West Nile Virus Restriction in Mosquito and Human Cells: A Virus under Confinement



Abstract
1. Introduction
1.1. West Nile Virus Incidence
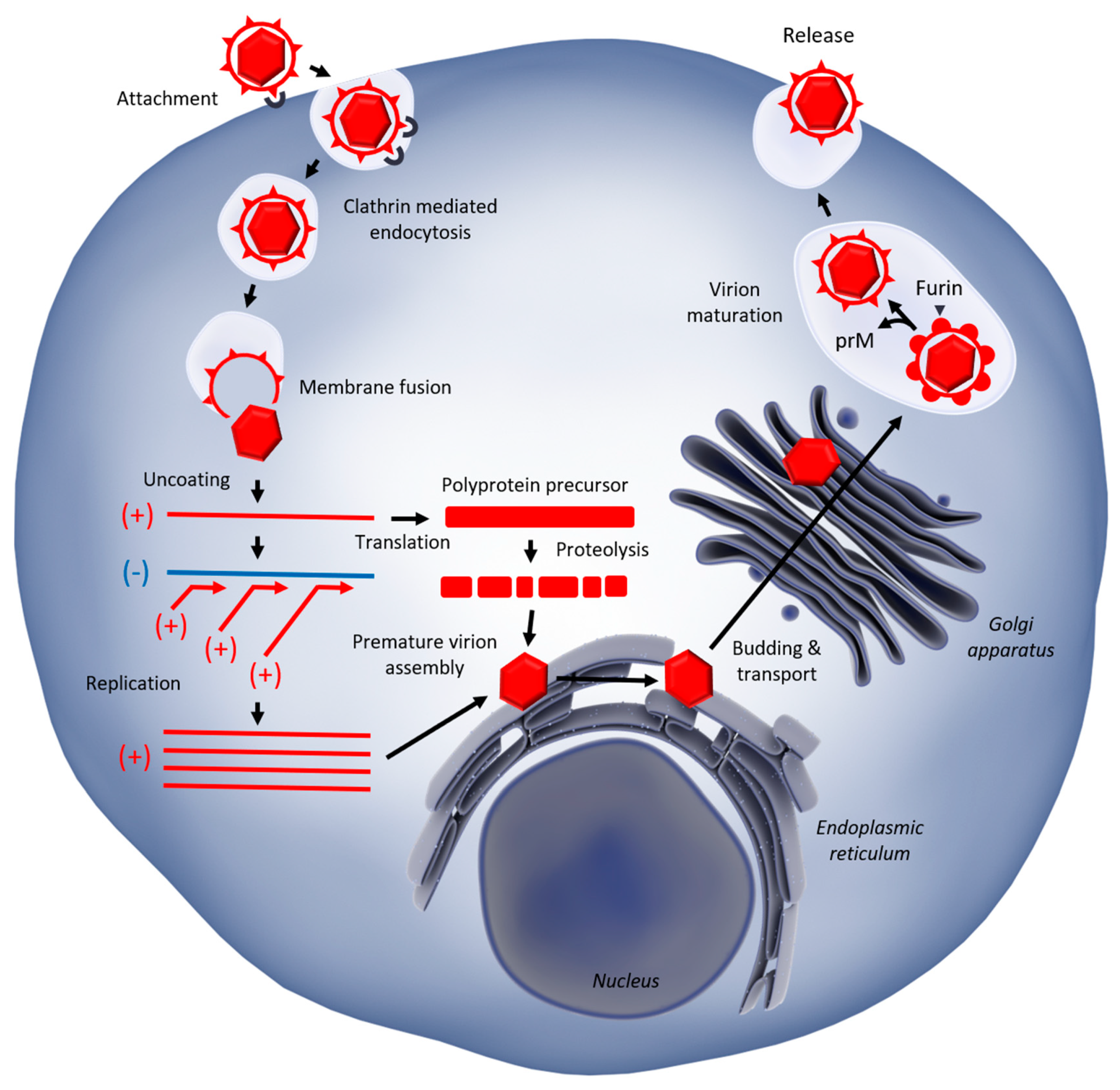
1.2. West Nile Virus Replication
2. Mosquitoes: Immune Defenses against WNV Infection and Viral Countermeasures
2.1. Immune Response
2.2. Antiviral Factors and Viral Countermeasures
3. Vertebrates: A General Insight into Anti-WNV Immunity
3.1. Immune Response to WNV Infection
3.2. Innate Immunity
4. Birds: Immune Defenses against WNV Infection
4.1. Importance of Birds in the WNV Enzootic Cycle
4.2. Immune Response to WNV Infection
5. Immune Defenses against WNV Infection in Mammals
5.1. WNV Infection
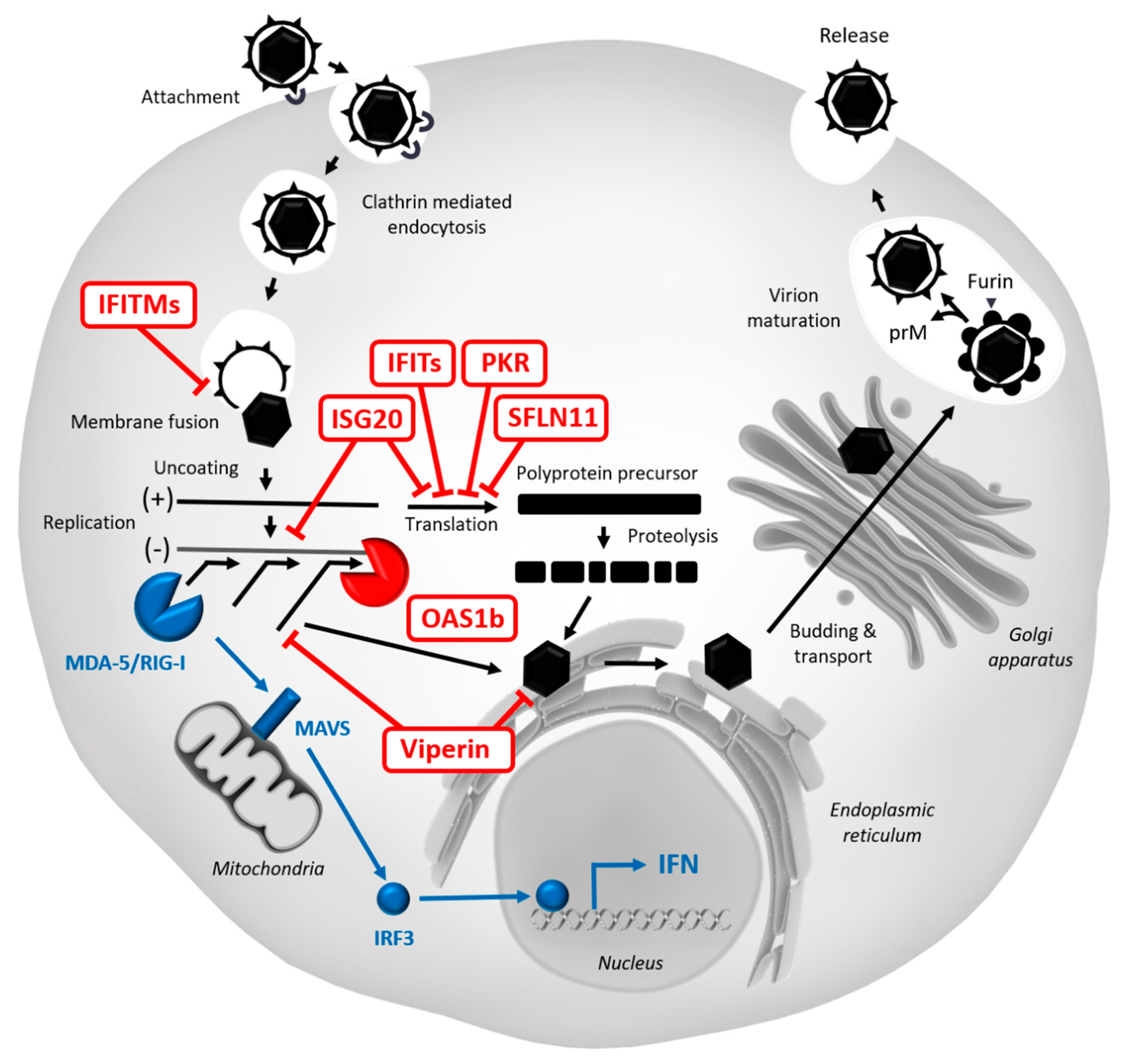
5.2. Restriction Factors Targeting WNV
5.2.1. Protein Kinase R (PKR)
5.2.2. Viperin
5.2.3. OAS/RNase L
5.2.4. IFITMs
5.2.5. ISG15/20
5.2.6. IFITs
5.2.7. IFI6, SC4MOL, DDX24, IFI44L, IFRD1, IL13RA1, MAFK, PAK3, and SAMD9L
5.2.8. IFI27l2a
5.2.9. TRIM6/VAMP8
5.2.10. SLFN11
5.2.11. RIPK3
5.2.12. Other Restriction Factors
5.3. Viral Countermeasures
5.3.1. Evasion from Innate Immune Recognition
5.3.2. Direct Inhibition of Innate Immune Sensors
5.3.3. Direct Inhibition of IFN Signaling
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smithburn, K.C.; Hughes, T.P.; Burke, A.W.; Paul, J.H. A Neurotropic Virus Isolated from the Blood of a Native of Uganda 1. Am. J. Trop. Med. Hyg. 1940, 20, 471–492. [Google Scholar] [CrossRef]
- Clé, M.; Beck, C.; Salinas, S.; Lecollinet, S.; Gutierrez, S.; Van de Perre, P.; Baldet, T.; Foulongne, V.; Simonin, Y. Usutu virus: A new threat? Epidemiol. Infect. 2019, 147. [Google Scholar] [CrossRef] [PubMed]
- Clé, M.; Barthelemy, J.; Desmetz, C.; Foulongne, V.; Lapeyre, L.; Bolloré, K.; Tuaillon, E.; Erkilic, N.; Kalatzis, V.; Lecollinet, S.; et al. Study of Usutu virus neuropathogenicity in mice and human cellular models. PLOS Negl. Trop. Dis. 2020, 14, e0008223. [Google Scholar] [CrossRef] [PubMed]
- Turell, M.J.; Sardelis, M.R.; Dohm, D.J.; O’guinn, M.L. Potential North American Vectors of West Nile Virus. Ann. N. Y. Acad. Sci. 2001, 951, 317–324. [Google Scholar] [CrossRef]
- Berthet, F.X.; Digoutte, J.P.; Zeller, H.G.; Deubel, V.; Rauzier, J.; Drouet, M.T. Extensive nucleotide changes and deletions within the envelope glycoprotein gene of Euro-African West Nile viruses. J. Gen. Virol. 1997, 78, 2293–2297. [Google Scholar] [CrossRef]
- Scherret, J.H.; Mackenzie, J.S.; Hall, R.A.; Deubel, V.; Gould, E.A. Phylogeny and Molecular Epidemiology of West Nile and Kunjin Viruses. In Japanese Encephalitis and West Nile Viruses; Mackenzie, J.S., Barrett, A.D.T., Deubel, V., Eds.; Current Topics in Microbiology and Immunology; Springer Berlin: Berlin/Heidelberg, Germany, 2002; Volume 267, pp. 373–390. ISBN 978-3-642-63966-1. [Google Scholar]
- May, F.J.; Davis, C.T.; Tesh, R.B.; Barrett, A.D.T. Phylogeography of West Nile Virus: From the Cradle of Evolution in Africa to Eurasia, Australia, and the Americas. J. Virol. 2011, 85, 2964–2974. [Google Scholar] [CrossRef]
- Martín-Acebes, M.A. West Nile virus: A re-emerging pathogen revisited. WJV 2012, 1, 51. [Google Scholar] [CrossRef]
- Dauphin, G.; Zientara, S. Infections par le virus du Nil occidental: Synthèse et actualités épidémiologiques. Virologie 2005, 9, 395–408. [Google Scholar]
- Hubálek, Z.; Halouzka, J.; Juricová, Z.; Sebesta, O. First isolation of mosquito-borne West Nile virus in the Czech Republic. Acta Virol. 1998, 42, 119–120. [Google Scholar]
- Lvov, D.K.; Butenko, A.M.; Gromashevsky, V.L.; Kovtunov, A.I.; Prilipov, A.G.; Kinney, R.; Aristova, V.A.; Dzharkenov, A.F.; Samokhvalov, E.I.; Savage, H.M.; et al. West Nile virus and other zoonotic viruses in Russia: Examples of emerging-reemerging situations. In Emergence and Control of Zoonotic Viral Encephalitides; Calisher, C.H., Griffin, D.E., Eds.; Springer: Vienna, Austria, 2004; pp. 85–96. ISBN 978-3-211-20454-2. [Google Scholar]
- Bondre, V.P.; Jadi, R.S.; Mishra, A.C.; Yergolkar, P.N.; Arankalle, V.A. West Nile virus isolates from India: Evidence for a distinct genetic lineage. J. Gen. Virol. 2007, 88, 875–884. [Google Scholar] [CrossRef]
- Bahuon, C.; Lecollinet, S.; Beck, C. West Nile Virus Infection. In eLS; American Cancer Society: Atlanta, GA, USA, 2015; pp. 1–11. ISBN 978-0-470-01590-2. [Google Scholar]
- Blitvich, B.J. Transmission dynamics and changing epidemiology of West Nile virus. Anim. Health Res. Rev. 2008, 9, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Shaman, J.; Day, J.F.; Stieglitz, M. Drought-Induced Amplification and Epidemic Transmission of West Nile Virus in Southern Florida. J. Med. Entomol. 2005, 42, 8. [Google Scholar] [CrossRef] [PubMed]
- Hindiyeh, M.; Shulman, L.M.; Mendelson, E.; Weiss, L.; Grossman, Z.; Bin, H. Isolation and Characterization of West Nile Virus from the Blood of Viremic Patients During the 2000 Outbreak in Israel. Emerg. Infect. Dis. 2001, 7, 3. [Google Scholar] [CrossRef]
- Lanciotti, R.S. Origin of the West Nile Virus Responsible for an Outbreak of Encephalitis in the Northeastern United States. Science 1999, 286, 2333–2337. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Ivanics, É.; Erdélyi, K.; Ursu, K.; Ferenczi, E.; Weissenböck, H.; Nowotny, N. Lineage 1 and 2 Strains of Encephalitic West Nile Virus, Central Europe. Emerg. Infect. Dis. 2006, 12, 618–623. [Google Scholar] [CrossRef]
- Hayes, E.B.; Komar, N.; Nasci, R.S.; Montgomery, S.P.; O’Leary, D.R.; Campbell, G.L. Epidemiology and Transmission Dynamics of West Nile Virus Disease. Emerg. Infect. Dis. 2005, 11, 1167–1173. [Google Scholar] [CrossRef]
- Campbell, G.L.; Marfin, A.A.; Lanciotti, R.S.; Gubler, D.J. West Nile virus. Lancet Infect. Dis. 2002, 2, 519–529. [Google Scholar] [CrossRef]
- Suen, W.; Prow, N.; Hall, R.; Bielefeldt-Ohmann, H. Mechanism of West Nile Virus Neuroinvasion: A Critical Appraisal. Viruses 2014, 6, 2796–2825. [Google Scholar] [CrossRef]
- Guarner, J.; Shieh, W.-J.; Hunter, S.; Paddock, C.D.; Morken, T.; Campbell, G.L.; Marfin, A.A.; Zaki, S.R. Clinicopathologic study and laboratory diagnosis of 23 cases with West Nile virus encephalomyelitis. Hum. Pathol. 2004, 35, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Morrey, J.D.; Diamond, M.S. Caspase 3-Dependent Cell Death of Neurons Contributes to the Pathogenesis of West Nile Virus Encephalitis. J. Virol. 2007, 81, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Epidemiological Update: West Nile Virus Transmission Season in Europe. 2018. Available online: http://ecdc.europa.eu/en/news-events/epidemiological-update-west-nile-virus-transmission-season-europe-2018 (accessed on 19 July 2019).
