemic methicillin-resistant staphylococcus aureus (mrsa) infection. Vaccine 2009, 27, 4867–4873. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct activation of sting in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. Sting agonist formulated cancer vaccines can cure established tumors resistant to pd-1 blockade. Sci. Transl. Med. 2015, 7, 283ra52. [Google Scholar] [CrossRef]
- Volckmar, J.; Knop, L.; Stegemann-Koniszewski, S.; Schulze, K.; Ebensen, T.; Guzman, C.A.; Bruder, D. The sting activator c-di-amp exerts superior adjuvant properties than the formulation poly(i:C)/cpg after subcutaneous vaccination with soluble protein antigen or dec-205-mediated antigen targeting to dendritic cells. Vaccine 2019, 37, 4963–4974. [Google Scholar] [CrossRef]
- Libanova, R.; Ebensen, T.; Schulze, K.; Bruhn, D.; Norder, M.; Yevsa, T.; Morr, M.; Guzman, C.A. The member of the cyclic di-nucleotide family bis-(3’, 5’)-cyclic dimeric inosine monophosphate exerts potent activity as mucosal adjuvant. Vaccine 2010, 28, 2249–2258. [Google Scholar] [CrossRef]
- Eriksson, K.; Fredriksson, M.; Nordstrom, I.; Holmgren, J. Cholera toxin and its b subunit promote dendritic cell vaccination with different influences on th1 and th2 development. Infect. Immun. 2003, 71, 1740–1747. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. As04, an aluminum salt- and tlr4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 2009, 183, 6186–6197. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Kuolee, R.; Yan, H. The potential of 3’,5’-cyclic diguanylic acid (c-di-gmp) as an effective vaccine adjuvant. Vaccine 2010, 28, 3080–3085. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, D.A.; Beatty, P.R.; Reiner, G.L.; Sivick, K.E.; Hix Glickman, L.; Dubensky, T.W., Jr.; Harris, E. Cyclic dinucleotide-adjuvanted dengue virus nonstructural protein 1 induces protective antibody and t cell responses. J. Immunol. 2019, 202, 1153–1162. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T cell dysfunction in cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T cell dysfunction and exhaustion in cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. Sting activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef]
- Ohkuri, T.; Kosaka, A.; Ishibashi, K.; Kumai, T.; Hirata, Y.; Ohara, K.; Nagato, T.; Oikawa, K.; Aoki, N.; Harabuchi, Y.; et al. Intratumoral administration of cgamp transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol. Immunother. 2017, 66, 705–716. [Google Scholar] [CrossRef]
- Nicolai, C.J.; Wolf, N.; Chang, I.C.; Kirn, G.; Marcus, A.; Ndubaku, C.O.; McWhirter, S.M.; Raulet, D.H. Nk cells mediate clearance of cd8(+) t cell-resistant tumors in response to sting agonists. Sci. Immunol. 2020, 5, eaaz2738. [Google Scholar] [CrossRef]
- Francica, B.J.; Ghasemzadeh, A.; Desbien, A.L.; Theodros, D.; Sivick, K.E.; Reiner, G.L.; Hix Glickman, L.; Marciscano, A.E.; Sharabi, A.B.; Leong, M.L.; et al. Tnfalpha and radioresistant stromal cells are essential for therapeutic efficacy of cyclic dinucleotide sting agonists in nonimmunogenic tumors. Cancer Immunol. Res. 2018, 6, 422–433. [Google Scholar] [CrossRef]
