Identification and Analysis of Unstructured, Linear B-Cell Epitopes in SARS-CoV-2 Virion Proteins for Vaccine Development


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of uBCELs
2.2. Structural and Accessory Analysis of Protein Antigens
2.3. Phylogenetic Analyses
2.4. Epitope Collection in SARS-CoV-2
2.5. Statistical Analyses
3. Results
3.1. SARS-CoV-2 Epitope Catalogue
3.2. Unstructured Epitope Selection to Design Antigenic Peptides and Chimera Proteins
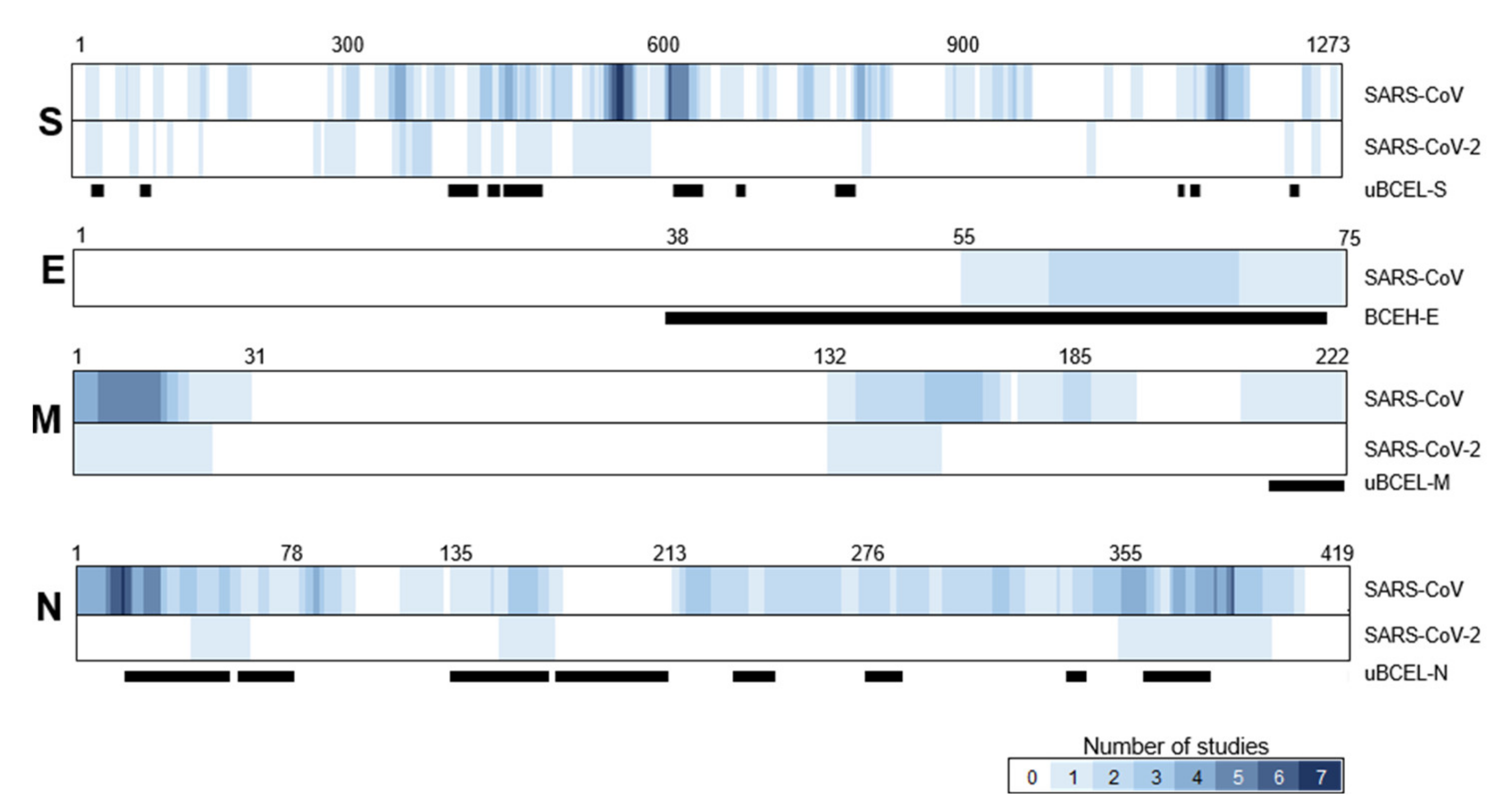
3.3. S Protein uBCEL Analysis
3.4. E Protein Epitope Analysis
3.5. M Protein Epitope Analysis
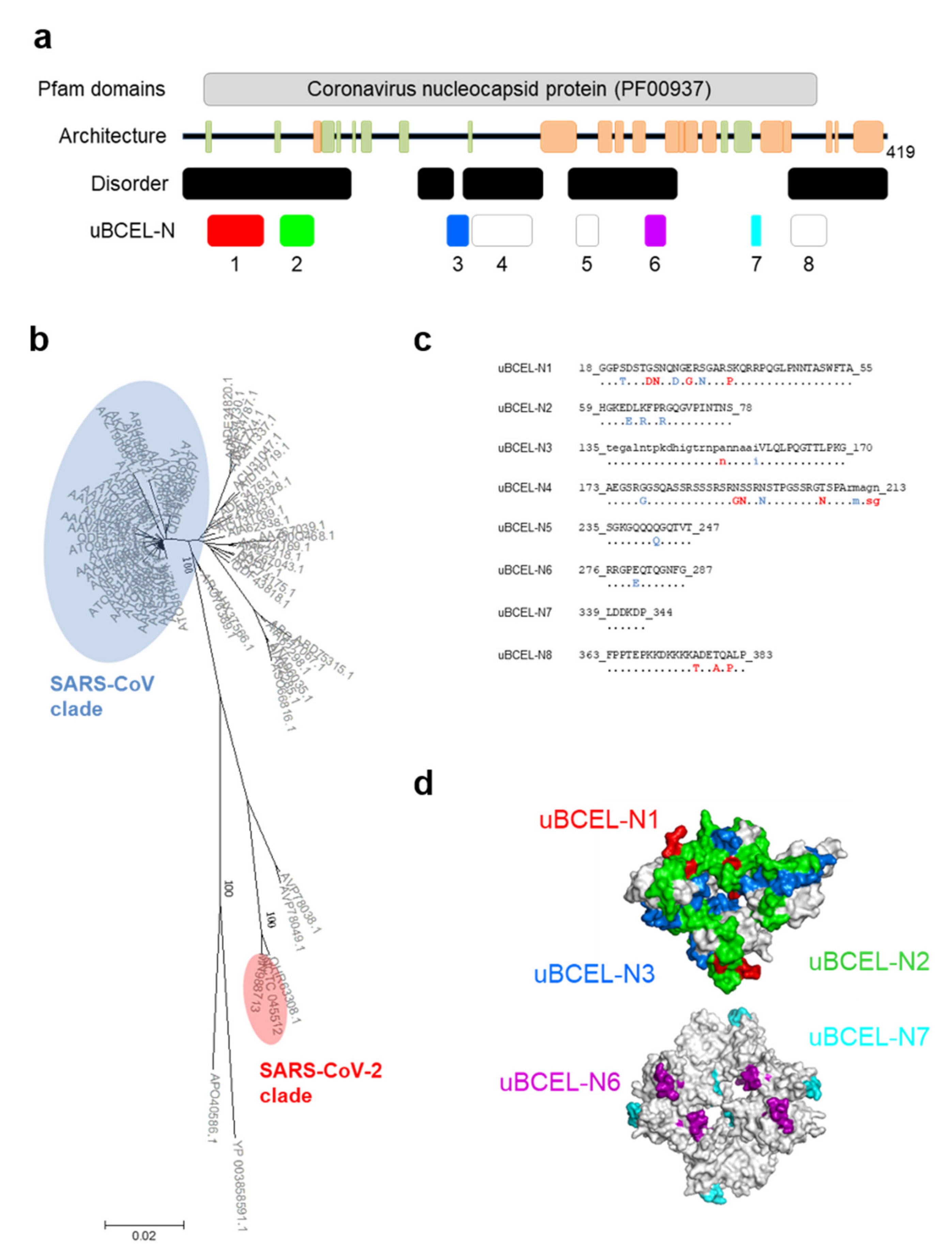
3.6. N Protein Epitope Analysis
3.7. Assessment of the Agreement between uBCELs in SARS-CoV-2 and Linear B-Cell Epitopes Previously Reported for SARS-CoV
3.8. Epitope Conservation in Bat Coronaviruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BCE | B-cell epitope |
| BCEH | B-cell epitope in an alpha-helix section |
| E | envelope protein |
| IEDB | immune epitope database |
| M | membrane protein |
| N | nucleocapsid protein |
| S | spike protein |
| SARS-CoV | Severe Acute Respiratory Syndrome Coronavirus |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| SDP | specificity determining position |
| TMH | transmembrane helix |
| uBCE | unstructured B-cell epitope |
| uBCEL | unstructured B-cell epitope-containing loop |
| UPR | unfolded protein response |
References
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.K.J.; Shan, J. Novel coronavirus: Where we are and what we know. Infection 2019. [Google Scholar] [CrossRef]
- Tyrrell, D.A.; Bynoe, M.L. Cultivation of viruses from a high proportion of patients with colds. Lancet 1966, 1, 76–77. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef]
- Assiri, A.; McGeer, A.; Perl, T.M.; Price, C.S.; Al Rabeeah, A.A.; Cummings, D.A.; Alabdullatif, Z.N.; Assad, M.; Almulhim, A.; Makhdoom, H.; et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 2013, 369, 407–416. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.Y.; Guo, D.Y. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- He, Y.X.; Li, J.J.; Du, L.Y.; Yan, X.X.; Hu, G.G.; Zhou, Y.S.; Jiang, S.B. Identification and characterization of novel neutralizing epitopes in the receptor-binding domain of SARS-CoV spike protein: Revealing the critical antigenic determinants in inactivated SARS-CoV vaccine. Vaccine 2006, 24, 5498–5508. [Google Scholar] [CrossRef]