- Davis, C.W.; Nguyen, H.-Y.; Hanna, S.L.; Sanchez, M.D.; Doms, R.W.; Pierson, T.C. West Nile Virus Discriminates between DC-SIGN and DC-SIGNR for Cellular Attachment and Infection. J. Virol. 2006, 80, 1290–1301. [Google Scholar] [CrossRef]
- Chu, J.J.; Ng, M.-L. Interaction of West Nile Virus with α v β 3 Integrin Mediates Virus Entry into Cells. J. Biol. Chem. 2004, 279, 54533–54541. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.J.H.; Ng, M.L. Infectious Entry of West Nile Virus Occurs through a Clathrin-Mediated Endocytic Pathway. J. Virol. 2004, 78, 10543–10555. [Google Scholar] [CrossRef]
- Thompson, B.S.; Moesker, B.; Smit, J.M.; Wilschut, J.; Diamond, M.S.; Fremont, D.H. A Therapeutic Antibody against West Nile Virus Neutralizes Infection by Blocking Fusion within Endosomes. PLoS Pathog. 2009, 5, e1000453. [Google Scholar] [CrossRef]
- Gollins, S.W.; Porterfield, J.T.S. The uncoating and infectivity of the flavivirus West Nile on interaction with cells: Effects of pH and ammonium chloride. J. Gen. Virol. 1986, 67, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Brinton, M.A. The Molecular Biology of West Nile Virus: A New Invader of the Western Hemisphere. Annu. Rev. Microbiol. 2002, 56, 371–402. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The Endoplasmic Reticulum Provides the Membrane Platform for Biogenesis of the Flavivirus Replication Complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed]
- Wengler, G.; Wengler, G. Cell-associated West Nile flavivirus is covered with E+pre-M protein heterodimers which are destroyed and reorganized by proteolytic cleavage during virus release. J. Virol. 1989, 63, 2521–2526. [Google Scholar] [CrossRef]
- Nowak, T.; Färber, P.M.; Wengler, G.; Wengler, G. Analyses of the terminal sequences of west nile virus structural proteins and of the in vitro translation of these proteins allow the proposal of a complete scheme of the proteolytic cleavages involved in their synthesis. Virology 1989, 169, 365–376. [Google Scholar] [CrossRef]
- Xu, J.; Cherry, S. Viruses and antiviral immunity in Drosophila. Dev. Comp. Immunol. 2014, 42, 67–84. [Google Scholar] [CrossRef]
- TenOever, B.R. The Evolution of Antiviral Defense Systems. Cell Host Microbe 2016, 19, 142–149. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Trinidad, L.; Voysey, R.; Duchemin, J.-B.; Walker, P.J. Secreted Vago restricts West Nile virus infection in Culex mosquito cells by activating the Jak-STAT pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 18915–18920. [Google Scholar] [CrossRef] [PubMed]
- Kingsolver, M.B.; Hardy, R.W. Making connections in insect innate immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 18639–18640. [Google Scholar] [CrossRef] [PubMed]
- Samuel, G.H.; Adelman, Z.N.; Myles, K.M. Antiviral immunity and virus-mediated antagonism in disease vector mosquitoes. Trends Microbiol. 2018, 26, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Slonchak, A.; Hussain, M.; Torres, S.; Asgari, S.; Khromykh, A.A. Expression of Mosquito MicroRNA Aae-miR-2940-5p is downregulated in response to west nile virus infection to restrict viral replication. J. Virol. 2014, 88, 8457–8467. [Google Scholar] [CrossRef]
- Lee, W.-S.; Webster, J.A.; Madzokere, E.T.; Stephenson, E.B.; Herrero, L.J. Mosquito antiviral defense mechanisms: A delicate balance between innate immunity and persistent viral infection. Parasites Vectors 2019, 12. [Google Scholar] [CrossRef]
- Styer, L.M.; Kent, K.A.; Albright, R.G.; Bennett, C.J.; Kramer, L.D.; Bernard, K.A. Mosquitoes Inoculate High Doses of West Nile Virus as They Probe and Feed on Live Hosts. PLoS Pathog. 2007, 3, e132. [Google Scholar] [CrossRef]
- CDC. Principles of Epidemiology in Public Health Practice, An Introduction to Applied Epidemiology and Biostatistics, 3rd ed.; CDC: Atlanta, GA, USA, 2006. [Google Scholar]
- Cheng, G.; Liu, Y.; Wang, P.; Xiao, X. Mosquito Defense Strategies against Viral Infection. Trends Parasitol. 2016, 32, 177–186. [Google Scholar] [CrossRef]
- Goic, B.; Vodovar, N.; Mondotte, J.A.; Monot, C.; Frangeul, L.; Blanc, H.; Gausson, V.; Vera-Otarola, J.; Cristofari, G.; Saleh, M.-C. RNA-mediated interference and reverse transcription control the persistence of RNA viruses in the insect model Drosophila. Nat. Immunol. 2013, 14, 396–403. [Google Scholar] [CrossRef]
- Poirier, E.Z.; Goic, B.; Tomé-Poderti, L.; Frangeul, L.; Boussier, J.; Gausson, V.; Blanc, H.; Vallet, T.; Loyd, H.; Levi, L.I.; et al. Dicer-2-Dependent Generation of Viral DNA from Defective Genomes of RNA Viruses Modulates Antiviral Immunity in Insects. Cell Host Microbe 2018, 23, 353–365.e8. [Google Scholar] [CrossRef]
- Goic, B.; Stapleford, K.A.; Frangeul, L.; Doucet, A.J.; Gausson, V.; Blanc, H.; Schemmel-Jofre, N.; Cristofari, G.; Lambrechts, L.; Vignuzzi, M.; et al. Virus-derived DNA drives mosquito vector tolerance to arboviral infection. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, A.; Hanna, S.L.; Li, J.; Cho, H.; Rose, P.P.; Spiridigliozzi, A.; Gold, B.; Diamond, M.S.; Cherry, S. Genome-Wide RNAi Screen Identifies Broadly-Acting Host Factors That Inhibit Arbovirus Infection. PLoS Pathog. 2014, 10, e1003914. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Côté, J. Structural and Functional Conservation of the NuA4 Histone Acetyltransferase Complex from Yeast to Humans. Mol. Cell. Biol. 2004, 24, 1884–1896. [Google Scholar] [CrossRef]
- Kakumani, P.K.; Ponia, S.S.; Shanmugan, R.K.; Sood, V.; Chinnappan, M.; Banerjea, A.C.; Medigeshi, G.R.; Malhotra, P.; Mukherjee, S.K.; Bhatnagar, R.K. Role of RNA Interference (RNAi) in Dengue Virus Replication and Identification of NS4B as an RNAi Suppressor. J. Virol. 2013, 87, 8870–8883. [Google Scholar] [CrossRef] [PubMed]
- Pompon, J.; Manuel, M.; Ng, G.K.; Wong, B.; Shan, C.; Manokaran, G.; Soto-Acosta, R.; Bradrick, S.S.; Ooi, E.E.; Missé, D.; et al. Dengue subgenomic flaviviral RNA disrupts immunity in mosquito salivary glands to increase virus transmission. PLoS Pathog. 2017, 13, e1006535. [Google Scholar] [CrossRef] [PubMed]
- Samuel, G.H.; Wiley, M.R.; Badawi, A.; Adelman, Z.N.; Myles, K.M. Yellow fever virus capsid protein is a potent suppressor of RNA silencing that binds double-stranded RNA. Proc. Natl. Acad. Sci. USA 2016, 113, 13863–13868. [Google Scholar] [CrossRef]
- Hussain, M.; Torres, S.; Schnettler, E.; Funk, A.; Grundhoff, A.; Pijlman, G.P.; Khromykh, A.A.; Asgari, S. West Nile virus encodes a microRNA-like small RNA in the 3′ untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res. 2012, 40, 2210–2223. [Google Scholar] [CrossRef]
- Moon, S.L.; Dodd, B.J.T.; Brackney, D.E.; Wilusz, C.J.; Ebel, G.D.; Wilusz, J. Flavivirus sfRNA suppresses antiviral RNA interference in cultured cells and mosquitoes and directly interacts with the RNAi machinery. Virology 2015, 485, 322–329. [Google Scholar] [CrossRef]