- Ebensen, T.; Delandre, S.; Prochnow, B.; Guzman, C.A.; Schulze, K. The combination vaccine adjuvant system alum/c-di-amp results in quantitative and qualitative enhanced immune responses post immunization. Front. Cell Infect. Microbiol. 2019, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, P.A.; Cu-Uvin, S.; Neutra, M.R.; Flanigan, T.P. Comparison of the oral, rectal, and vaginal immunization routes for induction of antibodies in rectal and genital tract secretions of women. Infect. Immun. 1997, 65, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, P.A.; Williams, S.B.; Lynch, R.M.; Flanigan, T.P.; Patterson, R.R.; Cu-Uvin, S.; Neutra, M.R. Differential induction of mucosal and systemic antibody responses in women after nasal, rectal, or vaginal immunization: Influence of the menstrual cycle. J. Immunol. 2002, 169, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Staats, H.F.; Montgomery, S.P.; Palker, T.J. Intranasal immunization is superior to vaginal, gastric, or rectal immunization for the induction of systemic and mucosal anti-hiv antibody responses. AIDS Res. Hum. Retroviruses 1997, 13, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, K.; Miller, C.J.; Kubota, M.; McChesney, M.B.; Lohman, B.; Yamamoto, M.; Fujihashi, K.; Someya, K.; Honda, M.; McGhee, J.R.; et al. Nasal immunization of nonhuman primates with simian immunodeficiency virus p55gag and cholera toxin adjuvant induces th1/th2 help for virus-specific immune responses in reproductive tissues. J. Immunol. 1998, 161, 5952–5958. [Google Scholar] [PubMed]
- Rudin, A.; Riise, G.C.; Holmgren, J. Antibody responses in the lower respiratory tract and male urogenital tract in humans after nasal and oral vaccination with cholera toxin b subunit. Infect. Immun. 1999, 67, 2884–2890. [Google Scholar] [CrossRef]
- Egan, M.A.; Chong, S.Y.; Hagen, M.; Megati, S.; Schadeck, E.B.; Piacente, P.; Ma, B.J.; Montefiori, D.C.; Haynes, B.F.; Israel, Z.R.; et al. A comparative evaluation of nasal and parenteral vaccine adjuvants to elicit systemic and mucosal hiv-1 peptide-specific humoral immune responses in cynomolgus macaques. Vaccine 2004, 22, 3774–3788. [Google Scholar] [CrossRef]
- Belyakov, I.M.; Derby, M.A.; Ahlers, J.D.; Kelsall, B.L.; Earl, P.; Moss, B.; Strober, W.; Berzofsky, J.A. Mucosal immunization with hiv-1 peptide vaccine induces mucosal and systemic cytotoxic t lymphocytes and protective immunity in mice against intrarectal recombinant hiv-vaccinia challenge. Proc. Natl. Acad. Sci. USA 1998, 95, 1709–1714. [Google Scholar] [CrossRef]
- Lycke, N. Recent progress in mucosal vaccine development: Potential and limitations. Nat. Rev. Immunol. 2012, 12, 592–605. [Google Scholar] [CrossRef]
- van Ginkel, F.W.; Jackson, R.J.; Yuki, Y.; McGhee, J.R. Cutting edge: The mucosal adjuvant cholera toxin redirects vaccine proteins into olfactory tissues. J. Immunol. 2000, 165, 4778–4782. [Google Scholar] [CrossRef]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of bell’s palsy in switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Couch, R.B. Nasal vaccination, escherichia coli enterotoxin, and bell’s palsy. N. Engl. J. Med. 2004, 350, 860–861. [Google Scholar] [CrossRef] [PubMed]
- Blaauboer, S.M.; Mansouri, S.; Tucker, H.R.; Wang, H.L.; Gabrielle, V.D.; Jin, L. The mucosal adjuvant cyclic di-gmp enhances antigen uptake and selectively activates pinocytosis-efficient cells in vivo. Elife 2015, 4, e06670. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, L.; Lemos, H.; Chandler, P.R.; Pacholczyk, G.; Baban, B.; Barber, G.N.; Hayakawa, Y.; McGaha, T.L.; Ravishankar, B.; et al. Cutting edge: DNA sensing via the sting adaptor in myeloid dendritic cells induces potent tolerogenic responses. J. Immunol. 2013, 191, 3509–3513. [Google Scholar] [CrossRef]
