1. Introduction
In India, Newcastle disease (ND) and infectious bursal disease (IBD) have been a constant threat to the poultry industry [
1,
2] which causes heavy economic losses in terms of mortality and immunosuppression in young chicks due to ND and IBD, respectively. Infections caused by Newcastle disease virus (NDV) may exacerbate infections with other etiological agents, and reduce the chicken’s ability to respond to vaccination. ND is a highly contagious and fatal viral disease affecting all species of birds, and so does IBD which is an immunosuppressive disease of immature chickens caused by a birnavirus, infectious bursal disease virus (IBDV). The actively dividing and differentiating lymphocytes of the B-cell lineage of the bursa of Fabricius of particularly young chickens get affected, resulting in morbidity, mortality and immune-suppression along with ineffective responses to vaccines. IBDV belongs to the Avibirnavirus genus of the Birnaviridae family, and has non-enveloped, icosahedral capsid consisting of double-stranded RNA segments, namely segment A and B [
3]. The larger ORF1 of segment A encodes for a 110-kDa polyprotein which auto-catalytically splices into mature viral proteins; VP2 (48 kDa), VP3 (33–35 kDa) and VP4 (24 kDa) [
4]. VP2 is the major capsid protein that carries the immunogenic determinants and the major host-protective antigen, able to elicit neutralizing antibodies [
5]. Very virulent strain of IBDV (vvIBDV) has emerged in the late 1980’s [
6] and the virus is continuously evolving in the field with changes in antigenicity and virulence. Currently, the disease is controlled by live attenuated or inactivated IBDV vaccines which may not give complete protection against vvIBDV strain [
7]. The inactivated or killed viruses are usually given to birds in pre-laying stage to induce higher levels of antibody production for at least 2 weeks. The commercially available live vaccines based on classical virulent strains induce both cellular and humoral immunity but may also show necrosis and lymphoid depletion. They may show reversion to virulence and vaccine associated reactions, resulting in clinical disease and production losses. Non-replicating antigens such as inactivated whole viruses, viral subunits or recombinant viral antigens are not efficacious unless combined with adjuvants and administered repeatedly [
8].
Newcastle disease virus (NDV), an avian paramyxovirus-1 is being developed as a vaccine vector candidate. NDV, strain F is a virus of low virulence originally reported in England [
9]. Since then, in several countries in Europe, Africa and Asia, the use of this virus as an immunizing agent in the form of a live vaccine has been studied. However, the complete genome of this virus was not determined to be made amenable for its usage as a vector. Therefore, we elucidated the complete genome sequence of this NDV strain F [
10], which allowed us to generate NDV in the laboratory from cDNAs with predefined genetic markers by reverse genetics technology. In this study, for the first time we have developed a reverse genetics system exclusively for the NDV vaccine strain F, incorporating the major capsid protein gene VP2 of a very virulent IBDV to generate a bivalent vaccine candidate. Previously, two bivalent vaccine candidates, utilizing NDV backbone with LaSota strain and a chimeric NDV LaSota virus with the L gene of clone-30, were generated expressing the VP2 gene of IBDV. The investigators reported that VP2 protein was not incorporated into the virions of recombinant virus [
11] and only the humoral immune responses in susceptible birds were evaluated in protection against these diseases [
11,
12]. Moreover, none of these investigators could unequivocally show the expression of VP2 protein by Western blot analysis using purified recombinant NDV and demonstrate protective cell mediated immune response. Therefore to overcome these pitfalls, we added the hemagglutinin (HN) signal sequence at the N-terminal of VP2 and generated a recombinant rNDV-F/VP2 virus, using reverse genetics. In this study, we evaluated the efficacy of NDV-vectored IBDV vaccine in affording protection against both NDV and vvIBDV challenges and the vaccine candidate induced both humoral and cellular immune responses.
2. Material and Methods
2.1. Virus and Cells
Very virulent IBDV strain, available at Indian Veterinary Research Institute, India was grown in eleven-days-old specific-pathogen-free (SPF) embryonated chicken eggs inoculated through the chorio-allantoic membrane (CAM) route. The plaque purified F strain seed virus, available with the viral repository of Indian Veterinary Research Institute, Izatnagar was propagated in eleven-days-old SPF embryonated chicken eggs via the allantoic route. The viral antigen was prepared and purified by ultracentrifugation procedure using sucrose gradient, as described [
13]. Vero cells were used for virus recovery and propagation. The cells were grown at 37 °C in the minimum essential medium containing 10% fetal bovine serum. Exogenous trypsin in the form of 10% allantoic fluid was added to Vero cells for viral rescue and subsequent passages of the virus in Vero cells.
