Hepatitis E Virus: How It Escapes Host Innate Immunity

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. HEV RNA Sensing by Infected Cells
2.1. Recognition by TLR Pattern-Recognition Receptors
2.2. Recognition by RLR Pattern-Recognition Receptors
2.3. HEV Motif Recognized
3. Innate Immune Response to HEV
3.1. IFN Response to HEV Infection
3.2. Inflammatory Response to a HEV Infection
4. Virus Evasion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines on hepatitis E virus infection. J. Hepatol. 2018. [Google Scholar] [CrossRef]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef]
- Kamar, N.; Izopet, J.; Pavio, N.; Aggarwal, R.; Labrique, A.; Wedemeyer, H.; Dalton, H.R. Hepatitis E virus infection. Nat. Rev. Dis. Prim. 2017, 3, 17086. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Kamar, N.; van Eijk, J.J.; McLean, B.N.; Cintas, P.; Bendall, R.P.; Jacobs, B.C. Hepatitis E virus and neurological injury. Nat. Rev. Neurol. 2016, 12, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Marion, O.; Abravanel, F.; Izopet, J.; Dalton, H.R. Extrahepatic manifestations of hepatitis E virus. Liver Int. 2016, 36, 467–472. [Google Scholar] [CrossRef]
- Kamar, N.; Rostaing, L.; Legrand-Abravanel, F.; Izopet, J. How should hepatitis E virus infection be defined in organ-transplant recipients? Am. J. Transplant. 2013, 13, 1935–1936. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P. Classification and genomic diversity of enterically transmitted hepatitis viruses. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Smith, D.B.; Izopet, J.; Nicot, F.; Simmonds, P.; Jameel, S.; Meng, X.J.; Norder, H.; Okamoto, H.; van der Poel, W.H.M.; Reuter, G.; et al. Update: Proposed reference sequences for subtypes of hepatitis E virus (species Orthohepevirus A). J. Gen. Virol 2020. [Google Scholar] [CrossRef]
- Sridhar, S.; Yip, C.C.Y.; Wu, S.; Cai, J.; Zhang, A.J.; Leung, K.H.; Chung, T.W.H.; Chan, J.F.W.; Chan, W.M.; Teng, J.L.L.; et al. Rat hepatitis E virus as cause of persistent hepatitis after liver transplant. Emerg. Infect. Dis. 2018, 24, 2241–2250. [Google Scholar] [CrossRef]
- Sridhar, S.; Yip, C.C.; Wu, S.; Chew, N.F.; Leung, K.H.; Chan, J.F.; Zhao, P.S.; Chan, W.M.; Poon, R.W.; Tsoi, H.W.; et al. Transmission of rat hepatitis E virus infection to humans in Hong Kong: A clinical and epidemiological analysis. Hepatology 2020. [Google Scholar] [CrossRef]
- Andonov, A.; Robbins, M.; Borlang, J.; Cao, J.; Hattchete, T.; Stueck, A.; Deschaumbault, Y.; Murnaghan, K.; Varga, J.; Johnston, B. Rat hepatitis E virus linked to severe acute hepatitis in an immunocompetent patient. J. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Harms, D.; Yang, X.L.; Bock, C.T. Orthohepevirus C: An expanding species of emerging hepatitis E virus variants. Pathogens 2020, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.P.; Meng, X.J. Hepatitis E virus genome structure and replication strategy. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- LeDesma, R.; Nimgaonkar, I.; Ploss, A. Hepatitis E virus replication. Viruses 2019, 11, 719. [Google Scholar] [CrossRef]
