Specific Antibodies Induced by Immunization with Hepatitis B Virus-Like Particles Carrying Hepatitis C Virus Envelope Glycoprotein 2 Epitopes Show Differential Neutralization Efficiency

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Leishmania tarentolae Cultivation and Protein Expression
2.3. SDS-PAGE and Western Blot
2.4. Cell Lysis and Ultracentrifugation
2.5. Electron Microscopy
2.6. Immunization Protocol
2.7. Analysis of the Antibody Response by ELISA
2.8. IgG Purification from Mouse Sera
2.9. Virus Stocks for HCVcc Neutralization Assay
2.10. HCVcc Neutralization Assay
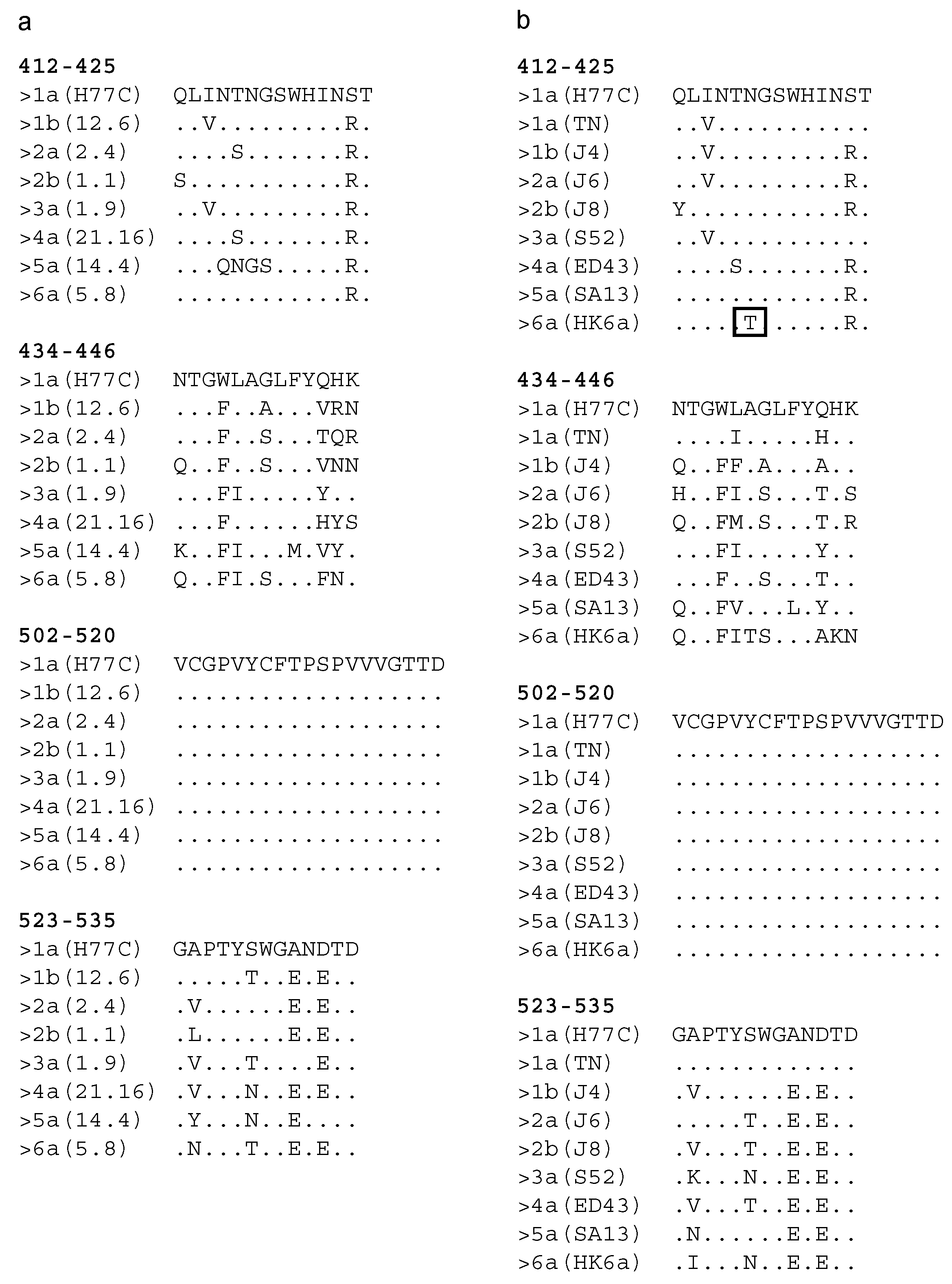
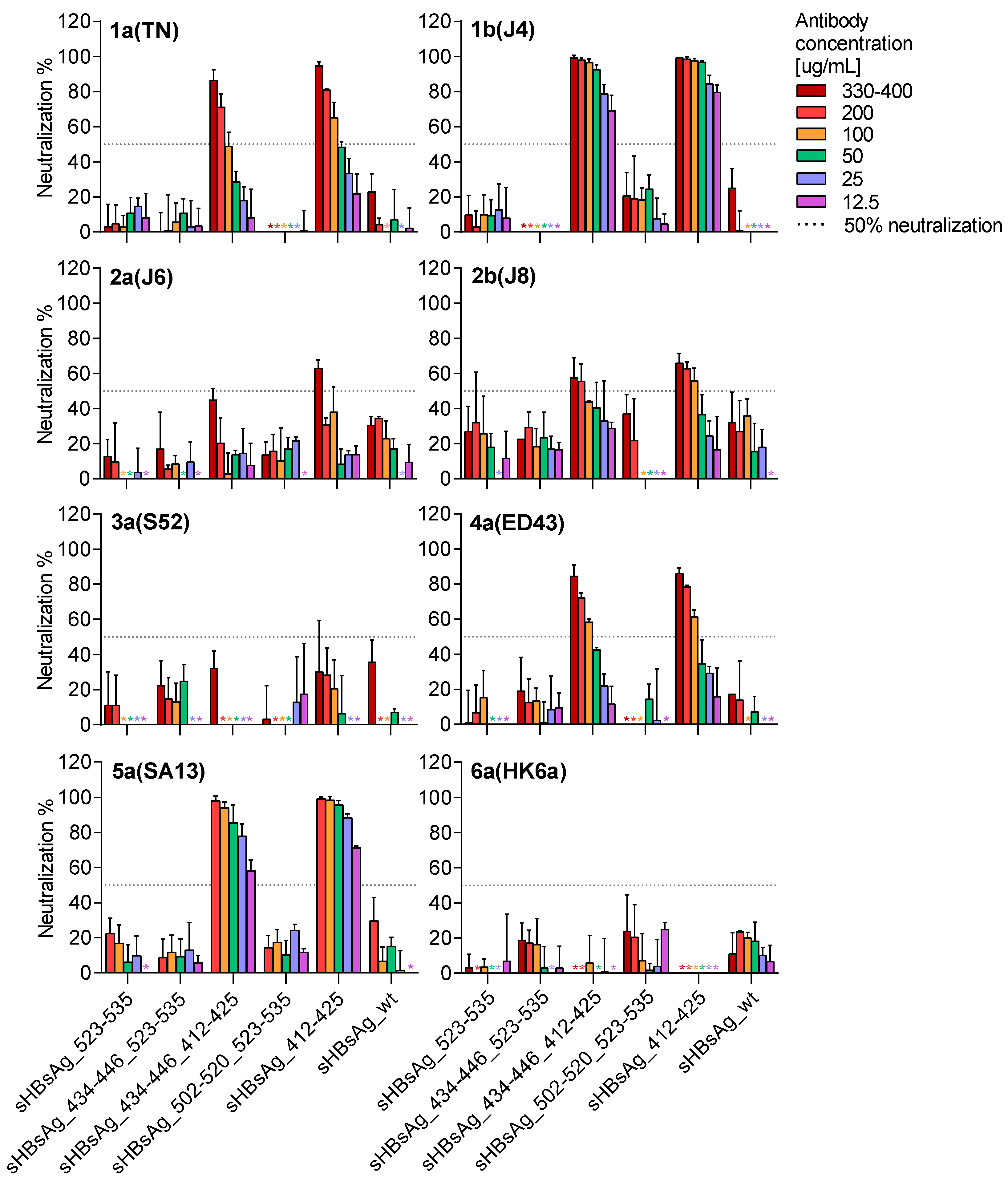
3. Results
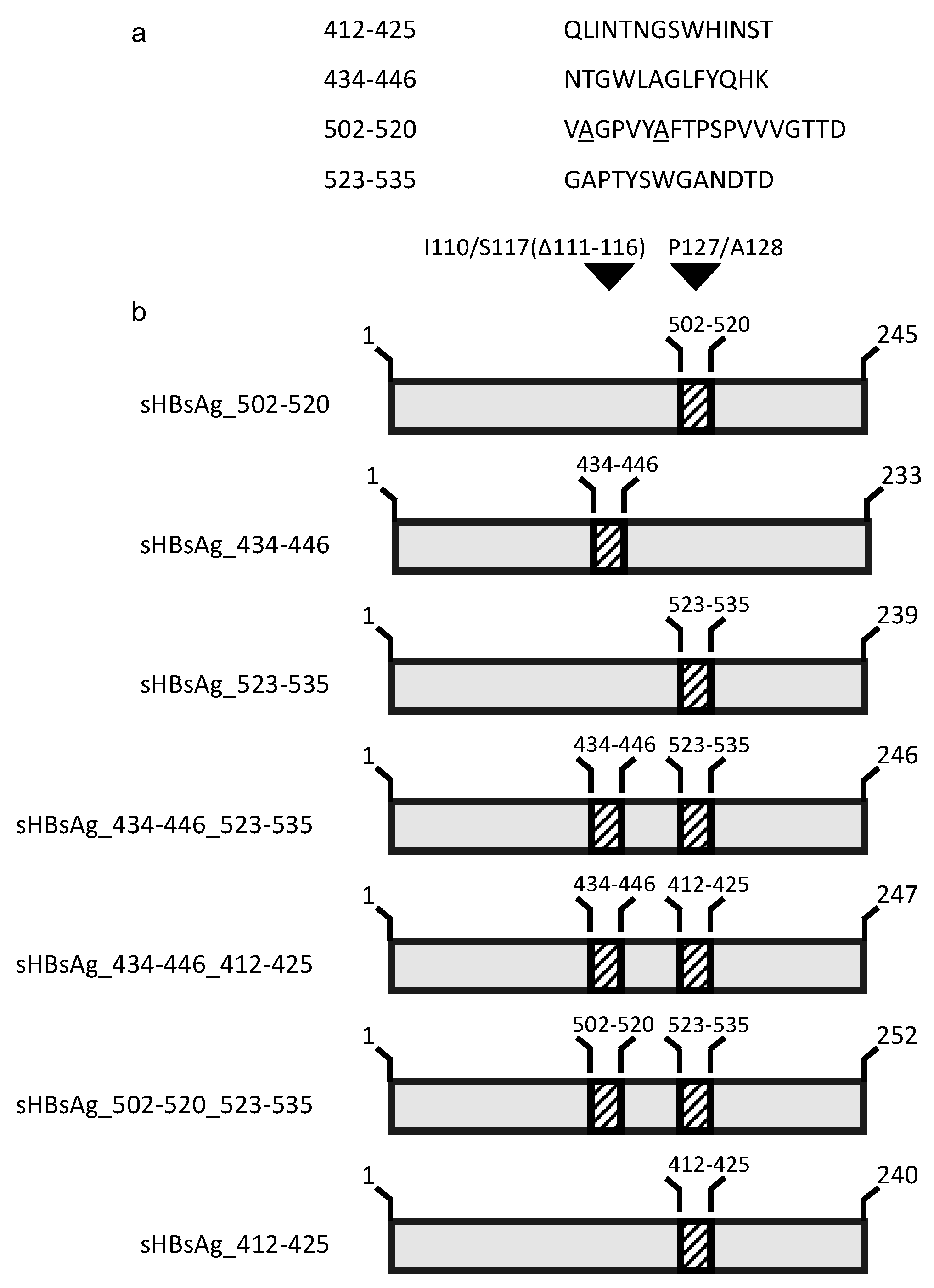
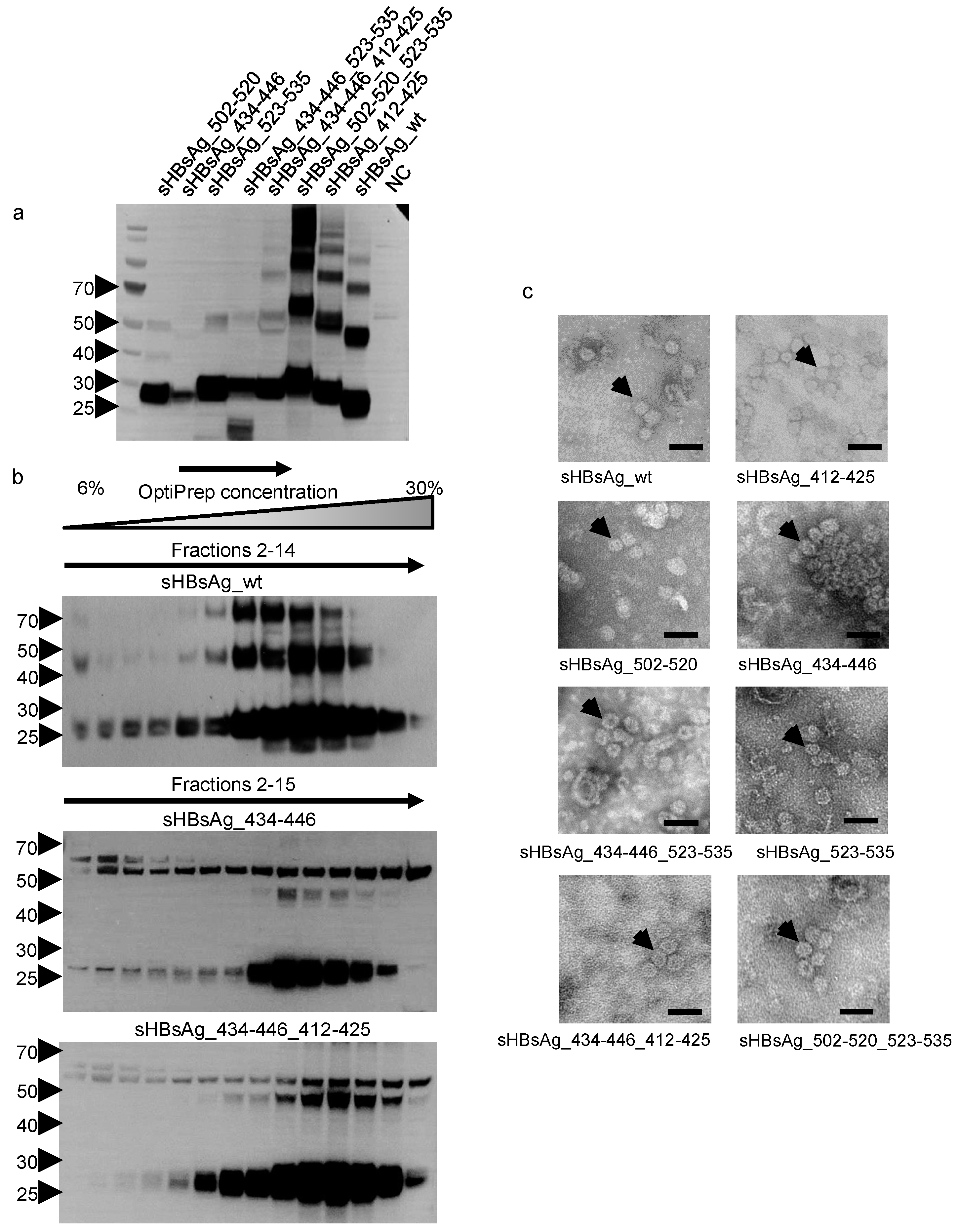
3.1. Expression of the Chimeric sHBsAg-Based Particles
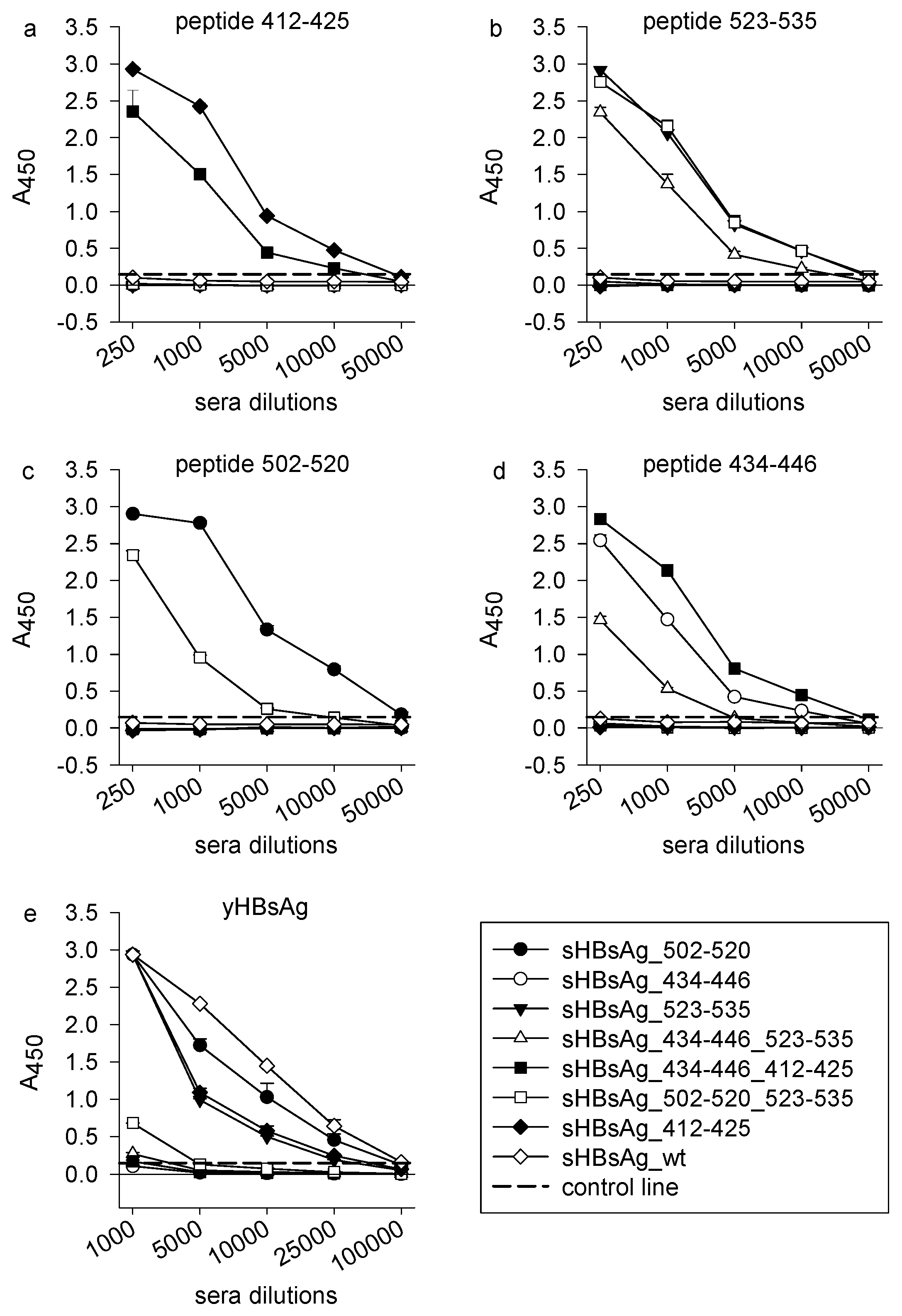
3.2. Immunogenicity of the Chimeric sHBsAg-Based Particles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Hepatitis Report; World Health Organization: Geneva, Switzerland, 2017; ISBN 978-92-4-156545-5. [Google Scholar]
- Spengler, U. Direct antiviral agents (DAAs)—A new age in the treatment of hepatitis C virus infection. Pharmacol. Ther. 2018, 183, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.-M. Retreatment of hepatitis C virus-infected patients with direct-acting antiviral failures. Semin. Liver Dis. 2019, 39, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Kinchen, V.J.; Cox, A.L.; Bailey, J.R. Can broadly neutralizing monoclonal antibodies lead us to a hepatitis C virus vaccine? Trends Microbiol. 2018, 26, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Fuerst, T.R.; Pierce, B.G.; Keck, Z.-Y.; Foung, S.K.H. Designing a B cell-based vaccine against a highly variable hepatitis C virus. Front. Microbiol. 2017, 8, 2692. [Google Scholar] [CrossRef] [PubMed]
- Fauvelle, C.; Colpitts, C.C.; Keck, Z.-Y.; Pierce, B.G.; Foung, S.K.H.; Baumert, T.F. Hepatitis C virus vaccine candidates inducing protective neutralizing antibodies. Expert Rev. Vaccines 2016, 15, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Plotkin, S.L. The development of vaccines: How the past led to the future. Nat. Rev. Microbiol. 2011, 9, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Ralston, R.; Weiner, A.; Chien, D.; Van Nest, G.; Han, J.; Berger, K.; Thudium, K.; Kuo, C. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc. Natl. Acad. Sci. USA 1994, 91, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Capone, S.; Antrobus, R.D.; Brown, A.; Richardson, R.; Newell, E.W.; Halliday, J.; Kelly, C.; Bowen, D.; Fergusson, J.; et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts and sustains functional HCV specific T-cell memory. Sci. Transl. Med. 2014, 6, 261ra153. [Google Scholar] [CrossRef] [PubMed]
- Trial Evaluating Experimental Hepatitis C Vaccine Concludes|NIH: National Institute of Allergy and Infectious Diseases. Available online: https://www.niaid.nih.gov/news-events/trial-evaluating-experimental-hepatitis-c-vaccine-concludes (accessed on 26 November 2019).
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a novel hepatitis C virus genotype from Punjab, India: Expanding classification of hepatitis C virus into 8 genotypes. J. Infect. Dis. 2018, 218, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J. The history of hepatitis C virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J. Hepatol. 2016, 65, S2–S21. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Aleman, F.; Tzarum, N.; Kong, L.; Nagy, K.; Zhu, J.; Wilson, I.A.; Law, M. Immunogenetic and structural analysis of a class of HCV broadly neutralizing antibodies and their precursors. Proc. Natl. Acad. Sci. USA 2018, 115, 7569–7574. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Keck, Z.-Y.; Lau, P.; Fauvelle, C.; Gowthaman, R.; Baumert, T.F.; Fuerst, T.R.; Mariuzza, R.A.; Foung, S.K.H. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: Implications for vaccine design. Proc. Natl. Acad. Sci. USA 2016, 113, E6946–E6954. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.T.; Gordon, F.D.; Curry, M.P.; Schiano, T.D.; Emre, S.; Corey, K.; Markmann, J.F.; Hertl, M.; Pomposelli, J.J.; Pomfret, E.A.; et al. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: A randomized controlled study. Am. J. Transplant. 2013, 13, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Owsianka, A.; Tarr, A.W.; Juttla, V.S.; Lavillette, D.; Bartosch, B.; Cosset, F.-L.; Ball, J.K.; Patel, A.H. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J. Virol. 2005, 79, 11095–11104. [Google Scholar] [CrossRef] [PubMed]