2.2. Construction of Full-Length Clone of NDV, Strain F
Viral RNA was extracted from the purified virus using TRIzol (Sigma, St. Louis, MO, USA), according to manufacturer’s instructions. Reverse transcription was carried out with the extracted RNA using the Superscript RT kit (Invitrogen, Carlsbad, CA, USA) to synthesize the first strand cDNA. Oligonucleotide primers were synthesized for amplifying the entire genome of NDV-F virus as overlapping fragments and the complete genome was elucidated [
10] and deposited in the GenBank (Accession No. KC987036.1). A full-length cDNA copy of the RNA genome was constructed by assembling three overlapping synthetic fragments (GenScript, Piscataway, NJ, USA) by standard cloning techniques. The fragments were ligated serially by natural or artificially created unique restriction sites. Two ribozyme sites were incorporated at both the ends of the full-length clone of the virus, namely hammerhead ribozyme (HHRz) at the 5′ end and hepatitis delta virus ribozyme (HdvRz) at the 3′ end. The HHRz sequence (5′-CTGATGAGTCCGTGAGGACGAAACTATAGGAAAGGAATTCCTATAGTC-3′) and HdvRzsequence (5′GGGTCGGCATGGCATCTCCACCTCCTCGCGGTCCGACCTGGGCATCCGAAGGAGGACAGACGTCCACTCGGATGGCTAAGGGAGAGCCA-3′) were fused with the first and third fragment by overlapping PCR [
14].
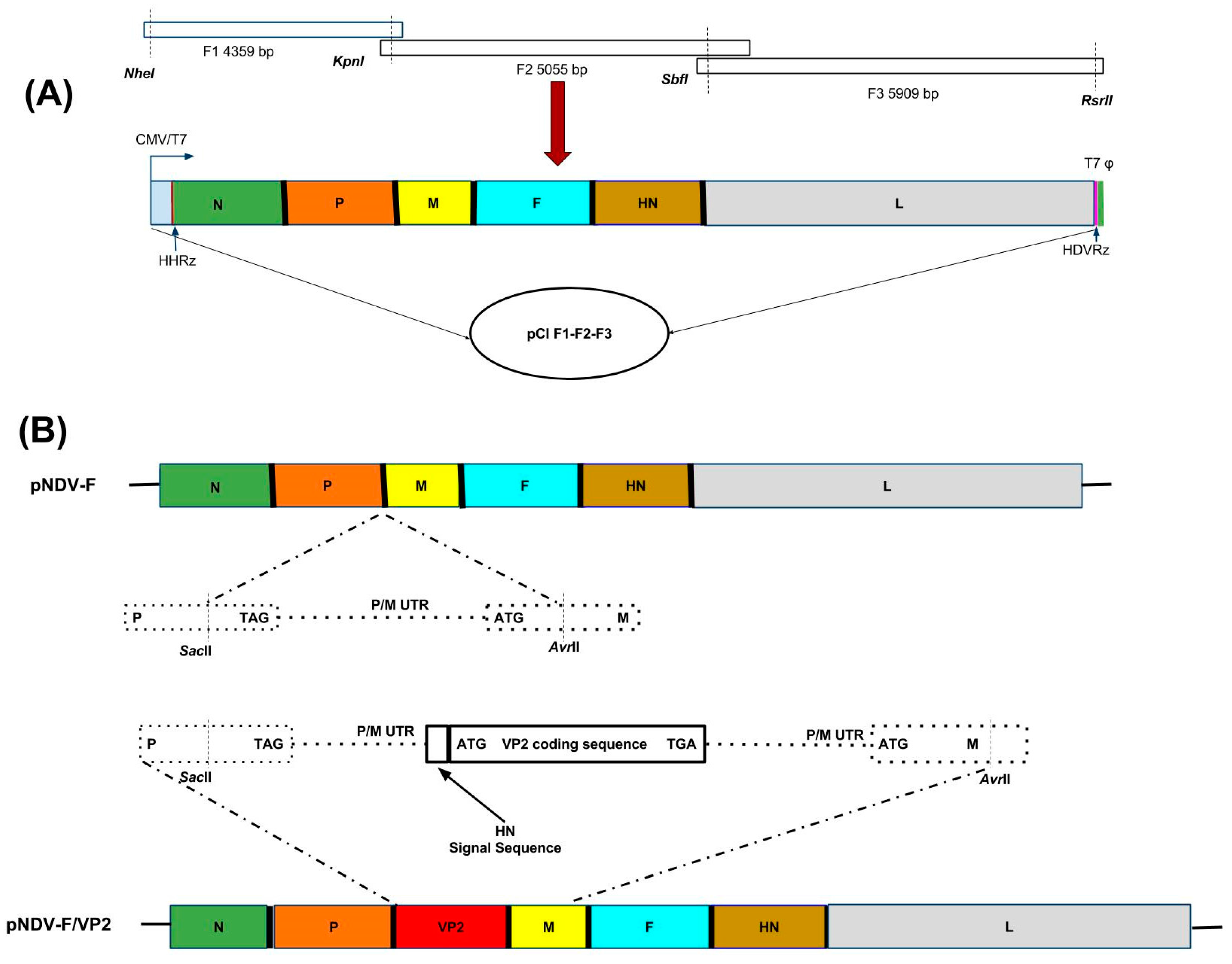
Three plasmids, namely F1, F2 and F3 with a size of 4359 bp, 5055 bp and 5909 bp, respectively and representing the entire genome complement of NDV strain ‘F’, were synthesized in pUC57 based vector having the NDV sequence (GenScript, USA). The restriction enzyme sites SacII (1661 bp position), AvrII (1736 bp position), RsrII (2662 bp position), NheI (10,168 bp and 14,530 bp positions), and AvrII (14,866 bp position) were deleted in the F1, F2 and F3 synthesized fragments. To assemble the full length clone, the F1 fragment was double digested with NheI and KpnI restriction enzymes and cloned between NheI and KpnI sites of the modified pCI vector harboring the HHRz and HdvRz sequences [
15]. Second, the F2 fragment was double digested with KpnI and RsrII restriction enzymes and cloned in the F1 pCI vector between KpnI site of F1 and RsrII sites of the Hdv sequence and named as F1-F2 pCI vector. Third, the F3 fragment was double digested with SbfI and RsrII enzymes and cloned into F1-F2 pCI vector and named as pCI F1-F2-F3. This entire cassette F1-F2-F3 inserted into the modified mammalian expression vector pCI (Promega, Madison, WI, USA) (
Figure 1A) was under the control of the cytomegalovirus promoter [
15].