- Martina, B.E.E.; Koraka, P.; van den Doel, P.; Rimmelzwaan, G.F.; Haagmans, B.L.; Osterhaus, A.D.M.E. DC-SIGN enhances infection of cells with glycosylated West Nile virus in vitro and virus replication in human dendritic cells induces production of IFN-α and TNF-α. Virus Res. 2008, 135, 64–71. [Google Scholar] [CrossRef]
- Shrestha, B.; Zhang, B.; Purtha, W.E.; Klein, R.S.; Diamond, M.S. Tumor Necrosis Factor Alpha Protects against Lethal West Nile Virus Infection by Promoting Trafficking of Mononuclear Leukocytes into the Central Nervous System. J. Virol. 2008, 82, 8956–8964. [Google Scholar] [CrossRef]
- Silva, M.C.; Guerrero-Plata, A.; Gilfoy, F.D.; Garofalo, R.P.; Mason, P.W. Differential Activation of Human Monocyte-Derived and Plasmacytoid Dendritic Cells by West Nile Virus Generated in Different Host Cells. J. Virol. 2007, 81, 13640–13648. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, M.; Daniel, S.; Huang, Y.; Chancey, C.; Huang, Q.; Lei, Y.F.; Grinev, A.; Mostowski, H.; Rios, M.; Dayton, A. Anti-West Nile virus activity of in vitro expanded human primary natural killer cells. BMC Immunol. 2010, 11. [Google Scholar] [CrossRef]
- Hershkovitz, O.; Rosental, B.; Rosenberg, L.A.; Navarro-Sanchez, M.E.; Jivov, S.; Zilka, A.; Gershoni-Yahalom, O.; Brient-Litzler, E.; Bedouelle, H.; Ho, J.W.; et al. NKp44 Receptor Mediates Interaction of the Envelope Glycoproteins from the West Nile and Dengue Viruses with NK Cells. J. Immunol. 2009, 183, 2610–2621. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Kong, K.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A Paradoxical Role for Neutrophils in the Pathogenesis of West Nile Virus. J. Infect. Dis. 2010, 202, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Scully, E.; Yin, Z.; Kim, J.H.; Wang, S.; Yan, J.; Mamula, M.; Anderson, J.F.; Craft, J.; Fikrig, E. IFN-γ-Producing γδ T Cells Help Control Murine West Nile Virus Infection. J. Immunol. 2003, 171, 2524–2531. [Google Scholar] [CrossRef]
- Suthar, M.S.; Diamond, M.S.; Gale Jr, M. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Ben-Nathan, D.; Huitinga, I.; Lustig, S.; van Rooijen, N.; Kobiler, D. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch. Virol. 1996, 141, 459–469. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B Cells and Antibody Play Critical Roles in the Immediate Defense of Disseminated Infection by West Nile Encephalitis Virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A Critical Role for Induced IgM in the Protection against West Nile Virus Infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar] [CrossRef]
- Chambers, T.J.; Droll, D.A.; Walton, A.H.; Schwartz, J.; Wold, W.S.M.; Nickells, J. West Nile 25A virus infection of B-cell-deficient (μMT) mice: Characterization of neuroinvasiveness and pseudoreversion of the viral envelope protein. J. Gen. Virol. 2008, 89, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Draves, K.E.; Young, L.B.; Roe, K.; Bryan, M.A.; Dresch, C.; Richner, J.M.; Diamond, M.S.; Gale, M.; Clark, E.A. Protection of mice deficient in mature B cells from West Nile virus infection by passive and active immunization. PLoS Pathog. 2017, 13, e1006743. [Google Scholar] [CrossRef]
- Brien, J.D.; Uhrlaub, J.L.; Nikolich–Žugich, J. Protective capacity and epitope specificity of CD8+ T cells responding to lethal West Nile virus infection. Eur. J. Immunol. 2007, 37, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Wang, T.; Samuel, M.A.; Whitby, K.; Craft, J.; Fikrig, E.; Diamond, M.S. Gamma Interferon Plays a Crucial Early Antiviral Role in Protection against West Nile Virus Infection. J. Virol. 2006, 80, 5338–5348. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T Cells in Control of West Nile Virus Infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef] [PubMed]
- Sitati, E.M.; Diamond, M.S. CD4+ T-Cell Responses Are Required for Clearance of West Nile Virus from the Central Nervous System. J. Virol. 2006, 80, 12060–12069. [Google Scholar] [CrossRef] [PubMed]
- Brien, J.D.; Uhrlaub, J.L.; Nikolich-Žugich, J. West Nile Virus-Specific CD4 T Cells Exhibit Direct Antiviral Cytokine Secretion and Cytotoxicity and Are Sufficient for Antiviral Protection. J. Immunol. 2008, 181, 8568–8575. [Google Scholar] [CrossRef]
- Lanteri, M.C.; O’Brien, K.M.; Purtha, W.E.; Cameron, M.J.; Lund, J.M.; Owen, R.E.; Heitman, J.W.; Custer, B.; Hirschkorn, D.F.; Tobler, L.H.; et al. Tregs control the development of symptomatic West Nile virus infection in humans and mice. J. Clin. Investig. 2009. [Google Scholar] [CrossRef]
- Carroll, M.C. The complement system in regulation of adaptive immunity. Nat. Immunol. 2004, 5, 981–986. [Google Scholar] [CrossRef]
- Mehlhop, E.; Whitby, K.; Oliphant, T.; Marri, A.; Engle, M.; Diamond, M.S. Complement Activation Is Required for Induction of a Protective Antibody Response against West Nile Virus Infection. J. Virol. 2005, 79, 7466–7477. [Google Scholar] [CrossRef]
- Mehlhop, E.; Diamond, M.S. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J. Exp. Med. 2006, 203, 1371–1381. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D. Intrinsic immunity: A front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Diamond, M.S. Alpha/Beta Interferon Protects against Lethal West Nile Virus Infection by Restricting Cellular Tropism and Enhancing Neuronal Survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef]
- Lazear, H.M.; Pinto, A.K.; Vogt, M.R.; Gale, M.; Diamond, M.S. Beta Interferon Controls West Nile Virus Infection and Pathogenesis in Mice. J. Virol. 2011, 85, 7186–7194. [Google Scholar] [CrossRef]
- Lazear, H.M.; Daniels, B.P.; Pinto, A.K.; Huang, A.C.; Vick, S.C.; Doyle, S.E.; Gale, M.; Klein, R.S.; Diamond, M.S. Interferon- restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci. Transl. Med. 2015, 7, 284ra59. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M. Establishment and Maintenance of the Innate Antiviral Response to West Nile Virus Involves both RIG-I and MDA5 Signaling through IPS-1. J. Virol. 2008, 82, 609–616. [Google Scholar] [CrossRef]
- Suthar, M.S.; Ma, D.Y.; Thomas, S.; Lund, J.M.; Zhang, N.; Daffis, S.; Rudensky, A.Y.; Bevan, M.J.; Clark, E.A.; Kaja, M.-K.; et al. IPS-1 Is Essential for the Control of West Nile Virus Infection and Immunity. PLoS Pathog. 2010, 6, e1000757. [Google Scholar] [CrossRef]
- Errett, J.S.; Suthar, M.S.; McMillan, A.; Diamond, M.S.; Gale, M. The Essential, Nonredundant Roles of RIG-I and MDA5 in Detecting and Controlling West Nile Virus Infection. J. Virol. 2013, 87, 11416–11425. [Google Scholar] [CrossRef]
- Lazear, H.M.; Pinto, A.K.; Ramos, H.J.; Vick, S.C.; Shrestha, B.; Suthar, M.S.; Gale, M.; Diamond, M.S. Pattern Recognition Receptor MDA5 Modulates CD8+ T Cell-Dependent Clearance of West Nile Virus from the Central Nervous System. J. Virol. 2013, 87, 11401–11415. [Google Scholar] [CrossRef]