- Krey, T.; Meola, A.; Keck, Z.; Damier-Piolle, L.; Foung, S.K.H.; Rey, F.A. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 2013, 9, e1003364. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.-Y.; Wang, Y.; Lau, P.; Lund, G.; Rangarajan, S.; Fauvelle, C.; Liao, G.C.; Holtsberg, F.W.; Warfield, K.L.; Aman, M.J.; et al. Affinity maturation of a broadly neutralizing human monoclonal antibody that prevents acute hepatitis C virus infection in mice. Hepatology 2016, 64, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Kachko, A.; Kochneva, G.; Sivolobova, G.; Grazhdantseva, A.; Lupan, T.; Zubkova, I.; Wells, F.; Merchlinsky, M.; Williams, O.; Watanabe, H.; et al. New neutralizing antibody epitopes in hepatitis C virus envelope glycoproteins are revealed by dissecting peptide recognition profiles. Vaccine 2011, 30, 69–77. [Google Scholar] [CrossRef]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Lavie, M.; Sarrazin, S.; Montserret, R.; Descamps, V.; Baumert, T.F.; Duverlie, G.; Séron, K.; Penin, F.; Dubuisson, J. Identification of conserved residues in hepatitis C virus envelope glycoprotein E2 that modulate virus dependence on CD81 and SRB1 entry factors. J. Virol. 2014, 88, 10584–10597. [Google Scholar] [CrossRef]
- Owsianka, A.M.; Timms, J.M.; Tarr, A.W.; Brown, R.J.P.; Hickling, T.P.; Szwejk, A.; Bienkowska-Szewczyk, K.; Thomson, B.J.; Patel, A.H.; Ball, J.K. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 2006, 80, 8695–8704. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide vaccine: Progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Marintcheva, B. Chapter 8—Viruses as tools for vaccine development. In Harnessing the Power of Viruses; Marintcheva, B., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 217–242. ISBN 978-0-12-810514-6. [Google Scholar]
- Roldão, A.; Mellado, M.C.M.; Castilho, L.R.; Carrondo, M.J.T.; Alves, P.M. Virus-like particles in vaccine development. Expert Rev. Vaccines 2010, 9, 1149–1176. [Google Scholar] [CrossRef] [PubMed]
- Bruss, V.; Gerhardt, E.; Vieluf, K.; Wunderlich, G. Functions of the large hepatitis B virus surface protein in viral particle morphogenesis. Intervirology 1996, 39, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Gavilanes, F.; Gonzalez-Ros, J.M.; Peterson, D.L. Structure of hepatitis B surface antigen. Characterization of the lipid components and their association with the viral proteins. J. Biol. Chem. 1982, 257, 7770–7777. [Google Scholar] [PubMed]