- Lemos, H.; Huang, L.; Chandler, P.R.; Mohamed, E.; Souza, G.R.; Li, L.; Pacholczyk, G.; Barber, G.N.; Hayakawa, Y.; Munn, D.H.; et al. Activation of the sting adaptor attenuates experimental autoimmune encephalitis. J. Immunol. 2014, 192, 5571–5578. [Google Scholar] [CrossRef]
- Lemos, H.; Mohamed, E.; Huang, L.; Ou, R.; Pacholczyk, G.; Arbab, A.S.; Munn, D.; Mellor, A.L. Sting promotes the growth of tumors characterized by low antigenicity via ido activation. Cancer Res. 2016, 76, 2076–2081. [Google Scholar] [CrossRef]
- Lemos, H.; Mohamed, E.; Huang, L.; Chandler, P.R.; Ou, R.; Pacholczyk, R.; Mellor, A.L. Stimulator of interferon genes agonists attenuate type i diabetes progression in nod mice. Immunology 2019, 158, 353–361. [Google Scholar] [CrossRef]
- Kasper, L.H.; Reder, A.T. Immunomodulatory activity of interferon-beta. Ann. Clin. Transl. Neurol. 2014, 1, 622–631. [Google Scholar] [CrossRef]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. Sting is a direct innate immune sensor of cyclic di-gmp. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef]
- Walker, M.M.; Kim, S.; Crisler, W.J.; Nguyen, K.; Lenz, L.L.; Cambier, J.C.; Getahun, A. Selective loss of responsiveness to exogenous but not endogenous cyclic-dinucleotides in mice expressing sting-r231h. Front. Immunol. 2020, 11, 238. [Google Scholar] [CrossRef]
- Patel, S.; Blaauboer, S.M.; Tucker, H.R.; Mansouri, S.; Ruiz-Moreno, J.S.; Hamann, L.; Schumann, R.R.; Opitz, B.; Jin, L. The common r71h-g230a-r293q human tmem173 is a null allele. J. Immunol. 2017, 198, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Blaauboer, S.M.; Gabrielle, V.D.; Jin, L. Mpys/sting-mediated tnf-alpha, not type i ifn, is essential for the mucosal adjuvant activity of (3’-5’)-cyclic-di-guanosine-monophosphate in vivo. J. Immunol. 2014, 192, 492–502. [Google Scholar] [CrossRef] [PubMed]
- El-Shabouri, M.H. Positively charged nanoparticles for improving the oral bioavailability of cyclosporin-a. Int. J. Pharm. 2002, 249, 101–108. [Google Scholar] [CrossRef]
- Hu, L.; Tang, X.; Cui, F. Solid lipid nanoparticles (slns) to improve oral bioavailability of poorly soluble drugs. J. Pharm. Pharmacol. 2004, 56, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Jang, H.E.; Kang, Y.Y.; Kim, J.; Ahn, J.H.; Mok, H. Submicron-sized hydrogels incorporating cyclic dinucleotides for selective delivery and elevated cytokine release in macrophages. Acta Biomater. 2016, 29, 271–281. [Google Scholar] [CrossRef]
- Wilson, D.R.; Sen, R.; Sunshine, J.C.; Pardoll, D.M.; Green, J.J.; Kim, Y.J. Biodegradable sting agonist nanoparticles for enhanced cancer immunotherapy. Nanomedicine 2018, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Watkins-Schulz, R.; Tiet, P.; Gallovic, M.D.; Junkins, R.D.; Batty, C.; Bachelder, E.M.; Ainslie, K.M.; Ting, J.P.Y. A microparticle platform for sting-targeted immunotherapy enhances natural killer cell- and cd8(+) t cell-mediated anti-tumor immunity. Biomaterials 2019, 205, 94–105. [Google Scholar] [CrossRef]