The IBDV VP2 gene cassette was synthesized by incorporating the haemagglutinin neuraminidase (HN) signal sequence (141 bp) of NDV to the 5′-end of the VP2 sequence (1326 bp) and the cassette is flanked by the restriction sites SacII and AvrII; NDV P gene coding (cds); P gene non-coding (NC); intergenic sequence (IGS); M gene NC; and P gene NC; IGS; M gene NC; M cds. A termination codon (TGA) was placed at the end of the VP2 gene. Four additional nucleotides TAAA were added after the termination codon to maintain the “rule of six”. The amplified fragment was digested with SacII and AvrII enzymes, then inserted into the P–M intergenic region at nucleotide position 2354 and 5251 of the NDV genome (
Figure 1B). The generated plasmid was designated as pNDV-F/VP2.
2.3. Construction of Supporting Plasmids
Using the full-length clone of pNDV-F/VP2 as template, the open reading frames (ORFs) of NP (1469 bp), P (1187 bp), and L (6614 bp) genes were amplified by PCR. A Kozak consensus sequence, GCCACC, was incorporated in front of the start codon of each ORF [
16]. The NP and P ORFs were cloned between EcoRI and NotI restriction sites; whereas L ORF was cloned between NheI and NotI restriction sites of the pCI vector (Promega, USA) by digesting with appropriate restriction enzymes. The pNP, pP, and pL support plasmids were sequenced using an automated DNA Sequencer, as described earlier. All these plasmids were under the control of cytomegalovirus promoter (CMV).
2.4. Rescue of Recombinant NDV-F/VP2 Virus in Vero Cells
Transfection was done using the full-length clone of the virus along with the support plasmids using standard protocols. Briefly, the Vero cells were grown upto 80% confluency before transfection. The media was removed from the cells and the cells were incubated with OptiMEM media for 1 h. The plasmids pNDV-F and/or pNDV-F/VP2 (5 µg), pCI-NP (2.5 µg), pCI-P (1.5 µg) and pCI-L (0.5 µg) were diluted in 500 µL OptiMEM medium to which Lipofectamine LTX reagent (Invitrogen, USA) was added and incubated for 30 min. The OptiMEM media was removed from the Vero cells and the plasmid-lipofectamine mixture was added onto the cells in a drop wise manner. One mL of OptiMEM was added to the cells and was left undisturbed at 37 °C incubator with 5% CO2 for 24 h. After 24 h, the transfected mixture was removed and the cells were incubated with M199 medium (Life Technologies, Carlsbad, CA, USA) with 10% allantoic fluid obtained from SPF embryonated chicken eggs. The cells were left undisturbed for 72 h. The cells were then freeze-thawed thrice and re-infected onto healthy Vero cells. The process was repeated thrice.