- Zhao, J.; Vijay, R.; Zhao, J.; Gale, M.; Diamond, M.S.; Perlman, S. MAVS Expressed by Hematopoietic Cells Is Critical for Control of West Nile Virus Infection and Pathogenesis. J. Virol. 2016, 90, 7098–7108. [Google Scholar] [CrossRef] [PubMed]
- Roe, K.; Giordano, D.; Young, L.B.; Draves, K.E.; Holder, U.; Suthar, M.S.; Gale, M.; Clark, E.A. Dendritic cell-associated MAVS is required to control West Nile virus replication and ensuing humoral immune responses. PLoS ONE 2019, 14, e0218928. [Google Scholar] [CrossRef] [PubMed]
- Szretter, K.J.; Daffis, S.; Patel, J.; Suthar, M.S.; Klein, R.S.; Gale, M.; Diamond, M.S. The Innate Immune Adaptor Molecule MyD88 Restricts West Nile Virus Replication and Spread in Neurons of the Central Nervous System. J. Virol. 2010, 84, 12125–12138. [Google Scholar] [CrossRef] [PubMed]
- McGuckin Wuertz, K.; Treuting, P.M.; Hemann, E.A.; Esser-Nobis, K.; Snyder, A.G.; Graham, J.B.; Daniels, B.P.; Wilkins, C.; Snyder, J.M.; Voss, K.M.; et al. STING is required for host defense against neuropathological West Nile virus infection. PLoS Pathog. 2019, 15, e1007899. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Samuel, M.A.; Keller, B.C.; Gale, M.; Diamond, M.S. Cell-Specific IRF-3 Responses Protect against West Nile Virus Infection by Interferon-Dependent and -Independent Mechanisms. PLoS Pathog. 2007, 3, e106. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Lancaster, A.; Wilkins, C.; Suthar, M.S.; Huang, A.; Vick, S.C.; Clepper, L.; Thackray, L.; Brassil, M.M.; Virgin, H.W.; et al. IRF-3, IRF-5, and IRF-7 Coordinately Regulate the Type I IFN Response in Myeloid Dendritic Cells Downstream of MAVS Signaling. PLoS Pathog. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Keller, B.C.; Gale, M.; Diamond, M.S. Interferon Regulatory Factor IRF-7 Induces the Antiviral Alpha Interferon Response and Protects against Lethal West Nile Virus Infection. J. Virol. 2008, 82, 8465–8475. [Google Scholar] [CrossRef]
- Thackray, L.B.; Shrestha, B.; Richner, J.M.; Miner, J.J.; Pinto, A.K.; Lazear, H.M.; Gale, M.; Diamond, M.S. Interferon Regulatory Factor 5-Dependent Immune Responses in the Draining Lymph Node Protect against West Nile Virus Infection. J. Virol. 2014, 88, 11007–11021. [Google Scholar] [CrossRef]
- Berthoux, L. The Restrictome of Flaviviruses. Virol. Sin. 2020. [Google Scholar] [CrossRef]
- Komar, N.; Langevin, S.; Hinten, S.; Nemeth, N.; Edwards, E.; Hettler, D.; Davis, B.; Bowen, R.; Bunning, M. Experimental Infection of North American Birds with the New York 1999 Strain of West Nile Virus. Emerg. Infect. Dis. 2003, 9, 311–322. [Google Scholar] [CrossRef]
- Ahlers, L.R.H.; Goodman, A.G. The Immune Responses of the Animal Hosts of West Nile Virus: A Comparison of Insects, Birds, and Mammals. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.D.; Meece, J.K.; Henkel, J.S.; Shukla, S.K. Birds, Migration and Emerging Zoonoses: West Nile Virus, Lyme Disease, Influenza A and Enteropathogens. Clin. Med. Res. 2003, 1, 5–12. [Google Scholar] [CrossRef] [PubMed]
- LaDeau, S.L.; Kilpatrick, A.M.; Marra, P.P. West Nile virus emergence and large-scale declines of North American bird populations. Nature 2007, 447, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Epidemiological Update: West Nile Virus Transmission Season in Europe. 2019. Available online: https://www.ecdc.europa.eu/en/news-events/epidemiological-update-west-nile-virus-transmission-season-europe-2019 (accessed on 21 May 2020).
- Gibbs, S.E.J.; Ellis, A.E.; Mead, D.G.; Allison, A.B.; Moulton, J.K.; Howerth, E.W.; Stallknecht, D.E. West nile virus detection in the organs of naturally infected blue jays (cyanocitta cristata). J. Wildl. Dis. 2005, 41, 354–362. [Google Scholar] [CrossRef]
- Panella, N.A.; Kerst, A.J.; Lanciotti, R.S.; Bryant, P.; Wolf, B.; Komar, N. Comparative West Nile Virus Detection in Organs of Naturally Infected American Crows (Corvus brachyrhynchos). Emerg. Infect. Dis. 2001, 7, 754. [Google Scholar] [CrossRef]
- Wünschmann, A.; Shivers, J.; Bender, J.; Carroll, L.; Fuller, S.; Saggese, M.; van Wettere, A.; Redig, P. Pathologic Findings in Red-Tailed Hawks (Buteo jamaicensis) and Cooper’s Hawks (Accipiter cooperi) Naturally Infected with West Nile Virus. avdi 2004, 48, 570–580. [Google Scholar] [CrossRef]
- Nemeth, N.M.; Kratz, G.E.; Bates, R.; Scherpelz, J.A.; Bowen, R.A.; Komar, N. Naturally Induced Humoral Immunity to West Nile Virus Infection in Raptors. EcoHealth 2008, 5, 298–304. [Google Scholar] [CrossRef]
- Nemeth, N.M.; Oesterle, P.T.; Bowen, R.A. Humoral Immunity to West Nile Virus Is Long-Lasting and Protective in the House Sparrow (Passer domesticus). Am. J. Trop. Med. Hyg. 2009, 80, 864–869. [Google Scholar] [CrossRef]
- Bowen, R.A.; Nemeth, N.M. Dynamics of Passive Immunity to West Nile Virus in Domestic Chickens (Gallus Gallus Domesticus). Am. J. Trop. Med. Hyg. 2007, 76, 310–317. [Google Scholar] [CrossRef]
- Baitchman, E.J.; Tlusty, M.F.; Murphy, H.W. Passive Transfer of Maternal Antibodies to West Nile Virus in Flamingo Chicks (Phoenicopterus Chilensis and Phoenicopterus Ruber Ruber). J. Zoo Wildl. Med. 2007, 38, 337–340. [Google Scholar] [CrossRef]
- Gibbs, S.E.J.; Hoffman, D.M.; Stark, L.M.; Marlenee, N.L.; Blitvich, B.J.; Beaty, B.J.; Stallknecht, D.E. Persistence of Antibodies to West Nile Virus in Naturally Infected Rock Pigeons (Columba livia). Clin. Diagn. Lab. Immunol. 2005, 12, 665–667. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hahn, D.C.; Nemeth, N.M.; Edwards, E.; Bright, P.R.; Komar, N. Passive West Nile Virus Antibody Transfer from Maternal Eastern Screech-Owls (Megascops asio) to Progeny. Avian Dis. 2006, 50, 454–455. [Google Scholar] [CrossRef] [PubMed]
- Sokawa, J.; Shimizu, N.; Sokawa, Y. Presence of (2′-5′)Oligoadenylate Synthetase in Avian Erythrocytes. J. Biochem. 1984, 96, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Fulton, R.W.; Morton, R.J.; Burge, L.J.; Short, E.C.; Payton, M.E. Action of Quail and Chicken Interferons on a Quail Cell Line, QT35. J. Interferon Cytokine Res. 1995, 15, 297–300. [Google Scholar] [CrossRef]
- Tag-El-Din-Hassan, H.T.; Sasaki, N.; Moritoh, K.; Torigoe, D.; Maeda, A.; Agui, T. The chicken 2′-5′ oligoadenylate synthetase A inhibits the replication of West Nile virus. Jpn. J. Vet. Res. 2012, 60, 95–103. [Google Scholar]
- Fair, J.M.; Nemeth, N.M.; Taylor-McCabe, K.J.; Shou, Y.; Marrone, B.L. Clinical and acquired immunologic responses to West Nile virus infection of domestic chickens (Gallus gallus domesticus). Poult. Sci. 2011, 90, 328–336. [Google Scholar] [CrossRef]
- Oncfs—Réseau SAGIR. Available online: http://www.oncfs.gouv.fr/Reseau-SAGIR-ru105 (accessed on 19 July 2019).