- Proudfoot, A.; Hyrina, A.; Holdorf, M.; Frank, A.O.; Bussiere, D. First crystal structure of a nonstructural hepatitis E viral protein identifies a putative novel zinc-binding protein. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Ahola, T.; Karlin, D.G. Sequence analysis reveals a conserved extension in the capping enzyme of the alphavirus supergroup, and a homologous domain in nodaviruses. Biol. Direct 2015, 10, 16. [Google Scholar] [CrossRef]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Mansuy, J.M.; Rostaing, L.; Kamar, N.; Izopet, J. Characterization of the polyproline region of the hepatitis E virus in immunocompromised patients. J. Virol. 2014, 88, 12017–12025. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Nicot, F.; Jeanne, N.; Dimeglio, C.; Roulet, A.; Lefebvre, C.; Carcenac, R.; Manno, M.; Dubois, M.; Peron, J.M.; et al. Insertions and duplications in the polyproline region of the hepatitis E Virus. Front. Microbiol. 2020, 11, 1. [Google Scholar] [CrossRef]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenroder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef]
- Gouttenoire, J.; Pollan, A.; Abrami, L.; Oechslin, N.; Mauron, J.; Matter, M.; Oppliger, J.; Szkolnicka, D.; Dao Thi, V.L.; van der Goot, F.G.; et al. Palmitoylation mediates membrane association of hepatitis E virus ORF3 protein and is required for infectious particle secretion. PLoS Pathog. 2018, 14, e1007471. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic reticulum stress induced synthesis of a novel viral factor mediates efficient replication of genotype-1 hepatitis E virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef]
- Feng, Z.; Lemon, S.M. Peek-a-boo: Membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 2014, 22, 59–64. [Google Scholar] [CrossRef]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked viruses that aren’t always naked: Quasi-enveloped agents of acute hepatitis. Annu Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef]
- Tanaka, T.; Takahashi, M.; Kusano, E.; Okamoto, H. Development and evaluation of an efficient cell-culture system for Hepatitis E virus. J. Gen. Virol. 2007, 88, 903–911. [Google Scholar] [CrossRef]
- Tanaka, T.; Takahashi, M.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Okamoto, H. Development and characterization of a genotype 4 hepatitis E virus cell culture system using a HE-JF5/15F strain recovered from a fulminant hepatitis patient. J. Clin. Microbiol. 2009, 47, 1906–1910. [Google Scholar] [CrossRef]
- Emerson, S.U.; Zhang, M.; Meng, X.J.; Nguyen, H.; St Claire, M.; Govindarajan, S.; Huang, Y.K.; Purcell, R.H. Recombinant hepatitis E virus genomes infectious for primates: Importance of capping and discovery of a cis-reactive element. Proc. Natl. Acad. Sci. USA 2001, 98, 15270–15275. [Google Scholar] [CrossRef]