- Lien, S.P.; Shih, Y.P.; Chen, H.W.; Tsai, J.P.; Leng, C.H.; Lin, M.H.; Lin, L.H.; Liu, H.Y.; Chou, A.H.; Chang, Y.W.; et al. Identification of synthetic vaccine candidates against SARS CoV infection. Biochem. Biophys. Res. Commun. 2007, 358, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Hua, R.H.; Zhou, Y.J.; Wang, Y.F.; Hua, Y.Z.; Tong, G.Z. Identification of two antigenic epitopes on SARS-CoV spike protein. Biochem. Biophys. Res. Commun. 2004, 319, 929–935. [Google Scholar] [CrossRef]
- Wang, X.H.; Xu, W.; Tong, D.Y.; Ni, J.; Gao, H.F.; Wang, Y.; Chu, Y.W.; Li, P.P.; Yang, X.M.; Xiong, S.D. A chimeric multi-epitope DNA vaccine elicited specific antibody response against severe acute respiratory syndrome-associated coronavirus which attenuated the virulence of SARS-CoV in vitro. Immunol. Lett. 2008, 119, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.F.; Liang, L.H.; She, M.; Liao, X.L.; Gu, J.; Li, Y.H.; Han, Z.C. Production of a monoclonal antibody against SARS-CoV spike protein with single intrasplenic immunization of plasmid DNA. Immunol. Lett. 2005, 100, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 019–1182. [Google Scholar] [CrossRef]
- Guo, J.P.; Petric, M.; Campbell, W.; McGeer, P.L. SARS corona virus peptides recognized by antibodies in the sera of convalescent cases. Virology 2004, 324, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, Y.; Qi, J.; Chu, F.; Wu, H.; Gao, F.; Li, T.; Yan, J.; Gao, G.F. The membrane protein of severe acute respiratory syndrome coronavirus acts as a dominant immunogen revealed by a clustering region of novel functionally and structurally defined cytotoxic T-lymphocyte epitopes. J. Infect. Dis. 2010, 202, 1171–1180. [Google Scholar] [CrossRef]
- Chow, S.C.S.; Ho, C.Y.S.; Tam, T.T.Y.; Wu, C.; Cheung, T.; Chan, P.K.S.; Ng, M.H.L.; Hui, P.K.; Ng, H.K.; Au, D.M.Y.; et al. Specific epitopes of the structural and hypothetical proteins elicit variable humoral responses in SARS patients. J. Clin. Pathol. 2006, 59, 468–476. [Google Scholar] [CrossRef]
- Qian, C.; Qin, D.; Tang, Q.; Zeng, Y.; Tang, G.X.; Lu, C. Identification of a B-cell antigenic epitope at the N-terminus of SARS-CoV M protein and characterization of monoclonal antibody against the protein. Virus Genes 2006, 33, 147–156. [Google Scholar] [CrossRef]
- He, Y.X.; Zhou, Y.S.; Siddiqui, P.; Niu, J.K.; Jiang, S.B. Identification of immunodominant epitopes on the membrane protein of the severe acute respiratory syndrome-associated coronavirus. J. Clin. Microbiol. 2005, 43, 3718–3726. [Google Scholar] [CrossRef]
- Kannan, S.; Ali, P.S.S.; Sheeza, A.; Hemalatha, K. COVID-19 (Novel Coronavirus 2019)—Recent trends. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2006–2011. [Google Scholar] [PubMed]