- Anonymous Animal Disease Notification System (ADNS). Available online: https://ec.europa.eu/food/animals/animal-diseases/not-system_en (accessed on 21 May 2020).
- Blau, D.M.; Rabe, I.B.; Bhatnagar, J.; Civen, R.; Trivedi, K.K.; Rollin, D.; Hocevar, S.N.; Kuehnert, M.; Staples, J.E.; Zaki, S.R.; et al. West Nile Virus RNA in Tissues from Donor Associated with Transmission to Organ Transplant Recipients. Emerg. Infect. Dis. 2013, 19, 1518–1520. [Google Scholar] [CrossRef]
- Armah, H.B.; Wang, G.; Omalu, B.I.; Tesh, R.B.; Gyure, K.A.; Chute, D.J.; Smith, R.D.; Dulai, P.; Vinters, H.V.; Kleinschmidt-DeMasters, B.K.; et al. Systemic Distribution of West Nile Virus Infection: Postmortem Immunohistochemical Study of Six Cases. Brain Pathol. 2007, 17, 354–362. [Google Scholar] [CrossRef]
- Iwamoto, M.; Jernigan, D.B.; Guasch, A.; Trepka, M.J.; Blackmore, C.G.; Hellinger, W.C.; Pham, S.M.; Zaki, S.; Lanciotti, R.S.; Lance-Parker, S.E.; et al. Transmission of West Nile Virus from an Organ Donor to Four Transplant Recipients. N. Engl. J. Med. 2003, 348, 2196–2203. [Google Scholar] [CrossRef]
- Pealer, L.N.; Marfin, A.A.; Petersen, L.R.; Lanciotti, R.S.; Page, P.L.; Stramer, S.L.; Stobierski, M.G.; Signs, K.; Newman, B.; Kapoor, H.; et al. Transmission of West Nile Virus through Blood Transfusion in the United States in 2002. New Engl. J. Med. 2003, 349, 1236–1245. [Google Scholar] [CrossRef]
- Lim, P.-Y.; Behr, M.J.; Chadwick, C.M.; Shi, P.-Y.; Bernard, K.A. Keratinocytes Are Cell Targets of West Nile Virus In Vivo. J. Virol. 2011, 85, 5197–5201. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Alout, H.; Diop, F.; Damour, A.; Bengue, M.; Weill, M.; Missé, D.; Lévêque, N.; Bodet, C. Innate Immune Response of Primary Human Keratinocytes to West Nile Virus Infection and Its Modulation by Mosquito Saliva. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Kovats, S.; Turner, S.; Simmons, A.; Powe, T.; Chakravarty, E.; Alberola-Ila, J. West Nile virus-infected human dendritic cells fail to fully activate invariant natural killer T cells: WNV-infected DCs fail to fully activate iNKTs. Clin. Exp. Immunol. 2016, 186, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Sejvar, J.J. Neurologic Manifestations and Outcome of West Nile Virus Infection. JAMA 2003, 290, 511. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Leis, A.A.; Stokic, D.S.; Van Gerpen, J.A.; Marfin, A.A.; Webb, R.; Haddad, M.B.; Tierney, B.C.; Slavinski, S.A.; Polk, J.L.; et al. Acute Flaccid Paralysis and West Nile Virus Infection. Emerg. Infect. Dis. 2003, 9, 788–793. [Google Scholar] [CrossRef]
- Petersen, L.R.; Marfin, A.A. West Nile Virus: A Primer for the Clinician. Ann. Int. Med. 2002, 137, 173–179. [Google Scholar] [CrossRef]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of Tissue Inflammation by the Immune Receptor Tim-3 Expressed on Innate Immune Cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- Mazaheri, F.; Breus, O.; Durdu, S.; Haas, P.; Wittbrodt, J.; Gilmour, D.; Peri, F. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM Families of Phosphatidylserine Receptors Mediate Dengue Virus Entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef]
- Beasley, D.W.C.; Whiteman, M.C.; Zhang, S.; Huang, C.Y.-H.; Schneider, B.S.; Smith, D.R.; Gromowski, G.D.; Higgs, S.; Kinney, R.M.; Barrett, A.D.T. Envelope Protein Glycosylation Status Influences Mouse Neuroinvasion Phenotype of Genetic Lineage 1 West Nile Virus Strains. J. Virol. 2005, 79, 8339–8347. [Google Scholar] [CrossRef]
- Hanna, S.L.; Pierson, T.C.; Sanchez, M.D.; Ahmed, A.A.; Murtadha, M.M.; Doms, R.W. N-Linked Glycosylation of West Nile Virus Envelope Proteins Influences Particle Assembly and Infectivity. J. Virol. 2005, 79, 13262–13274. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Wang, X.J.; Clark, D.C.; Lobigs, M.; Hall, R.A.; Khromykh, A.A. A Single Amino Acid Substitution in the West Nile Virus Nonstructural Protein NS2A Disables Its Ability To Inhibit Alpha/Beta Interferon Induction and Attenuates Virus Virulence in Mice. J. Virol. 2006, 80, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K. Viral envelope protein glycosylation is a molecular determinant of the neuroinvasiveness of the New York strain of West Nile virus. J. Gen. Virol. 2004, 85, 3637–3645. [Google Scholar] [CrossRef] [PubMed]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.B.; Kerr, I.M.; Williams, B.R.G.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Dzananovic, E.; Patel, T.R.; Deo, S.; McEleney, K.; Stetefeld, J.; McKenna, S.A. Recognition of viral RNA stem–loops by the tandem double-stranded RNA binding domains of PKR. RNA 2013, 19, 333–344. [Google Scholar] [CrossRef]
- Gilfoy, F.D.; Mason, P.W. West Nile Virus-Induced Interferon Production Is Mediated by the Double-Stranded RNA-Dependent Protein Kinase PKR. J. Virol. 2007, 81, 11148–11158. [Google Scholar] [CrossRef]
- Samuel, M.A.; Whitby, K.; Keller, B.C.; Marri, A.; Barchet, W.; Williams, B.R.G.; Silverman, R.H.; Gale, M.; Diamond, M.S. PKR and RNase L Contribute to Protection against Lethal West Nile Virus Infection by Controlling Early Viral Spread in the Periphery and Replication in Neurons. J. Virol. 2006, 80, 7009–7019. [Google Scholar] [CrossRef]
- Jiang, D.; Weidner, J.M.; Qing, M.; Pan, X.-B.; Guo, H.; Xu, C.; Zhang, X.; Birk, A.; Chang, J.; Shi, P.-Y.; et al. Identification of Five Interferon-Induced Cellular Proteins That Inhibit West Nile Virus and Dengue Virus Infections. J. Virol. 2010, 84, 8332–8341. [Google Scholar] [CrossRef]
- Jiang, D.; Guo, H.; Xu, C.; Chang, J.; Gu, B.; Wang, L.; Block, T.M.; Guo, J.-T. Identification of Three Interferon-Inducible Cellular Enzymes That Inhibit the Replication of Hepatitis C Virus. J. Virol. 2008, 82, 1665–1678. [Google Scholar] [CrossRef]
- Wang, X.; Hinson, E.R.; Cresswell, P. The Interferon-Inducible Protein Viperin Inhibits Influenza Virus Release by Perturbing Lipid Rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef]
- Hinson, E.R.; Cresswell, P. The N-terminal Amphipathic α-Helix of Viperin Mediates Localization to the Cytosolic Face of the Endoplasmic Reticulum and Inhibits Protein Secretion. J. Biol. Chem. 2009, 284, 4705–4712. [Google Scholar] [CrossRef] [PubMed]
- Hinson, E.R.; Cresswell, P. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic -helix. Proc. Natl Acad. Sci. USA 2009, 106, 20452–20457. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Satoh, T.; Yamamoto, N.; Uematsu, S.; Takeuchi, O.; Kawai, T.; Akira, S. Antiviral Protein Viperin Promotes Toll-like Receptor 7- and Toll-like Receptor 9-Mediated Type I Interferon Production in Plasmacytoid Dendritic Cells. Immunity 2011, 34, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The Interferon-Inducible Gene viperin Restricts West Nile Virus Pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, R.; Kurhade, C.; Gilthorpe, J.D.; Överby, A.K. Cell-type- and region-specific restriction of neurotropic flavivirus infection by viperin. J. Neuroinflammation 2018, 15. [Google Scholar] [CrossRef]
- Gizzi, A.S.; Grove, T.L.; Arnold, J.J.; Jose, J.; Jangra, R.K.; Garforth, S.J.; Du, Q.; Cahill, S.M.; Dulyaninova, N.G.; Love, J.D.; et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 2018, 558, 610–614. [Google Scholar] [CrossRef]
- Mashimo, T.; Lucas, M.; Simon-Chazottes, D.; Frenkiel, M.-P.; Montagutelli, X.; Ceccaldi, P.-E.; Deubel, V.; Guenet, J.-L.; Despres, P. A nonsense mutation in the gene encoding 2′-5′-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11311–11316. [Google Scholar] [CrossRef]
- Perelygin, A.A.; Scherbik, S.V.; Zhulin, I.B.; Stockman, B.M.; Li, Y.; Brinton, M.A. Positional cloning of the murine flavivirus resistance gene. Proc. Natl Acad. Sci. USA 2002, 99, 9322–9327. [Google Scholar] [CrossRef]
- Madden, J.C.; Cui, D.; Brinton, M.A. RNase L Antiviral Activity Is Not a Critical Component of the Oas1b-Mediated Flavivirus Resistance Phenotype. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Zhou, A. Expression cloning of 2-5A-dependent RNAase: A uniquely regulated mediator of interferon action. Cell 1993, 72, 753–765. [Google Scholar] [CrossRef]