- Shukla, P.; Nguyen, H.T.; Faulk, K.; Mather, K.; Torian, U.; Engle, R.E.; Emerson, S.U. Adaptation of a genotype 3 hepatitis E virus to efficient growth in cell culture depends on an inserted human gene segment acquired by recombination. J. Virol. 2012, 86, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Shukla, P.; Torian, U.; Faulk, K.; Emerson, S.U. Hepatitis E virus genotype 1 infection of swine kidney cells in vitro is inhibited at multiple levels. J. Virol. 2014, 88, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Nguyen, H.; Graff, J.; Stephany, D.A.; Brockington, A.; Purcell, R.H. In vitro replication of hepatitis E virus (HEV) genomes and of an HEV replicon expressing green fluorescent protein. J. Virol. 2004, 78, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Nguyen, H.; Kasorndorkbua, C.; Halbur, P.G.; St Claire, M.; Purcell, R.H.; Emerson, S.U. In vitro and in vivo mutational analysis of the 3′-terminal regions of hepatitis e virus genomes and replicons. J. Virol. 2005, 79, 1017–1026. [Google Scholar] [CrossRef]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell culture systems for the study of hepatitis E virus. Antiviral Res. 2019, 163, 34–49. [Google Scholar] [CrossRef]
- Fu, R.M.; Decker, C.C.; Dao Thi, V.L. Cell culture models for hepatitis E Virus. Viruses 2019, 11, 608. [Google Scholar] [CrossRef]
- Dao Thi, V.L.; Wu, X.; Belote, R.L.; Andreo, U.; Takacs, C.N.; Fernandez, J.P.; Vale-Silva, L.A.; Prallet, S.; Decker, C.C.; Fu, R.M.; et al. Stem cell-derived polarized hepatocytes. Nat. Commun. 2020, 11, 1677. [Google Scholar] [CrossRef]
- Capelli, N.; Marion, O.; Dubois, M.; Allart, S.; Bertrand-Michel, J.; Lhomme, S.; Abravanel, F.; Izopet, J.; Chapuy-Regaud, S. Vectorial release of hepatitis E virus in polarized human hepatocytes. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Sayed, I.M.; Elkhawaga, A.A.; El-Mokhtar, M.A. In vivo models for studying Hepatitis E virus infection; Updates and applications. Virus Res. 2019, 274, 197765. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.J. Hepatitis E virus: Animal models and zoonosis. Annu Rev. Anim. Biosci. 2019, 7, 427–448. [Google Scholar] [CrossRef]
- Li, Y.; Qu, C.; Yu, P.; Ou, X.; Pan, Q.; Wang, W. The interplay between host innate immunity and hepatitis E virus. Viruses 2019, 11, 541. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, Z.; Kato, N.; Gale, M., Jr.; Lemon, S.M. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 2005, 280, 16739–16747. [Google Scholar] [CrossRef]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.P.; Pan, Q. Transcriptional regulation of antiviral interferon-stimulated genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Innate immune responses to hepatitis C virus. Curr. Top. Microbiol. Immunol. 2013, 369, 219–242. [Google Scholar] [CrossRef]
- Devhare, P.B.; Desai, S.; Lole, K.S. Innate immune responses in human hepatocyte-derived cell lines alter genotype 1 hepatitis E virus replication efficiencies. Sci. Rep. 2016, 6, 26827. [Google Scholar] [CrossRef]
- Devhare, P.B.; Chatterjee, S.N.; Arankalle, V.A.; Lole, K.S. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS ONE 2013, 8, e63793. [Google Scholar] [CrossRef]
- Choi, Y.H.; Zhang, X.; Tran, C.; Skinner, B. Expression profiles of host immune response-related genes against HEV genotype 3 and genotype 1 infections in rhesus macaques. J. Viral Hepat. 2018. [Google Scholar] [CrossRef]