- Hanson, M.C.; Crespo, M.P.; Abraham, W.; Moynihan, K.D.; Szeto, G.L.; Chen, S.H.; Melo, M.B.; Mueller, S.; Irvine, D.J. Nanoparticulate sting agonists are potent lymph node-targeted vaccine adjuvants. J. Clin. Investig. 2015, 125, 2532–2546. [Google Scholar] [CrossRef]
- Aroh, C.; Wang, Z.; Dobbs, N.; Luo, M.; Chen, Z.; Gao, J.; Yan, N. Innate immune activation by cgmp-amp nanoparticles leads to potent and long-acting antiretroviral response against hiv-1. J. Immunol. 2017, 199, 3840–3848. [Google Scholar] [CrossRef]
- Leach, D.G.; Dharmaraj, N.; Piotrowski, S.L.; Lopez-Silva, T.L.; Lei, Y.L.; Sikora, A.G.; Young, S.; Hartgerink, J.D. Stingel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials 2018, 163, 67–75. [Google Scholar] [CrossRef]
- Miyabe, H.; Hyodo, M.; Nakamura, T.; Sato, Y.; Hayakawa, Y.; Harashima, H. A new adjuvant delivery system ‘cyclic di-gmp/ysk05 liposome’ for cancer immunotherapy. J. Control Release 2014, 184, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of therapeutic sting activation determines cd8(+) t cell-mediated anti-tumor immunity. Cell Rep. 2018, 25, 3074–3085. [Google Scholar] [CrossRef] [PubMed]
- Shae, D.; Becker, K.W.; Christov, P.; Yun, D.S.; Lytton-Jean, A.K.R.; Sevimli, S.; Ascano, M.; Kelley, M.; Johnson, D.B.; Balko, J.M.; et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide sting agonists to enhance cancer immunotherapy. Nat. Nanotechnol. 2019, 14, 269–278. [Google Scholar] [PubMed]
- Koshy, S.T.; Cheung, A.S.; Gu, L.; Graveline, A.R.; Mooney, D.J. Liposomal delivery enhances immune activation by sting agonists for cancer immunotherapy. Adv. Biosyst. 2017, 1, 1600013. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Yu, C.; Xi, J.; Reyes, J.; Mao, G.; Wei, W.Z.; Liu, H. Induction of necrotic cell death and activation of sting in the tumor microenvironment via cationic silica nanoparticles leading to enhanced antitumor immunity. Nanoscale 2018, 10, 9311–9319. [Google Scholar] [CrossRef]
- Liu, Y.; Crowe, W.N.; Wang, L.; Lu, Y.; Petty, W.J.; Habib, A.A.; Zhao, D. An inhalable nanoparticulate sting agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nat. Commun. 2019, 10, 5108. [Google Scholar] [CrossRef]
- Curran, E.; Chen, X.; Corrales, L.; Kline, D.E.; Dubensky, T.W., Jr.; Duttagupta, P.; Kortylewski, M.; Kline, J. Sting pathway activation stimulates potent immunity against acute myeloid leukemia. Cell Rep. 2016, 15, 2357–2366. [Google Scholar] [CrossRef]
- Downey, C.M.; Aghaei, M.; Schwendener, R.A.; Jirik, F.R. Dmxaa causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide sting agonist, 2’3’-cgamp, induces m2 macrophage repolarization. PLoS ONE 2014, 9, e99988. [Google Scholar] [CrossRef]
- Junkins, R.D.; Gallovic, M.D.; Johnson, B.M.; Collier, M.A.; Watkins-Schulz, R.; Cheng, N.; David, C.N.; McGee, C.E.; Sempowski, G.D.; Shterev, I.; et al. A robust microparticle platform for a sting-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J. Control Release 2018, 270, 1–13. [Google Scholar] [CrossRef]
- Ball, R.L.; Bajaj, P.; Whitehead, K.A. Achieving long-term stability of lipid nanoparticles: Examining the effect of ph, temperature, and lyophilization. Int. J. Nanomed. 2017, 12, 305–315. [Google Scholar] [CrossRef]