- Cheung, Y.K.; Cheng, S.C.; Sin, F.W.; Chan, K.T.; Xie, Y. Induction of T-cell response by a DNA vaccine encoding a novel HLA-A*0201 severe acute respiratory syndrome coronavirus epitope. Vaccine 2007, 25, 6070–6077. [Google Scholar] [CrossRef] [PubMed]
- Bussmann, B.M.; Reiche, S.; Jacob, L.H.; Braun, J.M.; Jassoy, C. Antigenic and cellular localisation analysis of the severe acute respiratory syndrome coronavirus nucleocapsid protein using monoclonal antibodies. Virus Res. 2006, 122, 119–126. [Google Scholar] [CrossRef]
- Shin, G.C.; Chung, Y.S.; Kim, I.S.; Cho, H.W.; Kang, C. Preparation and characterization of a novel monoclonal antibody specific to severe acute respiratory syndrome-coronavirus nucleocapsid protein. Virus Res. 2006, 122, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Che, X.Y.; Hao, W.; Wang, Y.; Di, B.; Yin, K.; Xu, Y.C.; Feng, C.S.; Wan, Z.Y.; Cheng, V.C.; Yuen, K.Y. Nucleocapsid protein as early diagnostic marker for SARS. Emerg. Infect. Dis. 2004, 10, 1947–1949. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Guo, S.; Sun, J.; Tan, L.; Lu, C.; Ma, Z. Advances in In-silico B-cell Epitope Prediction. Curr. Top. Med. Chem. 2019, 19, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Sher, G.; Zhi, D.; Zhang, S. DRREP: Deep ridge regressed epitope predictor. BMC Genom. 2017, 18, 55–65. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H.V. Mapping Epitope Structure and Activity: From One-Dimensional Prediction to Four-Dimensional Description of Antigenic Specificity. Methods 1996, 9, 465–472. [Google Scholar] [CrossRef]
- Carpentier, G.S.; Fleury, M.J.J.; Touzé, A.; Sadeyen, J.R.; Tourne, S.; Sizaret, P.Y.; Coursaget, P. Mutations on the FG Surface Loop of Human Papillomavirus Type 16 Major Capsid Protein Affect Recognition by Both Type-Specific Neutralizing Antibodies and Cross-Reactive Antibodies. J. Med. Virol. 2005, 77, 558–565. [Google Scholar] [CrossRef][Green Version]
- Qu, P.; Zhang, C.; Li, M.; Ma, W.; Xiong, P.; Liu, Q.; Zou, G.; Lavillette, D.; Yin, F.; Jin, X.; et al. A New Class of Broadly Neutralizing Antibodies That Target the Glycan Loop of Zika Virus Envelope Protein. Cell Discov. 2020, 6, 5. [Google Scholar] [CrossRef]
- Xu, L.; Zheng, Q.; Li, S.; He, M.; Wu, Y.; Li, Y.; Zhu, R.; Yu, H.; Hong, Q.; Jiang, J.; et al. Atomic Structures of Coxsackievirus A6 and Its Complex With a Neutralizing Antibody. Nat. Commun. 2017, 8, 505. [Google Scholar] [CrossRef] [PubMed]