- Pandey, S.; Kawai, T.; Akira, S. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 2014, 7, a016246. [Google Scholar] [CrossRef] [PubMed]
- Jagya, N.; Varma, S.P.; Thakral, D.; Joshi, P.; Durgapal, H.; Panda, S.K. RNA-seq based transcriptome analysis of hepatitis E virus (HEV) and hepatitis B virus (HBV) replicon transfected Huh-7 cells. PLoS ONE 2014, 9, e87835. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, W.; Li, Y.; Zhou, X.; Yin, Y.; Wang, Y.; de Man, R.A.; van der Laan, L.J.W.; Huang, F.; Kamar, N.; et al. RIG-I is a key antiviral interferon-stimulated gene against hepatitis E virus regardless of interferon production. Hepatology 2017, 65, 1823–1839. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, P.; Qu, C.; Li, P.; Li, Y.; Ma, Z.; Wang, W.; de Man, R.A.; Peppelenbosch, M.P.; Pan, Q. MDA5 against enteric viruses through induction of interferon-like response partially via the JAK-STAT cascade. Antiviral Res. 2020, 176, 104743. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, X.; Wang, W.; Wang, Y.; Yin, Y.; Laan, L.J.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. IFN regulatory factor 1 restricts hepatitis E virus replication by activating STAT1 to induce antiviral IFN-stimulated genes. FASEB J. 2016, 30, 3352–3367. [Google Scholar] [CrossRef]
- Yin, X.; Li, X.; Ambardekar, C.; Hu, Z.; Lhomme, S.; Feng, Z. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 2017, 13, e1006417. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Qu, C.; Wang, S.; Zhou, J.; Cao, W.; Xu, L.; Ma, B.; Hakim, M.S.; Yin, Y.; et al. The RNA genome of hepatitis E virus robustly triggers an antiviral interferon response. Hepatology 2018, 67, 2096–2112. [Google Scholar] [CrossRef]
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Bruggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust hepatitis E virus infection and transcriptional response in human hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731–1741. [Google Scholar] [CrossRef]
- Choi, Y.; Zhang, X.; Skinner, B. Analysis of IgG anti-HEV antibody protective levels during hepatitis E virus reinfection in experimentally infected rhesus macaques. J. Infect. Dis. 2019, 219, 916–924. [Google Scholar] [CrossRef]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Sooryanarain, H.; Heffron, C.L.; Meng, X.J. The U-rich untranslated region of the hepatitis E virus induces differential type I and type III interferon responses in a host cell-dependent manner. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Nguyen, H.T.; Torian, U.; Burke, D.; Engle, R.; Purcell, R.H. Release of genotype 1 hepatitis E virus from cultured hepatoma and polarized intestinal cells depends on open reading frame 3 protein and requires an intact PXXP motif. J. Virol. 2010, 84, 9059–9069. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dao Thi, V.L.; Liu, P.; Takacs, C.N.; Xiang, K.; Andrus, L.; Gouttenoire, J.; Moradpour, D.; Rice, C.M. Pan-genotype hepatitis E virus replication in stem cell-derived hepatocellular systems. Gastroenterology 2018, 154, 663–674.e7. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Boon, D.; McDonald, S.L.; Myers, T.G.; Tomioka, K.; Nguyen, H.; Engle, R.E.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: Similarities and differences. J. Virol. 2010, 84, 11264–11278. [Google Scholar] [CrossRef]