2.5. Confirmation of Virus Stability
The recombinant viruses namely, rNDV-F and rNDV-F/VP2 were passaged serially in SPF embryonated chicken eggs ten times and was analyzed for its stability. RNA from the recombinant virus present in the allantoic fluid was isolated by TRIzol reagent according to standard protocols. A reverse transcription polymerase chain reaction (RT-PCR) was set up with different sets of primers to confirm the presence of different regions of NDV and the VP2 gene of IBDV.
To confirm the presence of the IBDV VP2 gene cassette in the full-length NDV genome, RT-PCR was done using the forward primer (nt position 3064 at P cds of the viral genome) and the reverse primer with respect to the 3′-end of VP2 gene (1693 bp). The PCR product was cloned into a T/A cloning vector and the plasmid DNA was sequenced.
2.6. Characterization of Recombinant Viruses
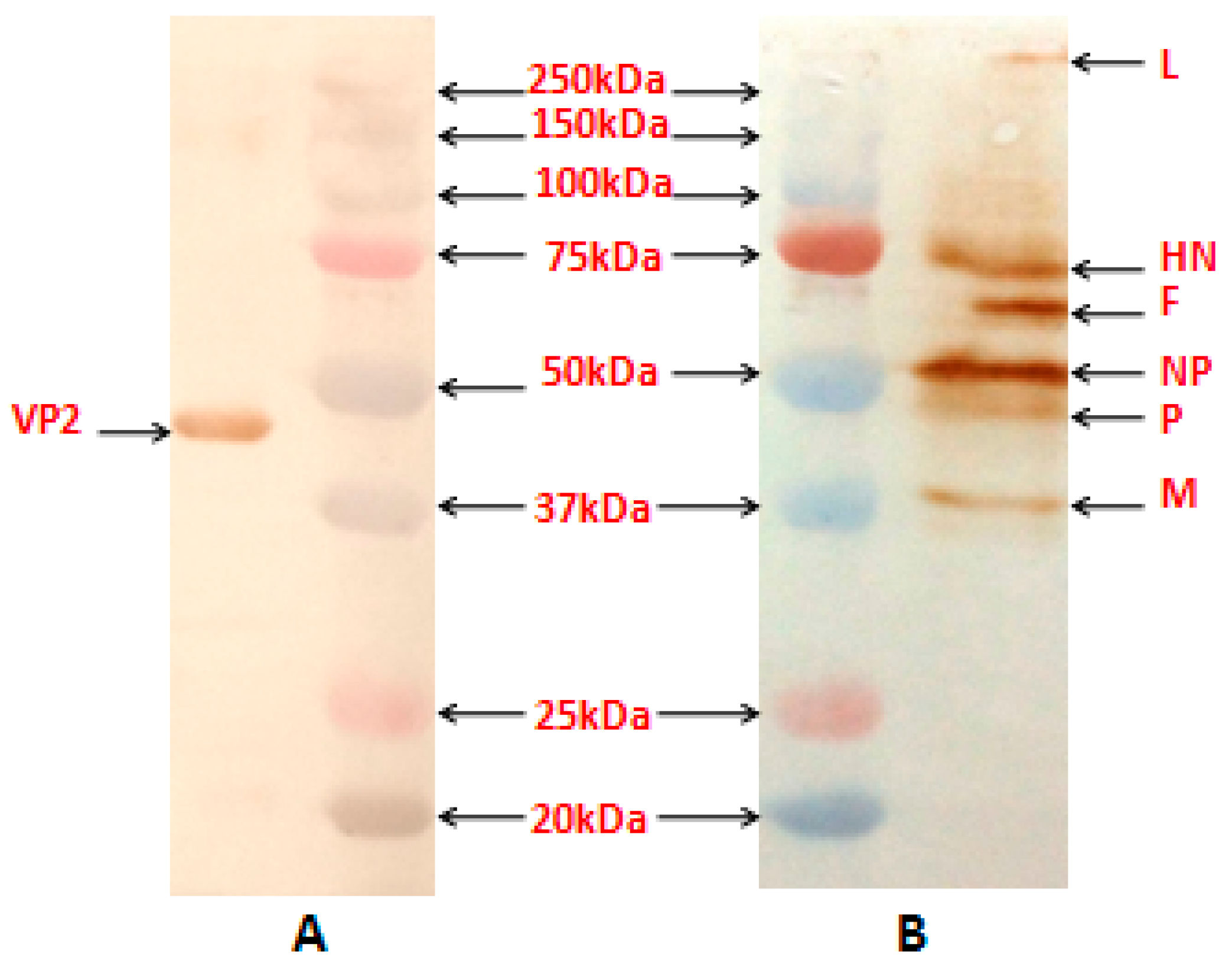
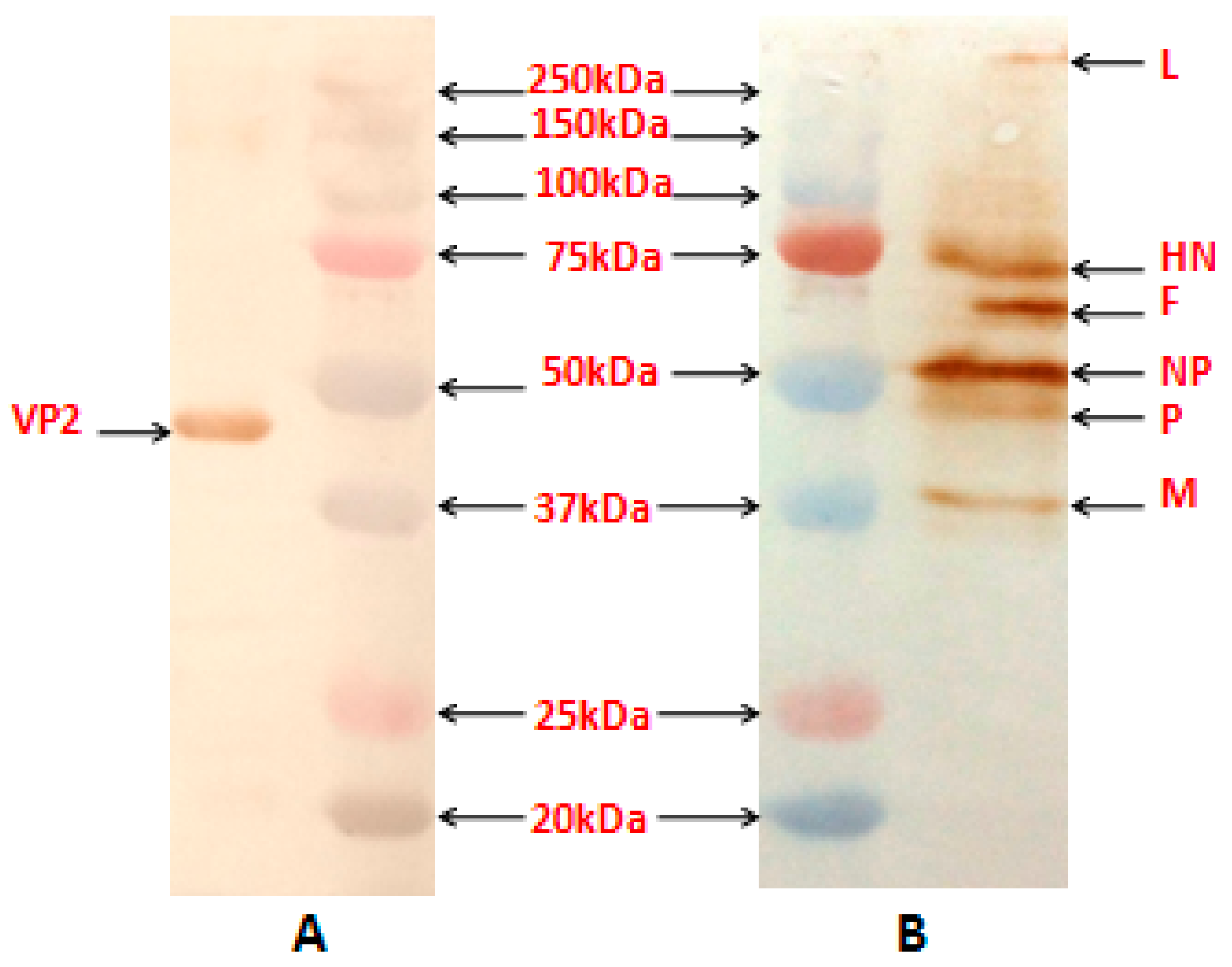
The reactivity of the NDV proteins and the VP2 protein in the purified recombinant virus, obtained by sucrose gradient centrifugation procedure [
13], was assessed using anti-NDV antibody (Abcam, Cambridge, MA, USA) and mouse anti-VP2 monoclonal antibody (Abcam, USA), respectively by Western blotting.