- Cheong, W.-S.; Reiseger, J.; Turner, S.J.; Boyd, R.; Netter, H.-J. Chimeric virus-like particles for the delivery of an inserted conserved influenza A-specific CTL epitope. Antivir. Res. 2009, 81, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Cheong, W.-S.; Drummer, H.E.; Netter, H.-J. Delivery of a foreign epitope by sharing amino acid residues with the carrier matrix. J. Virol. Methods 2009, 158, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Delpeyroux, F.; Chenciner, N.; Lim, A.; Malpièce, Y.; Blondel, B.; Crainic, R.; van der Werf, S.; Streeck, R.E. A poliovirus neutralization epitope expressed on hybrid hepatitis B surface antigen particles. Science 1986, 233, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Kotiw, M.; Johnson, M.; Pandey, M.; Fry, S.; Hazell, S.L.; Netter, H.J.; Good, M.F.; Olive, C. Immunological response to parenteral vaccination with recombinant hepatitis B virus surface antigen virus-like particles expressing Helicobacter pylori KatA epitopes in a murine H. pylori challenge model. Clin. Vaccine Immunol. 2012, 19, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Patient, R.; Hourioux, C.; Vaudin, P.; Pagès, J.-C.; Roingeard, P. Chimeric hepatitis B and C viruses envelope proteins can form subviral particles: Implications for the design of new vaccine strategies. New Biotechnol. 2009, 25, 226–234. [Google Scholar] [CrossRef]
- Vietheer, P.T.K.; Boo, I.; Drummer, H.E.; Netter, H.-J. Immunizations with chimeric hepatitis B virus-like particles to induce potential anti-hepatitis C virus neutralizing antibodies. Antivir. Ther. 2007, 12, 477–487. [Google Scholar]
- Czarnota, A.; Tyborowska, J.; Peszyńska-Sularz, G.; Gromadzka, B.; Bieńkowska-Szewczyk, K.; Grzyb, K. Immunogenicity of Leishmania-derived hepatitis B small surface antigen particles exposing highly conserved E2 epitope of hepatitis C virus. Microb. Cell Fact. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Netter, H.J.; Macnaughton, T.B.; Woo, W.P.; Tindle, R.; Gowans, E.J. Antigenicity and immunogenicity of novel chimeric hepatitis B surface antigen particles with exposed hepatitis C virus epitopes. J. Virol. 2001, 75, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, M.; Purcell, R.H.; Emerson, S.U.; Bukh, J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. USA 1997, 94, 8738–8743. [Google Scholar] [CrossRef] [PubMed]
- Lavillette, D.; Tarr, A.W.; Voisset, C.; Donot, P.; Bartosch, B.; Bain, C.; Patel, A.H.; Dubuisson, J.; Ball, J.K.; Cosset, F.-L. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 2005, 41, 265–274. [Google Scholar] [CrossRef]