- Murata, K.; Kang, J.H.; Nagashima, S.; Matsui, T.; Karino, Y.; Yamamoto, Y.; Atarashi, T.; Oohara, M.; Uebayashi, M.; Sakata, H.; et al. IFN-lambda3 as a host immune response in acute hepatitis E virus infection. Cytokine 2020, 125, 154816. [Google Scholar] [CrossRef]
- Marion, O.; Lhomme, S.; Nayrac, M.; Dubois, M.; Pucelle, M.; Requena, M.; Migueres, M.; Abravanel, F.; Peron, J.M.; Carrere, N.; et al. Hepatitis E virus replication in human intestinal cells. Gut 2019. [Google Scholar] [CrossRef]
- Abid, S.; Khan, A.H. Severe hemolysis and renal failure in glucose-6-phosphate dehydrogenase deficient patients with hepatitis E. Am. J. Gastroenterol. 2002, 97, 1544–1547. [Google Scholar] [CrossRef]
- Gouilly, J.; Chen, Q.; Siewiera, J.; Cartron, G.; Levy, C.; Dubois, M.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N.; El Costa, H. Genotype specific pathogenicity of hepatitis E virus at the human maternal-fetal interface. Nat. Commun. 2018, 9, 4748. [Google Scholar] [CrossRef]
- Knegendorf, L.; Drave, S.A.; Thi, V.L.D.; Debing, Y.; Brown, R.J.P.; Vondran, F.W.R.; Resner, K.; Friesland, M.; Khera, T.; Engelmann, M.; et al. Hepatitis E Virus replication and interferon responses in human placental cells. Hepatol. Commun. 2018, 2, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Verhoye, L.; Cocquerel, L.; Abravanel, F.; Foquet, L.; Montpellier, C.; Debing, Y.; Farhoudi, A.; Wychowski, C.; Dubuisson, J.; et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 2017, 66, 920–929. [Google Scholar] [CrossRef]
- Wang, W.; Xu, L.; Brandsma, J.H.; Wang, Y.; Hakim, M.S.; Zhou, X.; Yin, Y.; Fuhler, G.M.; van der Laan, L.J.; van der Woude, C.J.; et al. Convergent transcription of interferon-stimulated genes by TNF-alpha and IFN-alpha augments antiviral activity against HCV and HEV. Sci. Rep. 2016, 6, 25482. [Google Scholar] [CrossRef]
- Cao, D.; Cao, Q.M.; Subramaniam, S.; Yugo, D.M.; Heffron, C.L.; Rogers, A.J.; Kenney, S.P.; Tian, D.; Matzinger, S.R.; Overend, C.; et al. Pig model mimicking chronic hepatitis E virus infection in immunocompromised patients to assess immune correlates during chronicity. Proc. Natl. Acad. Sci. USA 2017, 114, 6914–6923. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 2007, 104, 7253–7258. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.J. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Myoung, J. Hepatitis E virus papain-like cysteine protease inhibits type I interferon induction by down-regulating melanoma differentiation-associated gene 5. J. Microbiol. Biotechnol. 2018, 28, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Myoung, J.; Min, K. Dose-dependent inhibition of melanoma differentiation-associated gene 5-mediated activation of type I interferon responses by methyltransferase of hepatitis E virus. J. Microbiol. Biotechnol. 2019, 29, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Myoung, J.; Lee, J.Y.; Min, K.S. Methyltransferase of a cell culture-adapted hepatitis E inhibits the MDA5 receptor signaling pathway. J. Microbiol. 2019, 57, 1126–1131. [Google Scholar] [CrossRef]