- Malathi, K.; Dong, B.; Gale, M.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Whitney, G.; Donovan, J.; Korennykh, A. Innate Immune Messenger 2-5A Tethers Human RNase L into Active High-Order Complexes. Cell Rep. 2012, 2, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Kajaste-Rudnitski, A.; Mashimo, T.; Frenkiel, M.-P.; Guénet, J.-L.; Lucas, M.; Desprès, P. The 2′,5′-Oligoadenylate Synthetase 1b Is a Potent Inhibitor of West Nile Virus Replication Inside Infected Cells. J. Biol. Chem. 2006, 281, 4624–4637. [Google Scholar] [CrossRef] [PubMed]
- Scherbik, S.V.; Kluetzman, K.; Perelygin, A.A.; Brinton, M.A. Knock-in of the Oas1br allele into a flavivirus-induced disease susceptible mouse generates the resistant phenotype. Virology 2007, 368, 232–237. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lim, J.K.; Lisco, A.; McDermott, D.H.; Huynh, L.; Ward, J.M.; Johnson, B.; Johnson, H.; Pape, J.; Foster, G.A.; Krysztof, D.; et al. Genetic Variation in OAS1 Is a Risk Factor for Initial Infection with West Nile Virus in Man. PLoS Pathog. 2009, 5, e1000321. [Google Scholar] [CrossRef]
- Bigham, A.W.; Buckingham, K.J.; Husain, S.; Emond, M.J.; Bofferding, K.M.; Gildersleeve, H.; Rutherford, A.; Astakhova, N.M.; Perelygin, A.A.; Busch, M.P.; et al. Host Genetic Risk Factors for West Nile Virus Infection and Disease Progression. PLoS ONE 2011, 6, e24745. [Google Scholar] [CrossRef]
- Deo, S.; Patel, T.R.; Dzananovic, E.; Booy, E.P.; Zeid, K.; McEleney, K.; Harding, S.E.; McKenna, S.A. Activation of 2′ 5′-Oligoadenylate Synthetase by Stem Loops at the 5′-End of the West Nile Virus Genome. PLoS ONE 2014, 9, e92545. [Google Scholar] [CrossRef]
- Ivanyi-Nagy, R.; Darlix, J.-L. Reprint of: Core protein-mediated 5′–3′ annealing of the West Nile virus genomic RNA in vitro. Virus Res. 2012, 169, 448–457. [Google Scholar] [CrossRef]
- Courtney, S.C.; Di, H.; Stockman, B.M.; Liu, H.; Scherbik, S.V.; Brinton, M.A. Identification of Novel Host Cell Binding Partners of Oas1b, the Protein Conferring Resistance to Flavivirus-Induced Disease in Mice. J. Virol. 2012, 86, 7953–7963. [Google Scholar] [CrossRef]
- Friedman, R.L.; Manly, S.P.; McMahon, M.; Kerr, I.M.; Stark, G.R. Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell 1984, 38, 745–755. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM Proteins Incorporated into HIV-1 Virions Impair Viral Fusion and Spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMs Restrict the Replication of Multiple Pathogenic Viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Xu, F.; Qian, J.; Yao, Y.; Miao, C.; Zheng, Y.-M.; Liu, S.-L.; Guo, F.; Geng, Y.; Qiao, W.; et al. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol. 2014, 16, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Chesarino, N.M.; Compton, A.A.; McMichael, T.M.; Kenney, A.D.; Zhang, L.; Soewarna, V.; Davis, M.; Schwartz, O.; Yount, J.S. IFITM3 requires an amphipathic helix for antiviral activity. EMBO Rep. 2017, 18, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.J.; Wu, W.-L.; Grotefend, C.R.; Radic, V.; Chung, C.; Chung, Y.-H.; Farzan, M.; Huang, I.-C. IFITM3 Polymorphism rs12252-C Restricts Influenza A Viruses. PLoS ONE 2014, 9, e110096. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sehgal, M.; Hou, Z.; Cheng, J.; Shu, S.; Wu, S.; Guo, F.; Le Marchand, S.J.; Lin, H.; Chang, J.; et al. Identification of Residues Controlling Restriction versus Enhancing Activities of IFITM Proteins on Entry of Human Coronaviruses. J. Virol. 2017, 92. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Pan, Q.; Ding, S.; Rong, L.; Liu, S.-L.; Geng, Y.; Qiao, W.; Liang, C. The N-Terminal Region of IFITM3 Modulates Its Antiviral Activity by Regulating IFITM3 Cellular Localization. J. Virol. 2012, 86, 13697–13707. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Roy, N.; Porrot, F.; Billet, A.; Casartelli, N.; Yount, J.S.; Liang, C.; Schwartz, O. Natural mutations in IFITM3 modulate post-translational regulation and toggle antiviral specificity. EMBO Rep. 2016, 17, 1657–1671. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.G.; Ribaudo, M.; Guo, J.-T.; Barik, S. Identification of Interferon-Stimulated Gene Proteins that Inhibit Human Parainfluenza Virus Type 3. J. Virol. 2016, 90, 11145–11156. [Google Scholar] [CrossRef]
- Gorman, M.J.; Poddar, S.; Farzan, M.; Diamond, M.S. The Interferon-Stimulated Gene Ifitm3 Restricts West Nile Virus Infection and Pathogenesis. J. Virol. 2016, 90, 8212–8225. [Google Scholar] [CrossRef] [PubMed]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.-M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 Inhibits Influenza A Virus Infection by Preventing Cytosolic Entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM Proteins Restrict Viral Membrane Hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef] [PubMed]
- Tartour, K.; Nguyen, X.-N.; Appourchaux, R.; Assil, S.; Barateau, V.; Bloyet, L.-M.; Burlaud Gaillard, J.; Confort, M.-P.; Escudero-Perez, B.; Gruffat, H.; et al. Interference with the production of infectious viral particles and bimodal inhibition of replication are broadly conserved antiviral properties of IFITMs. PLoS Pathog. 2017, 13, e1006610. [Google Scholar] [CrossRef] [PubMed]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.-C.; Farzan, M.; Jung, J.U. The Antiviral Effector IFITM3 Disrupts Intracellular Cholesterol Homeostasis to Block Viral Entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef]
- Spence, J.S.; He, R.; Hoffmann, H.-H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef]
- Yount, J.S.; Karssemeijer, R.A.; Hang, H.C. S -Palmitoylation and Ubiquitination Differentially Regulate Interferon-induced Transmembrane Protein 3 (IFITM3)-mediated Resistance to Influenza Virus. J. Biol. Chem. 2012, 287, 19631–19641. [Google Scholar] [CrossRef]
- Korant, B.D.; Blomstrom, D.C.; Jonak, G.J.; Knight, E. Interferon-induced proteins. Purification and characterization of a 15,000-dalton protein from human and bovine cells induced by interferon. J. Biol. Chem. 1984, 259, 14835–14839. [Google Scholar]
- Morales, D.J.; Lenschow, D.J. The Antiviral Activities of ISG15. J. Mol. Biol. 2013, 425, 4995–5008. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, D.-E. Interferon-Stimulated Gene 15 and the Protein ISGylation System. J. Interferon Cytokine Res. 2011, 31, 119–130. [Google Scholar] [CrossRef]
- Shi, H.-X.; Yang, K.; Liu, X.; Liu, X.-Y.; Wei, B.; Shan, Y.-F.; Zhu, L.-H.; Wang, C. Positive Regulation of Interferon Regulatory Factor 3 Activation by Herc5 via ISG15 Modification. Mol. Cell. Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Giannakopoulos, N.V.; Gunn, L.J.; Johnston, C.; O’Guin, A.K.; Schmidt, R.E.; Levine, B.; Virgin, H.W. Identification of Interferon-Stimulated Gene 15 as an Antiviral Molecule during Sindbis Virus Infection In Vivo. J. Virol. 2005, 79, 13974–13983. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Pan, W.; Wang, P. ISG15 facilitates cellular antiviral response to dengue and west nile virus infection in vitro. Virol. J. 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Espert, L.; Mechti, N.; Wilson, D.M. The Human Interferon- and Estrogen-Regulated ISG20/HEM45 Gene Product Degrades Single-Stranded RNA and DNA in Vitro. Biochemistry 2001, 40, 7174–7179. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Degols, G.; Gongora, C.; Blondel, D.; Williams, B.R.; Silverman, R.H.; Mechti, N. ISG20, a New Interferon-induced RNase Specific for Single-stranded RNA, Defines an Alternative Antiviral Pathway against RNA Genomic Viruses. J. Biol. Chem. 2003, 278, 16151–16158. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.M.; Trobaugh, D.W.; Sun, C.; Lucas, T.M.; Diamond, M.S.; Ryman, K.D.; Klimstra, W.B. The Interferon-Induced Exonuclease ISG20 Exerts Antiviral Activity through Upregulation of Type I Interferon Response Proteins. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Wu, N.; Nguyen, X.-N.; Wang, L.; Appourchaux, R.; Zhang, C.; Panthu, B.; Gruffat, H.; Journo, C.; Alais, S.; Qin, J.; et al. The interferon stimulated gene 20 protein (ISG20) is an innate defense antiviral factor that discriminates self versus non-self translation. PLoS Pathog. 2019, 15, e1008093. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Smith, M.; Katze, M.G.; Shi, P.-Y.; Gale, M. The Host Response to West Nile Virus Infection Limits Viral Spread through the Activation of the Interferon Regulatory Factor 3 Pathway. J. Virol. 2004, 78, 7737–7747. [Google Scholar] [CrossRef]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef]