- Gottwein, J.M.; Scheel, T.K.H.; Jensen, T.B.; Lademann, J.B.; Prentoe, J.C.; Knudsen, M.L.; Hoegh, A.M.; Bukh, J. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: Role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 2009, 49, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.H.; Gottwein, J.M.; Carlsen, T.H.R.; Li, Y.-P.; Jensen, T.B.; Spengler, U.; Weis, N.; Bukh, J. Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific Core-NS2 by using previously identified mutations. J. Virol. 2011, 85, 2891–2906. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.H.; Gottwein, J.M.; Jensen, T.B.; Prentoe, J.C.; Hoegh, A.M.; Alter, H.J.; Eugen-Olsen, J.; Bukh, J. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. USA 2008, 105, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.B.; Gottwein, J.M.; Scheel, T.K.H.; Hoegh, A.M.; Eugen-Olsen, J.; Bukh, J. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: Failure of homologous neutralizing-antibody treatment to control infection. J. Infect. Dis. 2008, 198, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Scheel, T.K.H.; Callendret, B.; Li, Y.-P.; Eccleston, H.B.; Engle, R.E.; Govindarajan, S.; Satterfield, W.; Purcell, R.H.; Walker, C.M.; et al. Novel infectious cDNA clones of hepatitis C virus genotype 3a (strain S52) and 4a (strain ED43): Genetic analyses and In Vivo pathogenesis studies. J. Virol. 2010, 84, 5277–5293. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, T.H.R.; Pedersen, J.; Prentoe, J.C.; Giang, E.; Keck, Z.-Y.; Mikkelsen, L.S.; Law, M.; Foung, S.K.H.; Bukh, J. Breadth of neutralization and synergy of clinically relevant human monoclonal antibodies against HCV genotypes 1a, 1b, 2a, 2b, 2c, and 3a. Hepatology 2014, 60, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.V.; Ramirez, S.; Gottwein, J.M.; Fahnøe, U.; Li, Y.-P.; Pedersen, J.; Bukh, J. HCV genotype 6a escape from and resistance to Velpatasvir, Pibrentasvir, and Sofosbuvir in robust infectious cell culture models. Gastroenterology 2018, 154, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pierce, B.G.; Wang, Q.; Keck, Z.-Y.; Fuerst, T.R.; Foung, S.K.H.; Mariuzza, R.A. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J. Biol. Chem. 2015, 290, 10117–10125. [Google Scholar] [CrossRef] [PubMed]