- Kang, S.; Choi, C.; Choi, I.; Han, K.N.; Rho, S.W.; Choi, J.; Kwon, J.; Park, M.K.; Kim, S.J.; Myoung, J. Hepatitis E virus methyltransferase inhibits type I interferon induction by targeting RIG-I. J. Microbiol. Biotechnol. 2018, 28, 1554–1562. [Google Scholar] [CrossRef]
- Ojha, N.K.; Lole, K.S. Hepatitis E virus ORF1 encoded macro domain protein interacts with light chain subunit of human ferritin and inhibits its secretion. Mol. Cell. Biochem. 2016, 417, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Bogunovic, D. ISG15: In sickness and in health. Trends Immunol. 2017, 38, 79–93. [Google Scholar] [CrossRef]
- Okumura, A.; Lu, G.; Pitha-Rowe, I.; Pitha, P.M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad Sci. USA 2006, 103, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Domingues, P.; Bamford, C.G.G.; Boutell, C.; McLauchlan, J. Inhibition of hepatitis C virus RNA replication by ISG15 does not require its conjugation to protein substrates by the HERC5 E3 ligase. J. Gen. Virol. 2015, 96, 3236–3242. [Google Scholar] [CrossRef] [PubMed]
- Broering, R.; Zhang, X.; Kottilil, S.; Trippler, M.; Jiang, M.; Lu, M.; Gerken, G.; Schlaak, J.F. The interferon stimulated gene 15 functions as a proviral factor for the hepatitis C virus and as a regulator of the IFN response. Gut 2010, 59, 1111–1119. [Google Scholar] [CrossRef]
- Sooryanarain, H.; Rogers, A.J.; Cao, D.; Haac, M.E.R.; Karpe, Y.A.; Meng, X.J. ISG15 modulates type I interferon signaling and the antiviral response during hepatitis E virus replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Lole, K.S. Deubiquitination activity associated with hepatitis E virus putative papain-like cysteine protease. J. Gen. Virol. 2011, 92, 2088–2092. [Google Scholar] [CrossRef]
- Bagdassarian, E.; Doceul, V.; Pellerin, M.; Demange, A.; Meyer, L.; Jouvenet, N.; Pavio, N. The amino-terminal region of hepatitis E virus ORF1 containing a methyltransferase (Met) and a papain-like cysteine protease (PCP) domain counteracts type I interferon response. Viruses 2018, 10, 726. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Fensterl, V.; Sen, G.C. Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef]
- Pingale, K.D.; Kanade, G.D.; Karpe, Y.A. Hepatitis E virus polymerase binds to IFIT1 to protect the viral RNA from IFIT1-mediated translation inhibition. J. Gen. Virol. 2019, 100, 471–483. [Google Scholar] [CrossRef]
- Lin, S.; Yang, Y.; Nan, Y.; Ma, Z.; Yang, L.; Zhang, Y.J. The capsid protein of hepatitis E virus inhibits interferon induction via its N-terminal arginine-rich motif. Viruses 2019, 11, 1050. [Google Scholar] [CrossRef] [PubMed]
- Hingane, S.; Joshi, N.; Surjit, M.; Ranjith-Kumar, C.T. Hepatitis E virus ORF2 inhibits RIG-I mediated interferon response. Front. Microbiol. 2020, 11, 656. [Google Scholar] [CrossRef] [PubMed]