- del Pozo-Rodriguez, A.; Solinis, M.A.; Gascon, A.R.; Pedraz, J.L. Short- and long-term stability study of lyophilized solid lipid nanoparticles for gene therapy. Eur. J. Pharm. Biopharm. 2009, 71, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, J.; Zhu, M.; Yang, Y.; Shen, J.; Gentile, E.; Paolino, D.; Fresta, M.; Nie, G.; Chen, C.; Shen, H.; et al. Safety of nanoparticles in medicine. Curr. Drug Targets 2015, 16, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Mishra, H.; Ekielski, A.; Talegaonkar, S.; Vaidya, B. Zinc oxide nanoparticles: A promising nanomaterial for biomedical applications. Drug Discov. Today 2017, 22, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S.M.; Barbalat, R.; Monroe, K.M.; Fontana, M.F.; Hyodo, M.; Joncker, N.T.; Ishii, K.J.; Akira, S.; Colonna, M.; Chen, Z.J.; et al. A host type i interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-gmp. J. Exp. Med. 2009, 206, 1899–1911. [Google Scholar] [CrossRef]
- Sauer, J.D.; Sotelo-Troha, K.; von Moltke, J.; Monroe, K.M.; Rae, C.S.; Brubaker, S.W.; Hyodo, M.; Hayakawa, Y.; Woodward, J.J.; Portnoy, D.A.; et al. The n-ethyl-n-nitrosourea-induced goldenticket mouse mutant reveals an essential function of sting in the in vivo interferon response to listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 2011, 79, 688–694. [Google Scholar] [CrossRef]
- Jin, L.; Hill, K.K.; Filak, H.; Mogan, J.; Knowles, H.; Zhang, B.; Perraud, A.L.; Cambier, J.C.; Lenz, L.L. Mpys is required for ifn response factor 3 activation and type i ifn production in the response of cultured phagocytes to bacterial second messengers cyclic-di-amp and cyclic-di-gmp. J. Immunol. 2011, 187, 2595–2601. [Google Scholar] [CrossRef]
- Mansouri, S.; Patel, S.; Katikaneni, D.S.; Blaauboer, S.M.; Wang, W.; Schattgen, S.; Fitzgerald, K.; Jin, L. Immature lung tnfr2(-) conventional dc 2 subpopulation activates modcs to promote cyclic di-gmp mucosal adjuvant responses in vivo. Mucosal Immunol. 2019, 12, 277–289. [Google Scholar] [CrossRef]
- Mansouri, S.; Katikaneni, D.S.; Gogoi, H.; Jin, L. Mucosal vaccine adjuvant cyclic di-GMP differentiates lung moDCs into Bcl6+ and Bcl6− mature moDCs to induce lung memory CD4+ TH cells and lung TFH cells respectively. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. Sting activation reprograms tumor vasculatures and synergizes with vegfr2 blockade. J. Clin. Investig. 2019, 129, 4350–4364. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. Sting-dependent cytosolic DNA sensing promotes radiation-induced type i interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. The host sting pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. Sting pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Lirussi, D.; Ebensen, T.; Schulze, K.; Trittel, S.; Duran, V.; Liebich, I.; Kalinke, U.; Guzman, C.A. Type i ifn and not tnf, is essential for cyclic di-nucleotide-elicited ctl by a cytosolic cross-presentation pathway. EBioMedicine 2017, 22, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Spanier, J.; Kasnitz, N.; Kroger, A.; Jin, L.; Brinkmann, M.M.; Kalinke, U.; Weiss, S.; Jablonska, J.; Lienenklaus, S. Growing tumors induce a local sting dependent type i ifn response in dendritic cells. Int. J. Cancer 2016, 139, 1350–1357. [Google Scholar] [CrossRef]