- Potocnakova, L.; Bhide, M.; Pulzova, L.B. An Introduction to B-Cell Epitope Mapping and In Silico Epitope Prediction. J. Immunol. Res. 2016, 2016, 6760830. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jiang, L.F.; Fang, D.Y.; Yan, H.J.; Zhou, J.J.; Zhou, J.M.; Liang, Y.; Gao, Y.; Zhao, W.; Long, B.G. Selection of SARS-Coronavirus-specific B cell epitopes by phage peptide library screening and evaluation of the immunological effect of epitope-based peptides on mice. Virology 2007, 359, 264–274. [Google Scholar] [CrossRef] [PubMed]
- He, Y.X.; Li, J.J.; Heck, S.; Lustigman, S.; Jiang, S.B. Antigenic and immunogenic characterization of recombinant baculovirus-expressed severe acute respiratory syndrome coronavirus spike protein: Implication for vaccine design. J. Virol. 2006, 80, 5757–5767. [Google Scholar] [CrossRef]
- Hu, H.B.; Li, L.; Kao, R.Y.; Kou, B.B.; Wang, Z.G.; Zhang, L.; Zhang, H.Y.; Hao, Z.Y.; Tsui, W.H.; Ni, A.P.; et al. Screening and identification of linear B-cell epitopes and entry-blocking peptide of severe acute respiratory syndrome (SARS)-associated coronavirus using synthetic overlapping peptide library. J. Comb. Chem. 2005, 7, 648–656. [Google Scholar] [CrossRef]
- Lu, W.; Wu, X.D.; De Shi, M.; Yang, R.F.; He, Y.Y.; Bian, C.; Shi, T.L.; Yang, S.; Zhu, X.L.; Jiang, W.H.; et al. Synthetic peptides derived from SARS coronavirus S protein with diagnostic and therapeutic potential. FEBS Lett. 2005, 579, 2130–2136. [Google Scholar] [CrossRef]
- Rubinchik, E.; Chow, A.W. Recombinant expression and neutralizing activity of an MHC class II binding epitope of toxic shock syndrome toxin-1. Vaccine 2000, 18, 2312–2320. [Google Scholar] [CrossRef]
- Qin, E.; Zhu, Q.; Yu, M.; Fan, B.; Chang, G.; Si, B.; Yang, B.; Peng, W.; Jiang, T.; Liu, B.; et al. A complete sequence and comparative analysis of a SARS-associated virus (Isolate BJ01). Chin. Sci. Bull. 2003, 48, 941–948. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef]
- Kumar, S.; Maurya, V.K.; Prasad, A.K.; Bhatt, M.L.B.; Saxena, S.K. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease 2020, 31, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Tilocca, B.; Soggiu, A.; Musella, V.; Britti, D.; Sanguinetti, M.; Urbani, A.; Roncada, P. Molecular basis of COVID-19 relationships in different species: A one health perspective. Microbes Infect. 2020, 22, 218–220. [Google Scholar] [CrossRef]