- John, L.; Thomas, S.; Herchenroder, O.; Putzer, B.M.; Schaefer, S. Hepatitis E virus ORF2 protein activates the pro-apoptotic gene CHOP and anti-apoptotic heat shock proteins. PLoS ONE 2011, 6, e25378. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Ma, Z.; Wang, R.; Yu, Y.; Kannan, H.; Fredericksen, B.; Zhang, Y.J. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol. 2014, 88, 8696–8705. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, M.; Huang, Y.; Peng, W.; Zheng, Z.; Xia, N.; Xu, J.; Tian, D. The ORF3 protein of genotype 1 hepatitis E virus suppresses TLR3-induced NF-kappaB signaling via TRADD and RIP1. Sci. Rep. 2016, 6, 27597. [Google Scholar] [CrossRef]
- Lei, Q.; Li, L.; Zhang, S.; Li, T.; Zhang, X.; Ding, X.; Qin, B. HEV ORF3 downregulates TLR7 to inhibit the generation of type I interferon via impairment of multiple signaling pathways. Sci. Rep. 2018, 8, 8585. [Google Scholar] [CrossRef]
- Dong, C.; Zafrullah, M.; Mixson-Hayden, T.; Dai, X.; Liang, J.; Meng, J.; Kamili, S. Suppression of interferon-alpha signaling by hepatitis E virus. Hepatology 2012, 55, 1324–1332. [Google Scholar] [CrossRef]
- Wang, M.; Huang, Y.; He, M.; Peng, W.J.; Tian, D.Y. Effects of hepatitis E virus infection on interferon production via ISG15. World J. Gastroenterol. 2018, 24, 2173–2180. [Google Scholar] [CrossRef]
- Sridhar, S. Use of S17 fragment containing hepatitis E virus infectious clones in cell culture experiments: The fine print does matter. J. Viral Hepat. 2018, 25, 1105. [Google Scholar] [CrossRef]



{kind=link}
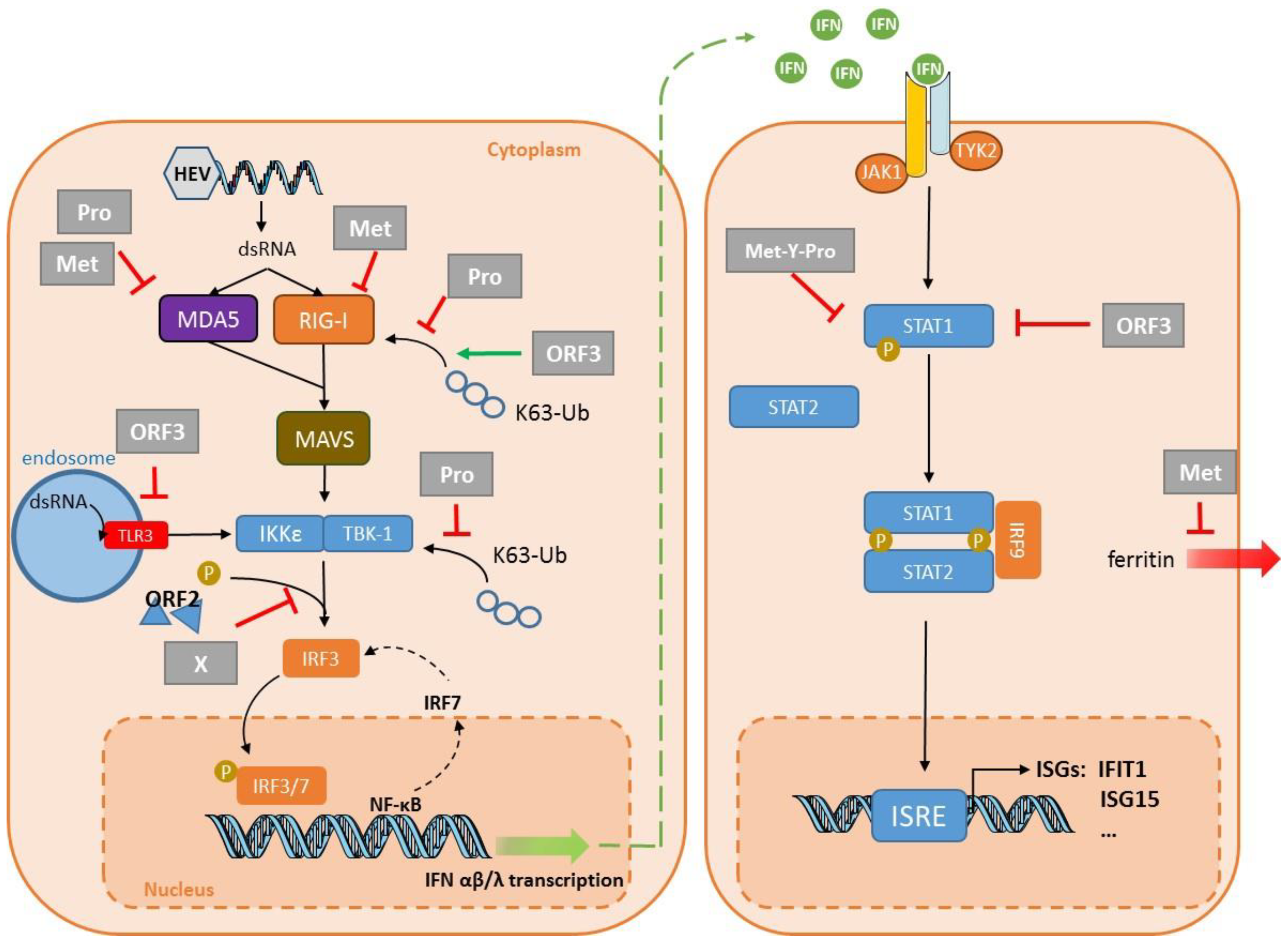{kind=link}
| Target | Cell Line | Strain | System | Key Finding | Reference |
|---|---|---|---|---|---|
| PRR signaling | HEK293T Huh7 S10-3 | HEV1–Sar55 | Transfection: plasmid encoding the domains of ORF1 Replicon | The protease can de-ubiquitinate RIG-I and TBK-1. Confirmed in a replicon system and S10-3 cells. | Nan, 2014 [76] |
| HEK293T | HEV3–47832c | Transfection: plasmid encoding Pro | The protease inhibits IFN-β induction by interfering with MDA5 signaling. | Kim, 2018 [77] | |
| HEK293T | HEV3–47832c | Transfection: plasmid encoding Met | The methyltransferase inhibits IFN-β induction by interfering with RIG-I signaling. | Kang, 2018 [80] | |
| HEK293T | HEV3–47832c | Transfection: plasmid encoding Met | The methyltransferase inhibits IFN-β induction by interfering with MDA5 signaling. | Myoung, 2019 [78] | |
| HEK293T | HEV3–47832c | Transfection: plasmid encoding Met | The methyltransferase inhibits IFN-β induction by inhibiting MDA5-mediated phosphorylation of NF-κB. | Myoung, 2019 [79] | |
| HEK293T Huh7 S10-3 | HEV1–Sar55 | Transfection: plasmid encoding the domains of ORF1 Replicon | The X domain impairs IRF3 phosphorylation. Confirmed in a replicon system in S10-3 cells. | Nan, 2014 [76] | |
| HEK293T HepG2/C3A | HEV1 Sar55 HEV3–kernow | Transfection: plasmid encoding ORF2 (HEV1/3) plus HEV3 infection | ORF2 interacts with TBK1 to impair IRF3 phosphorylation and its dissociation from MAVS. | Lin, 2019 [92] | |