- Arwert, E.N.; Milford, E.L.; Rullan, A.; Derzsi, S.; Hooper, S.; Kato, T.; Mansfield, D.; Melcher, A.; Harrington, K.J.; Sahai, E. Sting and irf3 in stromal fibroblasts enable sensing of genomic stress in cancer cells to undermine oncolytic viral therapy. Nat. Cell Biol. 2020, 22, 758–766. [Google Scholar] [CrossRef]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-derived cgamp triggers a sting-mediated interferon response in non-tumor cells to activate the nk cell response. Immunity 2018, 49, 754–763.e4. [Google Scholar] [CrossRef]
- Jin, L.; Xu, L.G.; Yang, I.V.; Davidson, E.J.; Schwartz, D.A.; Wurfel, M.M.; Cambier, J.C. Identification and characterization of a loss-of-function human mpys variant. Genes Immun. 2011, 12, 263–269. [Google Scholar] [CrossRef]
- Patel, S.; Jin, L. Tmem173 variants and potential importance to human biology and disease. Genes Immun. 2018, 20, 82–89. [Google Scholar] [CrossRef]
- Patel, S.; Blaauboer, S.M.; Tucker, H.R.; Mansouri, S.; Ruiz-Moreno, J.S.; Hamann, L.; Schumann, R.R.; Opitz, B.; Jin, L. Response to comment on “the common r71h-g230a-r293q human tmem173 is a null allele”. J. Immunol. 2017, 198, 4185–4188. [Google Scholar] [CrossRef]
- Sebastian, M.; Hsiao, C.J.; Futch, H.S.; Eisinger, R.S.; Dumeny, L.; Patel, S.; Gobena, M.; Katikaneni, D.S.; Cohen, J.; Carpenter, A.M.; et al. Obesity and sting1 genotype associate with 23-valent pneumococcal vaccination efficacy. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Kennedy, R.B.; Haralambieva, I.H.; Ovsyannikova, I.G.; Voigt, E.A.; Larrabee, B.R.; Schaid, D.J.; Zimmermann, M.T.; Oberg, A.L.; Poland, G.A. Polymorphisms in sting affect human innate immune responses to poxviruses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yi, G.; Brendel, V.P.; Shu, C.; Li, P.; Palanathan, S.; Cheng Kao, C. Single nucleotide polymorphisms of human sting can affect innate immune response to cyclic dinucleotides. PLoS ONE 2013, 8, e77846. [Google Scholar] [CrossRef] [PubMed]
- Darling, R.J.; Senapati, S.; Kelly, S.M.; Kohut, M.L.; Narasimhan, B.; Wannemuehler, M.J. Sting pathway stimulation results in a differentially activated innate immune phenotype associated with low nitric oxide and enhanced antibody titers in young and aged mice. Vaccine 2019, 37, 2721–2730. [Google Scholar] [CrossRef] [PubMed]
- Vassilieva, E.V.; Taylor, D.W.; Compans, R.W. Combination of sting pathway agonist with saponin is an effective adjuvant in immunosenescent mice. Front. Immunol. 2019, 10, 3006. [Google Scholar] [CrossRef] [PubMed]
- Gogoi, H.; Mansouri, S.; Katikaneni, D.S.; Jin, L. New modc-targeting tnf fusion proteins enhance cyclic di-gmp vaccine adjuvanticity in middle-aged and aged mice. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P. Immunosenescence and vaccine failure in the elderly: Strategies for improving response. Immunol. Lett. 2014, 162, 346–353. [Google Scholar] [CrossRef]
- Eaton, S.M.; Burns, E.M.; Kusser, K.; Randall, T.D.; Haynes, L. Age-related defects in cd4 t cell cognate helper function lead to reductions in humoral responses. J. Exp. Med. 2004, 200, 1613–1622. [Google Scholar] [CrossRef]
- Maue, A.C.; Haynes, L. Cd4+ t cells and immunosenescence—A mini-review. Gerontology 2009, 55, 491–495. [Google Scholar] [CrossRef]
- Lefebvre, J.S.; Haynes, L. Aging of the cd4 t cell compartment. Open Longev. Sci. 2012, 6, 83–91. [Google Scholar] [CrossRef]