Expression of the VP2 protein in rNDV-F/VP2 virus was examined by IFA using IBDV-specific monoclonal antibody (mAb) against the VP2 protein (Abcam, USA; 1:100) and an NDV-specific polyclonal antibody (Abcam, USA; 1:50). Briefly, confluent monolayers of Vero cells were infected with recombinant viruses at a multiplicity of infection (MOI) of 0.01. After 24 h, the infected cells and control cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (Affymetrix Inc., Cleveland, OH, USA) for 1 h at room temperature, followed by addition of 0.5% Triton X-100 (Sigma, USA) to permeabilize the cells at room temperature for 10 min. The permeabilized cells were blocked with 5% bovine serum albumin (Sigma, USA) for 30 min at 37 °C. After blocking, the cells were incubated for 1 h with a mixture of antibodies. Cells were washed with PBS and incubated with a mixture of the FITC labeled sheep anti-mouse IgG (Sigma, USA, 1:250 dilution) and Alexa Fluor(R) 568-labeled goat anti-chicken IgG (Thermo Fisher Scientific, Carlsbad, CA, USA, 1:200 dilutions) for 1 h at 37 °C. Nuclear staining was carried out using DAPI (Thermo Fisher Scientific, USA, 1:200 dilutions). Fluorescence images were photographed using a FluoViewFV1000 confocal microscope (Olympus, Waltham, MA, USA) with matching excitation/emission filters for FITC and Alexa Fluor 568.
The recombinant viruses were further characterized by inoculating eleven-days-old SPF embryonated chicken eggs and incubating at 37 °C. The embryos were observed for 96 h and at the end of incubation period, the lesions were evaluated.
The growth kinetics of the parent and the recombinant virus in tissue culture was analyzed using a multistep growth curve analysis. Monolayer of Vero cells (in triplicate) were infected with each virus at a multiplicity of infection (MOI) of 0.01 and supernatant was collected and replaced with an equal volume of fresh medium at 12 h interval until 72 h. The viral titers of these samples were determined by Reed and Muench method [
17].
2.7. Titration of Recombinant NDV-F Virus by Real Time PCR
A fragment of 113 bp of F gene of NDV was amplified using the primer pairs (Forward: 5′- TAA GCT CCT CCC GAA TCT-3′ at position 4732–4749 and reverse: 5′-TAC GGA TAG AGT CAC CAA GG-3′ at position 4825–4844 of the NDV viral genome) and the PCR product was cloned in the T/A vector. The OD value of the recombinant plasmid DNA concentrations was measured at 260 nm/280 nm (600 ng/µL) and plasmid copy number was calculated using the formula described by Adams, 2006. The standard plasmid DNA was diluted 10-fold serially from 10−1 to 10−12 and was used for making the standard curve along with the sample cDNA. The real-time PCR was performed on CFX96 real time system (Bio-Rad, Hercules, CA, USA) in duplicate for each sample. The reaction mixture was prepared in a total volume of 20 µL consisting of 2 µL cDNA, 10 µL of QuantiTect SYBR Green Master Mix (Qiagen, Hilden, Germany), and primers 0.5 µL each. Real time PCR was carried out with the following program: 1 cycle at 95 °C for 5 min, followed by 40 cycles of 94 °C for 30 s, 60 °C for 45 s, 70 °C for 45 s and 1 cycle of 94 °C for 30 s. The final step was to obtain a melt curve for the PCR products to determine the specificity of the amplicons.
2.8. Biological Characterization of the Recombinant Viruses
The recombinant viruses were characterized biologically by mean death time (MDT) and intra cerebral pathogenicity index (ICPI) analysis. The MDT analysis was carried out in eleven-days-old embryonated chicken eggs and ICPI was carried out in one-day-old SPF chickens, according to standard procedures [
18].
2.9. Vaccine Efficacy Testing in Chickens
2.9.1. Dose Optimization and Safety Test
A total of 42 day-old SPF chicks were randomly allocated to seven groups. The first group was kept as control; the remaining groups were vaccinated intranasally with rNDV-F and rNDV-F/VP2 recombinant viruses at different doses viz., 105 EID50/bird, 106 EID50/bird and 107 EID50/bird, respectively. Blood was collected for serum analysis from all the birds at days 0, 7, 14, 21 days post-immunization. The antibody response was checked by ELISA and cellular immune response was checked by antigen specific lympho-proliferative assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye. Following dose optimization, a 10-fold dose of the optimized dose (107 EID50/bird) of recombinant viruses were given intranasally to 10 SPF chickens and observed for 21 days for any abnormality.