| HEK293T Huh7 | HEV1–Sar5 HEV3–Kernow | Transfection: plasmid encoding the ORF2 protein | ORF2 inhibits IFN production by blocking TLR and RIG-I signaling pathways. | Hingane, 2020 [93] | |
| HEK293T | HEV1–Sar55 HEV3–Kernow | Transfection: plasmid encoding the ORF3 protein | ORF3 from HEV1 and HEV3 interact with RIG-I to increase its ubiquitination. | Nan, 2014 [95] | |
| A549 | HEV1–Sar55 | Transfection: plasmid encoding the ORF3 protein | ORF3 blocks TLR3-mediated NF-κB activity. | He, 2016 [96] | |
| THP1 Lo2 | HEV1–Sar55 | Transfection: plasmid encoding the ORF3 protein | ORF3 reduces TLR3 and TLR7 expression in these two cell lines. | Lei, 2018 [97] | |
| IFN signaling | A549 | HEV3–JN837481 | Infection | HEV ORF3 protein blocks IFN-α-induced STAT1 phosphorylation and impairs IFNα-induced gene expression. | Dong, 2012 [98] |
| HEK293T | HEV3–MG197988 | Transfection: plasmid encoding Met-Y–Pro | Met-Y-Pro domain interferes with STAT1 phosphorylation and subsequent nuclear translocation. HEV1 Met-Y-Pro domain interferes less efficiently than HEV3 Met-Y-Pro domain. | Bagdassarian, 2018 [88] | |
| Interferon Stimulated Genes | in vitro and HepG2 | HEV1–DQ459342 | Transfection: plasmid encoding the Met Pro domain | The protease domain has a deISGylation activity. | Karpe,2011 [87] |
| Huh7 S10-3 | HEV3–Kernow | Transfection: in vitro capped RNA transcript replicons | HEV induces ISG15 in vitro and in liver tissues of infected pigs. ISG15 is immunomodulatory—enhances HEV replication. | Sooryanarain, 2017 [86] | |
| HepG2/C3A | HEV3–Kernow without S17 fragment | Transfection: in vitro capped RNA transcripts Transfection: plasmid encoding the ORF3 protein | ORF3 enhances ISG15 production, hence HEV replication. | Wang, 2018 [99] | |
| Huh7 S10-3 | HEV1–Sar55 | Transfection: plasmid encoding RdRp | RdRp sequesters IFIT1 to inhibit its anti-translational activity. | Pingale, 2019 [91] | |
| Other | Huh7 S10-3 | HEV1–Sar55 | Transfection: plasmid encoding X-domain replicon | X domain interacts with the light chain to prevent its secretion, restraining innate immunity. | Ojha, 2016 [81] |
| Huh7 | HEV1–Sar55 | Transfection: plasmid encoding the ORF2 protein | ORF2 impairs apoptosis, allowing HEV lifecycle completion. | John, 2011 [94] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lhomme, S.; Migueres, M.; Abravanel, F.; Marion, O.; Kamar, N.; Izopet, J. Hepatitis E Virus: How It Escapes Host Innate Immunity. Vaccines 2020, 8, 422. https://doi.org/10.3390/vaccines8030422
Lhomme S, Migueres M, Abravanel F, Marion O, Kamar N, Izopet J. Hepatitis E Virus: How It Escapes Host Innate Immunity. Vaccines. 2020; 8(3):422. https://doi.org/10.3390/vaccines8030422
Chicago/Turabian StyleLhomme, Sébastien, Marion Migueres, Florence Abravanel, Olivier Marion, Nassim Kamar, and Jacques Izopet. 2020. "Hepatitis E Virus: How It Escapes Host Innate Immunity" Vaccines 8, no. 3: 422. https://doi.org/10.3390/vaccines8030422
APA StyleLhomme, S., Migueres, M., Abravanel, F., Marion, O., Kamar, N., & Izopet, J. (2020). Hepatitis E Virus: How It Escapes Host Innate Immunity. Vaccines, 8(3), 422. https://doi.org/10.3390/vaccines8030422