- Pantua, H.; Diao, J.; Ultsch, M.; Hazen, M.; Mathieu, M.; McCutcheon, K.; Takeda, K.; Date, S.; Cheung, T.K.; Phung, Q.; et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J. Mol. Biol. 2013, 425, 1899–1914. [Google Scholar] [CrossRef] [PubMed]
- Prentoe, J.; Velázquez-Moctezuma, R.; Foung, S.K.H.; Law, M.; Bukh, J. Hypervariable region 1 shielding of hepatitis C virus is a main contributor to genotypic differences in neutralization sensitivity. Hepatology 2016, 64, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.-Y.; Li, T.-K.; Xia, J.; Bartosch, B.; Cosset, F.-L.; Dubuisson, J.; Foung, S.K.H. Analysis of a highly flexible conformational immunogenic domain A in hepatitis C virus E2. J. Virol. 2005, 79, 13199–13208. [Google Scholar] [CrossRef] [PubMed]
- Giang, E.; Dorner, M.; Prentoe, J.C.; Dreux, M.; Evans, M.J.; Bukh, J.; Rice, C.M.; Ploss, A.; Burton, D.R.; Law, M. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2012, 109, 6205–6210. [Google Scholar] [CrossRef] [PubMed]
- Tarr, A.W.; Urbanowicz, R.A.; Jayaraj, D.; Brown, R.J.P.; McKeating, J.A.; Irving, W.L.; Ball, J.K. Naturally occurring antibodies that recognize linear epitopes in the amino terminus of the hepatitis C virus E2 protein confer noninterfering, additive neutralization. J. Virol. 2012, 86, 2739–2749. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.; Wang, W.; Wang, Y.; Lau, P.; Carlsen, T.H.R.; Prentoe, J.; Xia, J.; Patel, A.H.; Bukh, J.; Foung, S.K.H. Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J. Virol. 2013, 87, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, C.G.; Mihalik, K.; Virata-Theimer, M.L.; Yu, M.W.; Alter, H.J.; Feinstone, S.M. Hepatitis C virus epitope-specific neutralizing antibodies in Igs prepared from human plasma. Proc. Natl. Acad. Sci. USA 2007, 104, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhong, L.; Struble, E.B.; Watanabe, H.; Kachko, A.; Mihalik, K.; Virata-Theimer, M.L.; Alter, H.J.; Feinstone, S.; Major, M. Depletion of interfering antibodies in chronic hepatitis C patients and vaccinated chimpanzees reveals broad cross-genotype neutralizing activity. Proc. Natl. Acad. Sci. USA 2009, 106, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Kachko, A.; Frey, S.E.; Sirota, L.; Ray, R.; Wells, F.; Zubkova, I.; Zhang, P.; Major, M.E. Antibodies to an interfering epitope in hepatitis C virus E2 can mask vaccine-induced neutralizing activity. Hepatology 2015, 62, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Tzarum, N.; Wilson, I.A.; Law, M. The neutralizing face of hepatitis C virus E2 envelope glycoprotein. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Ball, J.K.; Tarr, A.W.; McKeating, J.A. The past, present and future of neutralizing antibodies for hepatitis C virus. Antivir. Res. 2014, 105, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Vasiliauskaite, I.; Owsianka, A.; England, P.; Khan, A.G.; Cole, S.; Bankwitz, D.; Foung, S.K.H.; Pietschmann, T.; Marcotrigiano, J.; Rey, F.A.; et al. Conformational flexibility in the immunoglobulin-like domain of the hepatitis C virus glycoprotein E2. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Krey, T.; d’Alayer, J.; Kikuti, C.M.; Saulnier, A.; Damier-Piolle, L.; Petitpas, I.; Johansson, D.X.; Tawar, R.G.; Baron, B.; Robert, B.; et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010, 6, e1000762. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Lei, Y.; Yang, J.; Wang, X.; Shu, F.; Wei, X.; Lin, F.; Li, B.; Cui, Y.; Zhang, H.; et al. Neutralization effects of antibody elicited by chimeric HBV S antigen viral-like particles presenting HCV neutralization epitopes. Vaccine 2018, 36, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Tarr, A.W.; Backx, M.; Hamed, M.R.; Urbanowicz, R.A.; McClure, C.P.; Brown, R.J.P.; Ball, J.K. Immunization with a synthetic consensus hepatitis C virus E2 glycoprotein ectodomain elicits virus-neutralizing antibodies. Antivir. Res. 2018, 160, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Tarr, A.W.; Owsianka, A.M.; Jayaraj, D.; Brown, R.J.P.; Hickling, T.P.; Irving, W.L.; Patel, A.H.; Ball, J.K. Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J. Gen. Virol. 2007, 88, 2991–3001. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OD600 | |
|---|---|
| sHBsAg_502–520 | 4.9 |
| sHBsAg_434–446 | 2.4 |
| sHBsAg_523–535 | 4.8 |
| sHBsAg_434–446_523–535 | 4.8 |
| sHBsAg_434–446_412–425 | 4.5 |
| sHBsAg_502–520_523–535 | 5.0 |
| sHBsAg_412–425 | 5.3 |
| sHBsAg_wt | 4.9 |
| (a) | ||||||||
| Reduced/Denatured | Genotype (Isolate) | |||||||
| MOUSE GROUP | 1a (H77C) | 1b (12.6) | 2a (2.4) | 2b (1.1) | 3a (1.9) | 4a (21.16) | 5a (14.4) | 6a (5.8) |
| sHBsAg_502–520 | + | + | + | + | + | + | + | + |
| sHBsAg_434–446 | - | - | - | - | - | - | - | - |
| sHBsAg_523–535 | + | + | + | + | + | + | + | + |