- Zheng, M.; Song, L. Novel antibody epitopes dominate the antigenicity of spike glycoprotein in SARS-CoV-2 compared to SARS-CoV. Cell. Mol. Immunol. 2020, 17, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Baruah, V.; Bose, S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J. Med. Virol. 2020, 92, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Robson, B. Computers and viral diseases. Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Comput. Biol. Med. 2020, 119, 26. [Google Scholar] [CrossRef]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020, 12, 30166–30169. [Google Scholar] [CrossRef]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. Npj Vaccines 2020, 5, 1–3. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Marra, M.A.; Jones, S.J.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The Genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chang, C.K.; Chang, Y.W.; Sue, S.C.; Bai, H.I.; Riang, L.; Hsiao, C.D.; Huang, T.H. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 2007, 368, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Buus, S.; Rockberg, J.; Forsstrom, B.; Nilsson, P.; Uhlen, M.; Schafer-Nielsen, C. High-resolution mapping of linear antibody epitopes using ultrahigh-density peptide microarrays. Mol. Cell. Proteom. 2012, 11, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, O.V.; Mironov, A.A.; Gelfand, M.S.; Rakhmaninova, A.B. Automated selection of positions determining functional specificity of proteins by comparative analysis of orthologous groups in protein families. Protein Sci. 2004, 13, 443–456. [Google Scholar] [CrossRef]
- Goo, L.; DeMaso, C.R.; Pelc, R.S.; Ledgerwood, J.E.; Graham, B.S.; Kuhn, R.J.; Pierson, T.C. The Zika virus envelope protein glycan loop regulates virion antigenicity. Virology 2018, 515, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Zhu, C.; Ai, L.; He, T.; Wang, Y.; Ye, F.; Yang, L.; Ding, C.; Zhu, X.; Lv, R.; et al. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerg. Microbes Infect. 2018, 7, 154. [Google Scholar] [CrossRef]
- Ng, O.W.; Keng, C.T.; Leung, C.S.W.; Peiris, J.S.M.; Poon, L.L.M.; Tan, Y.J. Substitution at Aspartic Acid 1128 in the SARS Coronavirus Spike Glycoprotein Mediates Escape from a S2 Domain-Targeting Neutralizing Monoclonal Antibody. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New Vaccine Technologies to Combat Outbreak Situations. Front. Immunol. 2018, 9, 1963. [Google Scholar] [CrossRef]
- Li, Z.; Song, S.; He, M.; Wang, D.; Shi, J.; Liu, X.; Li, Y.; Chi, X.; Wei, S.; Yang, Y.; et al. Rational design of a triple-type human papillomavirus vaccine by compromising viral-type specificity. Nat. Commun. 2018, 9, 018–07199. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef]
- Du, L.; Tai, W.; Yang, Y.; Zhao, G.; Zhu, Q.; Sun, S.; Liu, C.; Tao, X.; Tseng, C.K.; Perlman, S.; et al. Introduction of neutralizing immunogenicity index to the rational design of MERS coronavirus subunit vaccines. Nat. Commun. 2016, 7, 13473. [Google Scholar] [CrossRef]