- Wacher, C.; Muller, M.; Hofer, M.J.; Getts, D.R.; Zabaras, R.; Ousman, S.S.; Terenzi, F.; Sen, G.C.; King, N.J.C.; Campbell, I.L. Coordinated Regulation and Widespread Cellular Expression of Interferon-Stimulated Genes (ISG) ISG-49, ISG-54, and ISG-56 in the Central Nervous System after Infection with Distinct Viruses. J. Virol. 2007, 81, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Raychoudhuri, A.; Shrivastava, S.; Steele, R.; Kim, H.; Ray, R.; Ray, R.B. ISG56 and IFITM1 Proteins Inhibit Hepatitis C Virus Replication. J. Virol. 2011, 85, 12881–12889. [Google Scholar] [CrossRef] [PubMed]
- Saikia, P.; Fensterl, V.; Sen, G.C. The Inhibitory Action of P56 on Select Functions of E1 Mediates Interferon’s Effect on Human Papillomavirus DNA Replication. J. Virol. 2010, 84, 13036–13039. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Y.M.; Pichlmair, A.; Górna, M.W.; Superti-Furga, G.; Nagar, B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 2013, 494, 60–64. [Google Scholar] [CrossRef]
- Guo, J.; Peters, K.L.; Sen, G.C. Induction of the Human Protein P56 by Interferon, Double-Stranded RNA, or Virus Infection. Virology 2000, 267, 209–219. [Google Scholar] [CrossRef]
- Hui, D.J.; Bhasker, C.R.; Merrick, W.C.; Sen, G.C. Viral Stress-inducible Protein p56 Inhibits Translation by Blocking the Interaction of eIF3 with the Ternary Complex eIF2·GTP·Met-tRNAi. J. Biol. Chem. 2003, 278, 39477–39482. [Google Scholar] [CrossRef]
- Guo, J.; Hui, D.J.; Merrick, W.C.; Sen, G.C. A new pathway of translational regulation mediated by eukaryotic initiation factor 3. Embo J. 2000, 19, 6891–6899. [Google Scholar] [CrossRef]
- Diamond, M.S. IFIT1: A dual sensor and effector molecule that detects non-2′-O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev. 2014, 25, 543–550. [Google Scholar] [CrossRef]
- Kumar, P.; Sweeney, T.R.; Skabkin, M.A.; Skabkina, O.V.; Hellen, C.U.T.; Pestova, T.V. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res. 2014, 42, 3228–3245. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale, M.; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2′-O Methylation of the Viral mRNA Cap by West Nile Virus Evades Ifit1-Dependent and -Independent Mechanisms of Host Restriction In Vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, F.; Hui, D.J.; Merrick, W.C.; Sen, G.C. Distinct Induction Patterns and Functions of Two Closely Related Interferon-inducible Human Genes, ISG54 and ISG56. J. Biol. Chem. 2006, 281, 34064–34071. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liang, H.; Zhou, Q.; Li, Y.; Chen, H.; Ye, W.; Chen, D.; Fleming, J.; Shu, H.; Liu, Y. Crystal structure of ISG54 reveals a novel RNA binding structure and potential functional mechanisms. Cell Res. 2012, 22, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- TenOever, B.R.; Ng, S.-L.; Chua, M.A.; McWhirter, S.M.; Garcia-Sastre, A.; Maniatis, T. Multiple Functions of the IKK-Related Kinase IKK in Interferon-Mediated Antiviral Immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef]
- Perwitasari, O.; Cho, H.; Diamond, M.S.; Gale, M. Inhibitor of κB Kinase ϵ (IKKϵ), STAT1, and IFIT2 Proteins Define Novel Innate Immune Effector Pathway against West Nile Virus Infection. J. Biol. Chem. 2011, 286, 44412–44423. [Google Scholar] [CrossRef]
- Stawowczyk, M.; Van Scoy, S.; Kumar, K.P.; Reich, N.C. The Interferon Stimulated Gene 54 Promotes Apoptosis. J. Biol. Chem. 2011, 286, 7257–7266. [Google Scholar] [CrossRef]
- Reich, N.C. A Death-Promoting Role for ISG54/IFIT2. J. Interferon Cytokine Res. 2013, 33, 199–205. [Google Scholar] [CrossRef]
- Li, J.; Ding, S.C.; Cho, H.; Chung, B.C.; Gale, M.; Chanda, S.K.; Diamond, M.S. A Short Hairpin RNA Screen of Interferon-Stimulated Genes Identifies a Novel Negative Regulator of the Cellular Antiviral Response. mBio 2013, 4. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; De La Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol. 2018, 3, 1214–1223. [Google Scholar] [CrossRef]
- Yu, L.; Takeda, K.; Gao, Y. Characterization of virus-specific vesicles assembled by West Nile virus non-structural proteins. Virology 2017, 506, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.M.; Richner, J.M.; Diamond, M.S. The Interferon-Stimulated Gene Ifi27l2a Restricts West Nile Virus Infection and Pathogenesis in a Cell-Type- and Region-Specific Manner. J. Virol. 2016, 90, 2600–2615. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, S.; Atkins, C.; Bharaj, P.; Johnson, K.N.; Hage, A.; Freiberg, A.N.; Rajsbaum, R. VAMP8 Contributes to the TRIM6-Mediated Type I Interferon Antiviral Response during West Nile Virus Infection. J. Virol. 2019, 94. [Google Scholar] [CrossRef]
- Chiramel, A.I.; Meyerson, N.R.; McNally, K.L.; Broeckel, R.M.; Montoya, V.R.; Méndez-Solís, O.; Robertson, S.J.; Sturdevant, G.L.; Lubick, K.J.; Nair, V.; et al. TRIM5α Restricts Flavivirus Replication by Targeting the Viral Protease for Proteasomal Degradation. Cell Rep. 2019, 27, 3269–3283.e6. [Google Scholar] [CrossRef]
- Taylor, R.T.; Lubick, K.J.; Robertson, S.J.; Broughton, J.P.; Bloom, M.E.; Bresnahan, W.A.; Best, S.M. TRIM79α, an Interferon-Stimulated Gene Product, Restricts Tick-Borne Encephalitis Virus Replication by Degrading the Viral RNA Polymerase. Cell Host Microbe 2011, 10, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Valdez, F.; Salvador, J.; Palermo, P.M.; Mohl, J.E.; Hanley, K.A.; Watts, D.; Llano, M. Schlafen 11 Restricts Flavivirus Replication. J. Virol. 2019, 93, 19. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Deng, X.-Y.; Li, Y.-S.; Ma, X.-C.; Feng, J.-X.; Yu, B.; Chen, Y.; Luo, Y.-L.; Wang, X.; Chen, M.-L.; et al. Structure of Schlafen13 reveals a new class of tRNA/rRNA- targeting RNase engaged in translational control. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Newton, K. RIPK1 and RIPK3: Critical regulators of inflammation and cell death. Trends Cell Biol. 2015, 25, 347–353. [Google Scholar] [CrossRef]
- Moriwaki, K.; Chan, F.K.-M. The Inflammatory Signal Adaptor RIPK3: Functions Beyond Necroptosis. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 328, pp. 253–275. ISBN 978-0-12-812220-4. [Google Scholar]
- Daniels, B.P.; Snyder, A.G.; Olsen, T.M.; Orozco, S.; Oguin, T.H.; Tait, S.W.G.; Martinez, J.; Gale, M.; Loo, Y.-M.; Oberst, A. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell 2017, 169, 301–313.e11. [Google Scholar] [CrossRef]
- Daniels, B.P.; Kofman, S.B.; Smith, J.R.; Norris, G.T.; Snyder, A.G.; Kolb, J.P.; Gao, X.; Locasale, J.W.; Martinez, J.; Gale, M.; et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity 2019, 50, 64–76.e4. [Google Scholar] [CrossRef] [PubMed]