2.9.2. Immunization Trial
The immunization and protection studies of the recombinant viruses were evaluated in SPF chicks maintained under standard management practices at biosafety level 2 where chicks were provided with autoclaved feed and water. A total of ninety SPF day-old chicks were distributed randomly in 7 groups as follows: unvaccinated (no challenge), unvaccinated (IBDV and NDV challenge, 10 birds in each group), rNDV-F/VP2 virus (20 birds), rNDV-F virus (10 birds), commercially available NDV ‘F’ (RDF-Ranikhet disease ‘F’ strain), killed and live IBDV vaccines (10 birds in each group). At one day of age, the birds were vaccinated with rNDV-F/VP2 virus and rNDV-F virus through the intra-nasal route at a dose of 106 EID50/bird and the same dose was repeated two weeks post primary vaccination;RDF, killed and live IBDV vaccines were vaccinated with the recommended dose and route. The unvaccinated groups were given PBS. Two weeks following booster immunization, 10 chickens in each group were subdivided into two factions: control, rNDV-F/VP2 virus, rNDV-F virus and commercially available RDF groups, which were challenged with 105 ELD50 virulent NDV through intramuscular route (i/m), and the second faction: control, rNDV-F/VP2 virus, killed and live IBDV vaccines groups that were challenged with 103 ELD50 very virulent IBDV strain through intra-ocular route. All birds were observed daily for two weeks for any clinical signs and mortality.
2.9.3. Determination of Protection from Challenge
Protection against IBDV challenge was assessed by studying the occurrence of mortality, presence of viral antigen in bursa four days post-challenge as determined by agar gel precipitation test, bursal gross lesions, and bursa to body weight ratio (bursa/body weight B/B%) ten days post-challenge. Clinical signs and mortality were observed up to a period of 14 days post-challenge. The dead and surviving chickens were sacrificed and bursa of Fabricius was collected for gross and histological examinations with haematoxylin and eosin (H&E) staining. The bursae from all the groups were examined histologically to determine the severity of bursal damage. The bursal lesions were graded according to a scale of 0 to 4: (0) no lesions; (1) mild scattered cell depletion in a few follicles; (2) moderate with 1/31/2 of the follicle atrophied or with depleted cells; (3) diffuse with atrophy in all follicles; and (4) acute inflammation and acute necrosis, typical of IBDV infection. Protection against NDV was assessed for two weeks post-challenge for the presence of clinical signs and mortality.
2.9.4. Determination of Humoral Immune Response by ELISA and HI Assay
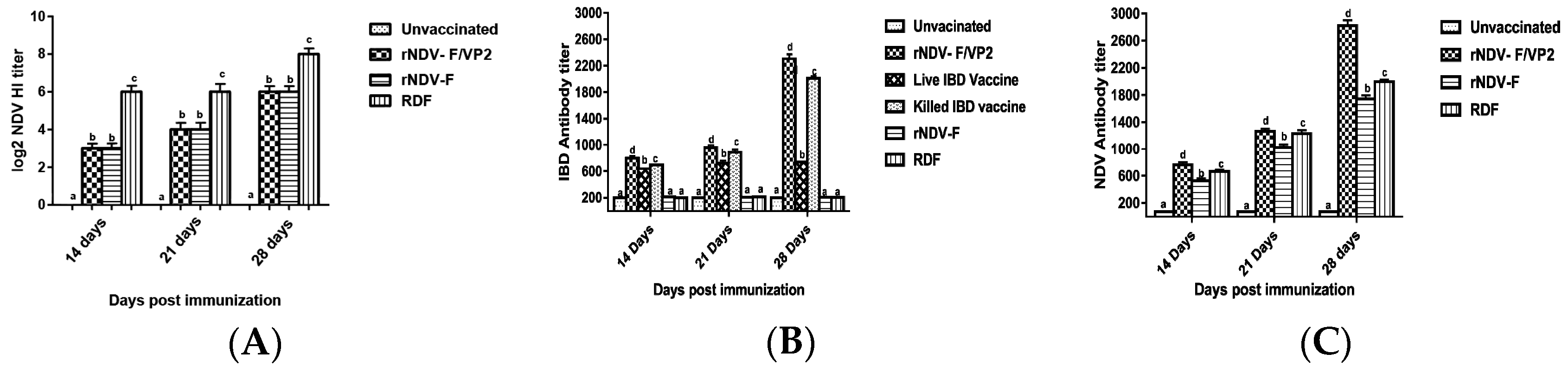
Briefly, 96-well ELISA plates were coated with recombinant VP2 antigen of IBDV or recombinant nucleoprotein (NP) antigen of NDV, diluted in the coating buffer (100 mM bicarbonate buffer, pH 9.0) at 4 °C overnight and blocked with 2% bovine serum albumin (BSA). The sera samples, diluted 1:200 in PBS were added on to the plate and incubated at 37 °C for 1 h. The relative antibody titers were calculated by sample to positive (S/P) ratio and end point titer was determined, as described previously [
19]. Serum samples with an end point titer of more than 500 and 200 were considered positive for IBD and ND, respectively. HI was performed to analyze the NDV antibody titers as per standard protocol.