- Enjuanes, L.; Zuniga, S.; Castano-Rodriguez, C.; Gutierrez-Alvarez, J.; Canton, J.; Sola, I. Molecular Basis of Coronavirus Virulence and Vaccine Development. Adv. Virus Res. 2016, 96, 245–286. [Google Scholar] [PubMed]
- Wrapp, D.; Wang, N.S.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Kumar, R.; Deshpande, S.; Samal, S.; Shrivastava, T.; Boliar, S.; Bansal, M.; Chaudhary, N.K.; Srikrishnan, A.K.; Murugavel, K.G.; et al. Conformational Epitope-Specific Broadly Neutralizing Plasma Antibodies Obtained from an HIV-1 Clade C-Infected Elite Neutralizer Mediate Autologous Virus Escape through Mutations in the V1 Loop. J. Virol. 2016, 90, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- Plant, E.P.; Manukyan, H.; Sanchez, J.L.; Laassri, M.; Ye, Z. Immune Pressure on Polymorphous Influenza B Populations Results in Diverse Hemagglutinin Escape Mutants and Lineage Switching. Vaccines 2020, 8, 125. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Frenz, B.; Snijder, J.; Li, W.; Rey, F.A.; DiMaio, F.; Bosch, B.J.; Veesler, D. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat. Struct. Mol. Biol. 2016, 23, 899–905. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | uBECL or BCEH a | uBCE b Location | uBCEL or BCEH Location | Flanking SS c | uBCEL Sequence d |
|---|---|---|---|---|---|
| S | uBCEL-S1 | 21–28 | 16–28 | SP-B1 | vnlttRTQLPPAY |
| uBCEL-S2 | 71–81 | 68–85 | B3-B4 | ihvSGTNGTKRFDNpvlp | |
| uBCEL-S3 | 404–412 | 402–429 | B25-B26 | irGDEVRQIAPgqtgkiadynyklpddf | |
| uBCEL-S4 | 440–445 | 440–450 | B26-B27 | NLDSKVggnyn | |
| uBCEL-S5 | 459–470 473–480 | 455–491 | B27-B28 | lfrkSNLKPFERDISTeiYQAGSTPCngvegfncyfp | |
| uBCEL-S6 | 615–630 | 615–642 | B38-B39 | VNCTEVPVAIHADQLTptwrvystgsnv | |
| uBCEL-S7 | 676–687 | 676–689 | B43-B44 | TQTNSPRRARSVas | |
| uBCEL-S8 | 783–797 | 783–803 | H3-B48 | AQVKQIYKTPPIKDFggfnfs | |
| uBCEL-S9 | 1125–1131 | 1125–1131 | B60-B61 | NCDVVIG | |
| uBCEL-S10 | 1137–1147 | 1136–1147 | B61-H12 | TVYDPLQPELDS | |
| uBCEL-S11 | 1240–1246 | 1238–1246 | H15p-H16p | tsCCSCLKG | |
| E | BCEH-E1 | 57–68 | 38–75 | H3p | rlcayccnivnvslvkpsfYVYSRVKNLNSSRvpdllv |
| M | uBCEL-M1 | 209–215 | 209–222 | B10-Ct | DHSSSSDniallvq |
| N | uBCEL-N1 | 16–48 | 18–55 | B1p-B2 | GGPSDSTGSNQNGERSGARSKQRRPQGLPNNTASWFTA |
| uBCEL-N2 | 59–78 | 59–78 | B2-H1 | HGKEDLKFPRGQGVPINTNS | |
| uBCEL-N3 | 158–170 | 135–170 | B8-B9 | tegalntpkdhigtrnpannaaiVLQLPQGTTLPKG | |
| uBCEL-N4 | 173–208 | 173–213 | B9-H2p | AEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPArmagn | |
| uBCEL-N5 | 235–247 | 235–247 | H2p-H3p | SGKGQQQQGQTVT | |