- Bian, P.; Ye, C.; Zheng, X.; Luo, C.; Yang, J.; Li, M.; Wang, Y.; Yang, J.; Zhou, Y.; Zhang, F.; et al. RIPK3 Promotes JEV Replication in Neurons via Downregulation of IFI44L. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. Corrigendum: A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2015, 505. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Olagnier, D.; Lin, R. Host and Viral Modulation of RIG-I-Mediated Antiviral Immunity. Front. Immunol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Den Boon, J.A.; Ahlquist, P. Organelle-Like Membrane Compartmentalization of Positive-Strand RNA Virus Replication Factories. Annu. Rev. Microbiol. 2010, 64, 241–256. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, B.; Shi, P.-Y. Flavivirus methyltransferase: A novel antiviral target. Antivir. Res. 2008, 80, 1–10. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.-Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Habjan, M.; Hubel, P.; Lacerda, L.; Benda, C.; Holze, C.; Eberl, C.H.; Mann, A.; Kindler, E.; Gil-Cruz, C.; Ziebuhr, J.; et al. Sequestration by IFIT1 Impairs Translation of 2′O-unmethylated Capped RNA. PLoS Pathog. 2013, 9, e1003663. [Google Scholar] [CrossRef]
- Chung, K.M.; Liszewski, M.K.; Nybakken, G.; Davis, A.E.; Townsend, R.R.; Fremont, D.H.; Atkinson, J.P.; Diamond, M.S. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc. Natl Acad. Sci. USA 2006, 103, 19111–19116. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Dutta, S.; Konwerski, J.; Jose, J.; Jurkiw, T.J.; DelProposto, J.; Ogata, C.M.; Skiniotis, G.; Kuhn, R.J.; et al. Flavivirus NS1 Structures Reveal Surfaces for Associations with Membranes and the Immune System. Science 2014, 343, 881–885. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Ye, H.-Q.; Liu, S.-Q.; Deng, C.-L.; Li, X.-D.; Shi, P.-Y.; Zhang, B. West Nile Virus NS1 Antagonizes Interferon Beta Production by Targeting RIG-I and MDA5. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; de Sessions, P.F.; Leon, M.A.; Scholle, F. West Nile Virus Nonstructural Protein 1 Inhibits TLR3 Signal Transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Gack, M.U. A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 2016, 17, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Chen, H.B.; Wang, X.J.; Huang, H.; Khromykh, A.A. Analysis of Adaptive Mutations in Kunjin Virus Replicon RNA Reveals a Novel Role for the Flavivirus Nonstructural Protein NS2A in Inhibition of Beta Interferon Promoter-Driven Transcription. J. Virol. 2004, 78, 12225–12235. [Google Scholar] [CrossRef]
- Dalrymple, N.A.; Cimica, V.; Mackow, E.R. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. mBio 2015, 6. [Google Scholar] [CrossRef]
- Arjona, A.; Ledizet, M.; Anthony, K.; Bonafé, N.; Modis, Y.; Town, T.; Fikrig, E. West Nile Virus Envelope Protein Inhibits dsRNA-Induced Innate Immune Responses. J. Immunol. 2007, 179, 8403–8409. [Google Scholar] [CrossRef]
- Schuessler, A.; Funk, A.; Lazear, H.M.; Cooper, D.A.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile Virus Noncoding Subgenomic RNA Contributes to Viral Evasion of the Type I Interferon-Mediated Antiviral Response. J. Virol. 2012, 86, 5708–5718. [Google Scholar] [CrossRef]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.-Y.; Hall, R.A.; et al. A Highly Structured, Nuclease-Resistant, Noncoding RNA Produced by Flaviviruses Is Required for Pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef]
- Zhang, A.P.P.; Bornholdt, Z.A.; Liu, T.; Abelson, D.M.; Lee, D.E.; Li, S.; Woods, V.L.; Saphire, E.O. The Ebola Virus Interferon Antagonist VP24 Directly Binds STAT1 and Has a Novel, Pyramidal Fold. PLoS Pathog. 2012, 8, e1002550. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 Protein of the Virulent West Nile Virus NY99 Strain Is a Potent Antagonist of Type I Interferon-Mediated JAK-STAT Signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-T.; Hayashi, J.; Seeger, C. West Nile Virus Inhibits the Signal Transduction Pathway of Alpha Interferon. J. Virol. 2005, 79, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale, M. Resistance to Alpha/Beta Interferon Is a Determinant of West Nile Virus Replication Fitness and Virulence. J. Virol. 2006, 80, 9424–9434. [Google Scholar] [CrossRef]
- Evans, J.D.; Seeger, C. Differential Effects of Mutations in NS4B on West Nile Virus Replication and Inhibition of Interferon Signaling. J. Virol. 2007, 81, 11809–11816. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.L.; Johnson, N.; Cosby, S.L.; Solomon, T.; Fooks, A.R. Transcriptional Upregulation of SOCS 1 and Suppressors of Cytokine Signaling 3 mRNA in the Absence of Suppressors of Cytokine Signaling 2 mRNA After Infection with West Nile Virus or Tick-Borne Encephalitis Virus. Vector-Borne Zoonotic Dis. 2010, 10, 649–653. [Google Scholar] [CrossRef]
- Evans, J.D.; Crown, R.A.; Sohn, J.A.; Seeger, C. West Nile Virus Infection Induces Depletion of IFNAR1 Protein Levels. Viral Immunol. 2011, 24, 253–263. [Google Scholar] [CrossRef]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.-Y.; Randall, R.; Khromykh, A.A. Inhibition of Interferon Signaling by the New York 99 Strain and Kunjin Subtype of West Nile Virus Involves Blockage of STAT1 and STAT2 Activation by Nonstructural Proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar] [CrossRef]
- Roby, J.A.; Esser-Nobis, K.; Dewey-Verstelle, E.C.; Fairgrieve, M.R.; Schwerk, J.; Lu, A.Y.; Soveg, F.W.; Hemann, E.A.; Hatfield, L.D.; Keller, B.C.; et al. Flavivirus Nonstructural Protein NS5 Dysregulates HSP90 to Broadly Inhibit JAK/STAT Signaling. Cells 2020, 9, 899. [Google Scholar] [CrossRef]
- Suthar, M.S.; Brassil, M.M.; Blahnik, G.; Gale, M. Infectious Clones of Novel Lineage 1 and Lineage 2 West Nile Virus Strains WNV-TX02 and WNV-Madagascar. J. Virol. 2012, 86, 7704–7709. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Restriction Factor | Affected Step | Mechanism | Model | References |
|---|---|---|---|---|
| PKR | Translation | Phosphorylation of the translation initiation factor EIF2a | Human cells & Mice | [135,137] |
| Viperin | Assembly Secretion | Catalyzes the formation of chain terminators for flaviviral RNA polymerases | Human cells & Mice | [137,139,142,144] |
| Oas1b | Sensing | Catalyzes the formation of 2′-5′-oligos / Belongs to an antiviral complex | Mice | [147,151,152,155,157] |
| IFITM2 | Entry | Inhibits viral fusion with host membranes | Human cells | [136] |
| IFITM3 | Entry | Inhibits viral fusion with host membranes | Human cells & Mice | [136,159,169,174] |
| ISG15 | Replication | Up-regulation of the IFN response | Human & mice cells | [136,181,184] |
| ISG20 | Replication, translation | Viral RNA degradation, translation inhibition, IFN response up-regulation | Human cells & mice | [182,183,184,185] |
| IFITs | Translation | Recognition of the 5′-end 2′-O-unmethylated RNA and 5′-PPP RNA / Binds eIF3 / Competes with eIF4E | Mice | [190,192,199,200,201] |
| IFI27l2a | Unknown | Unknown | Mice | [210] |
| TRIM6/VAMP8 | Unknown | Regulation of the IFN response | Human cells | [211] |
| Schlaffen 11 | Translation | Regulation of tRNA abundance | Human cells | [215] |
| IFI6 | Unknown | Interferes with the membrane of replication organelles | Human cells | [206,207,208] |
| DDX24 | Unknown | Unknown | Human cells | [206] |
| IFI44L | Unknown | Unknown | Human cells | [206] |
| IFRD1 | Unknown | Unknown | Human cells | [206] |
| IL13RA1 | Unknown | Unknown | Human cells | [206] |
| MAFK | Unknown | Unknown | Human cells | [206] |
| PAK3 | Unknown | Unknown | Human cells | [206] |
| SAMD9L | Unknown | Unknown | Human cells | [206] |
| SC4MOL | Unknown | Unknown | Human cells | [206] |
| RIPK3 | Unknown | Promotes inflammatory chemokines expression | Mice | [219] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, M.-F.; Nisole, S. West Nile Virus Restriction in Mosquito and Human Cells: A Virus under Confinement. Vaccines 2020, 8, 256. https://doi.org/10.3390/vaccines8020256
Martin M-F, Nisole S. West Nile Virus Restriction in Mosquito and Human Cells: A Virus under Confinement. Vaccines. 2020; 8(2):256. https://doi.org/10.3390/vaccines8020256
Chicago/Turabian StyleMartin, Marie-France, and Sébastien Nisole. 2020. "West Nile Virus Restriction in Mosquito and Human Cells: A Virus under Confinement" Vaccines 8, no. 2: 256. https://doi.org/10.3390/vaccines8020256
APA StyleMartin, M.-F., & Nisole, S. (2020). West Nile Virus Restriction in Mosquito and Human Cells: A Virus under Confinement. Vaccines, 8(2), 256. https://doi.org/10.3390/vaccines8020256