| sHBsAg_434–446_523–535 | + | + | + | + | + | + | + | + |
| sHBsAg_434–446_412–425 | + | + | + | + | + | + | - | + |
| sHBsAg_502–520_523–535 | + | + | + | + | + | + | + | + |
| sHBsAg_412–425 | + | + | + | + | + | + | - | + |
| (b) | ||||||||
| Native | Genotype (Isolate) | |||||||
| MOUSE GROUP | 1a (H77C) | 1b (12.6) | 2a (2.4) | 2b (1.1) | 3a (1.9) | 4a (21.16) | 5a (14.4) | 6a (5.8) |
| sHBsAg_502–520 | - | - | - | - | - | - | - | - |
| sHBsAg_434–446 | - | - | - | - | - | - | - | - |
| sHBsAg_523–535 | + | + | + | +/− | - | - | + | +/− |
| sHBsAg_434–446_523–535 | + | - | + | +/− | +/− | - | + | +/− |
| sHBsAg_434–446_412–425 | + | + | + | + | +/− | + | - | +/− |
| sHBsAg_502–520_523–535 | + | + | + | + | - | - | + | - |
| sHBsAg_412–425 | + | + | + | + | + | + | - | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnota, A.; Offersgaard, A.; Pihl, A.F.; Prentoe, J.; Bukh, J.; Gottwein, J.M.; Bieńkowska-Szewczyk, K.; Grzyb, K. Specific Antibodies Induced by Immunization with Hepatitis B Virus-Like Particles Carrying Hepatitis C Virus Envelope Glycoprotein 2 Epitopes Show Differential Neutralization Efficiency. Vaccines 2020, 8, 294. https://doi.org/10.3390/vaccines8020294
Czarnota A, Offersgaard A, Pihl AF, Prentoe J, Bukh J, Gottwein JM, Bieńkowska-Szewczyk K, Grzyb K. Specific Antibodies Induced by Immunization with Hepatitis B Virus-Like Particles Carrying Hepatitis C Virus Envelope Glycoprotein 2 Epitopes Show Differential Neutralization Efficiency. Vaccines. 2020; 8(2):294. https://doi.org/10.3390/vaccines8020294
Chicago/Turabian StyleCzarnota, Anna, Anna Offersgaard, Anne Finne Pihl, Jannick Prentoe, Jens Bukh, Judith Margarete Gottwein, Krystyna Bieńkowska-Szewczyk, and Katarzyna Grzyb. 2020. "Specific Antibodies Induced by Immunization with Hepatitis B Virus-Like Particles Carrying Hepatitis C Virus Envelope Glycoprotein 2 Epitopes Show Differential Neutralization Efficiency" Vaccines 8, no. 2: 294. https://doi.org/10.3390/vaccines8020294
APA StyleCzarnota, A., Offersgaard, A., Pihl, A. F., Prentoe, J., Bukh, J., Gottwein, J. M., Bieńkowska-Szewczyk, K., & Grzyb, K. (2020). Specific Antibodies Induced by Immunization with Hepatitis B Virus-Like Particles Carrying Hepatitis C Virus Envelope Glycoprotein 2 Epitopes Show Differential Neutralization Efficiency. Vaccines, 8(2), 294. https://doi.org/10.3390/vaccines8020294