| uBCEL-N6 | 276–287 | 276–287 | H4-H5 | RRGPEQTQGNFG | |
| uBCEL-N7 | 339–344 | 339–344 | B11-H9 | LDDKDP | |
| uBCEL-N8 | 363–383 | 363–383 | H10-H11p | FPPTEPKKDKKKKADETQALP |
| uBCEL | Change(s) | n | Date of First Isolation | Geolocation |
|---|---|---|---|---|
| uBCEL-S2 | I68- | 11 | 15/03/2020 | USA: WA |
| N74K | 2 | 20/01/2020 | Brasil; China | |
| D80Y | 2 | 31/03/2020 | USA: WA | |
| uBCEL-S5 | G476S | 7 | 10/03/2020 | USA: WA |
| V483A | 11 | 05/03/2020 | USA: WA | |
| uBCEL-S7 | Q677H | 2 | 19/03/2020 | USA: UT |
| uBCEL-S8 | T791I | 6 | 26/02/2020 | Taiwan |
| BCEH-E1 | P71L | 2 | 19/03/2020 | USA: WA |
| uBCEL-N2 | P67S | 2 | 17/03/2020 | USA: NY; USA: WA |
| uBCEL-N3 | A152S | 2 | 13/03/2020 | USA: UT |
| uBCEL-N4 | S180I | 2 | 31/03/2020 | USA: WA |
| S183Y | 4 | 17/03/2020 | USA | |
| R185C | 5 | 15/03/2020 | USA | |
| R185L | 2 | 19/03/2020 | USA | |
| S188L | 3 | 18/03/2020 | USA | |
| S188P | 2 | 13/03/2020 | Taiwan | |
| S190I | 3 | 17/03/2020 | USA: NY | |
| S196L | 6 | 29/02/2020 | USA | |
| S197L | 17 | 26/02/2020 | Greece; Spain; USA | |
| S202N | 7 | 30/01/2020 | China; USA | |
| R203K,G204R | 62 | 27/02/2020 | Czech Republic; Greece; India; Israel; Peru; Spain; Sri Lanka; Taiwan; USA | |
| T205I | 10 | 29/01/2020 | China; USA | |
| A208G | 4 | 16/03/2020 | USA: WA; USA: NY | |
| uBCEL-N7 | P344S | 2 | ?/01/2020 | Japan |
| uBCEL-N8 | E367- | 2 | 16/03/2020 | SA: UT; USA: WA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corral-Lugo, A.; López-Siles, M.; López, D.; McConnell, M.J.; Martin-Galiano, A.J. Identification and Analysis of Unstructured, Linear B-Cell Epitopes in SARS-CoV-2 Virion Proteins for Vaccine Development. Vaccines 2020, 8, 397. https://doi.org/10.3390/vaccines8030397
Corral-Lugo A, López-Siles M, López D, McConnell MJ, Martin-Galiano AJ. Identification and Analysis of Unstructured, Linear B-Cell Epitopes in SARS-CoV-2 Virion Proteins for Vaccine Development. Vaccines. 2020; 8(3):397. https://doi.org/10.3390/vaccines8030397
Chicago/Turabian StyleCorral-Lugo, Andrés, Mireia López-Siles, Daniel López, Michael J. McConnell, and Antonio J. Martin-Galiano. 2020. "Identification and Analysis of Unstructured, Linear B-Cell Epitopes in SARS-CoV-2 Virion Proteins for Vaccine Development" Vaccines 8, no. 3: 397. https://doi.org/10.3390/vaccines8030397
APA StyleCorral-Lugo, A., López-Siles, M., López, D., McConnell, M. J., & Martin-Galiano, A. J. (2020). Identification and Analysis of Unstructured, Linear B-Cell Epitopes in SARS-CoV-2 Virion Proteins for Vaccine Development. Vaccines, 8(3), 397. https://doi.org/10.3390/vaccines8030397