- Ruiz-Moreno, J.S.; Hamann, L.; Shah, J.A.; Verbon, A.; Mockenhaupt, F.P.; Puzianowska-Kuznicka, M.; Naujoks, J.; Sander, L.E.; Witzenrath, M.; Cambier, J.C.; et al. The common haq sting variant impairs cgas-dependent antibacterial responses and is associated with susceptibility to legionnaires’ disease in humans. PLoS Pathog. 2018, 14, e1006829. [Google Scholar] [CrossRef]
- Nissen, S.K.; Pedersen, J.G.; Helleberg, M.; Kjaer, K.; Thavachelvam, K.; Obel, N.; Tolstrup, M.; Jakobsen, M.R.; Mogensen, T.H. Multiple homozygous variants in the sting-encoding tmem173 gene in hiv long-term nonprogressors. J. Immunol. 2018, 200, 3372–3382. [Google Scholar] [CrossRef] [PubMed]
- Bader, C.S.; Barreras, H.; Lightbourn, C.O.; Copsel, S.N.; Wolf, D.; Meng, J.; Ahn, J.; Komanduri, K.V.; Blazar, B.R.; Jin, L.; et al. Sting differentially regulates experimental gvhd mediated by cd8 versus cd4 t cell subsets. Sci. Transl. Med. 2020, 12, eaay5006. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hong, C.; Yan, E.Z.; Fletcher, S.J.; Zhu, G.; Yang, M.; Li, Y.; Sun, X.; Irvine, D.J.; Li, J.; et al. Self-assembled cgamp-stingdeltatm signaling complex as a bioinspired platform for cgamp delivery. Sci. Adv. 2020, 6, eaba7589. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Adjuvant | Composition | Immune Responses |
|---|---|---|
| Alum | Aluminum oxyhydroxide, Potassium aluminum sulfate [1,2,3] | Antibody, Th2 response |
| MF59 | Squalene, polysorbate [2,3] sorbitan trioleate | Antibody, Mixed Th1/Th2 responses with Th2 biased, CD8+ T cell response |
| AS01 | MPL, QS21, liposome [2,3] | Antibody, Th1 response, CD8+ T cell response |
| AS03 | Squalene, polysorbate [2,3], α-tocopherol | Antibody, Th1/Th2 |
| AS04 | MPL, aluminum hydroxide [2,3] | Antibody, Th1/Th2 responses |
| Cyclic Dinucleotide | Immune Responses |
|---|---|
| RpRp-SS-CDG/ML-RpRp-SS-cGAMP in Addvax [Squalene based oil-in-water nano emulsion] (s.c.) or in PBS (i.n.) [11] | Th1, Th17 responses; protection against Mtb infection |
| cGAMP in pulmonary surfactant mimicking liposome (i.n.) [10] | CD8+ T cell response; 100% protection against a 10LD50 virulent strain of CA09 H1N1 |
| CDG in PEG lipid nanoparticles (s.c.) [58] | Tfh and GC B cell induction |
| cGAMP in ultra-pH sensitive nanoparticle ex vivo human PBMC [59] | IL-6, IL-8, G-CSF, TNF-a, type I IFNs, MIP-1α production from HIV infected PBMC, and inhibited HIV I replication |
| CDG in PBS (i.n., i.m. s.c., i.p.) [4,5,8,9,13,15,43] | Systemic and mucosal humoral and cellular immune responses that protect from bacterial infections |
| CDA with alum (i.m.) or in PBS (s.c. or i.n.) [7,18,31] | Balanced IgG1 and IgG2a responses; Th1/Th2/Th17 responses |
| (STINGel) Synthetic ML-RpRp-SS-CDA in multidomain peptide hydrogel (s.c.) [60] | Slow-release of CDN and highly effective in maintaining tumor clearance and rejecting tumor growth and provide durable anti-tumor immunity |
| CDG in YSK05 liposome (s.c.) [61] | Activation of antigen-specific CTL activity and restrict murine tumor growth completely |
| ADU-S100 (Synthetic ML-RpRp-SS-CDA) in combination with anti-PD1 or anti-CTLA4 antibody (intratumoral) [62] | Low-dosage promotes local STING activation primarily in monocytic cell lineages whereas a high-dose resulted in cellular activation at distal sites A combination of ADU-S100 and α-CPI provide durable immunity even against cold tumors |
| Synthetic RpRp-SS-CDG and ML-RpRp-CDA in PBAE nanoparticles and anti-PD1 antibody (intratumoral) [56] | Enhanced shelf-life stability for up to nine months. A 10-fold lower dose of PBAE/CDN nanoparticles with α-PD1 antibody resulted in significantly reduced tumor growth and reduction as compared to soluble CDN |
| cGAMP in acetalated dextran mps (i.p., i.m., i.v., and intratumoral) [57] | Tumor reduction at 100-1000 fold lower dose as compared to soluble cGAMP The synergistic effect of NK cells and CD8+ T cells resulted in early anti-tumor efficacy in a triple-negative breast cancer model |
| cGAMP in pH-responsive polymersomes (intratumoral) [63] | Recruitment of active neutrophil and neutrophil chemokine Cxcl1 in TME, the polarization of M2 macrophage towards inflammatory profile; enhanced CDN accumulation in lymph nodes and inhibited contralateral tumor growth. Increased CD8+ and CD4+ T cells with enhanced CD8+/CD4+ T cell ratio in TME |
| cGAMP in cationic liposomes (variable PEG levels) (i.v.) [64] | Durable anti-tumor immunity against a B16F10 orthotopic skin melanoma model The anti-tumor activity of cGAMP in liposome was obtained at 10-100-fold lower concentration than soluble cGAMP |
| CDG in cationic silica nps (intratumoral) [65] | A single intratumoral dose showed 37.5% protection and 100% protection upon tumor re-challenge Intratumoral treatment with CDG/NP in OVA expressing B16-F10 melanoma cell line induced IFNγ and TNFα producing OVA-specific CD8+ T cells in PBMC |
| ADU-S100 (Synthetic ML-RpRp-SS-CDA) in saline (intratumoral) [29,30] | NK cell-mediated and CD8+ T cell dispensable tumor regression and clearance in local and distal tumor sites. Increased cytokines and chemokines and innate immune cells (macrophages, neutrophils) in the TME and tumor dLNs leading to complete tumor clearance |
| cGAMP in phosphatidylserine liposome/radiotherapy (inhalation) [66] | Enhanced cytosolic release of cGAMP, efficient STING signaling, and type I IFN production in APCs resulting in a pro-inflammatory TME and suppressed Tregs production in TME. Liposome/cGAMP and radiation therapy-induced complete regression of lung metastasis and inhibited metastasis in both the irradiated and non-irradiated lung |
| STINGVAX Soluble CDA/Synthetic RR-S2-CDA/Synthetic ML-RR-S2-CDA (s.c.) [17] | Enhanced IFN-γ+ tumor-infiltrating CD8+T cells. STINGVAX in combination with synthetic CDA-induced type I IFN, MHC I, CD80, CD83, CD86 in monocytes and DC obtained from donor PBMCs |
| Synthetic DMXAA (i.p., i.v., intratumoral) [16,67,68] | Tumor-specific CD8+ T cell, and long term protection in a C1498.SIY acute leukemia model Anti-tumor immunity in distal tumor sites Macrophage repolarization in non-small cell lung cancer model |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogoi, H.; Mansouri, S.; Jin, L. The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines 2020, 8, 453. https://doi.org/10.3390/vaccines8030453
Gogoi H, Mansouri S, Jin L. The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines. 2020; 8(3):453. https://doi.org/10.3390/vaccines8030453
Chicago/Turabian StyleGogoi, Himanshu, Samira Mansouri, and Lei Jin. 2020. "The Age of Cyclic Dinucleotide Vaccine Adjuvants" Vaccines 8, no. 3: 453. https://doi.org/10.3390/vaccines8030453
APA StyleGogoi, H., Mansouri, S., & Jin, L. (2020). The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines, 8(3), 453. https://doi.org/10.3390/vaccines8030453