2.10. Determination of Cellular Immune Response
2.10.1. Lymphocyte Proliferation Assay
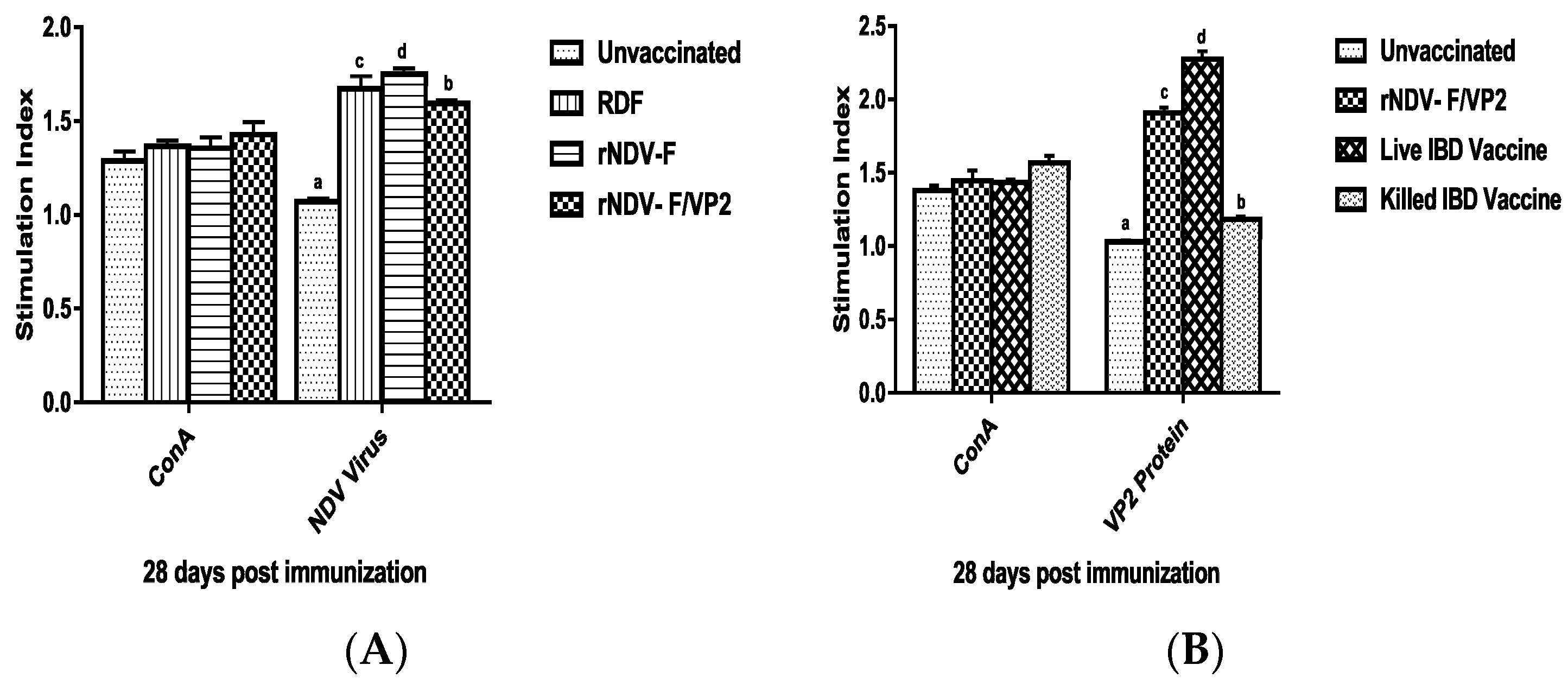
At 28 days post immunization (dpi), peripheral blood mononuclear cells (PBMCs) were collected from both vaccinated and unvaccinated control groups (n = 6) by using ficoll-hypaque (specific gravity 1.077 g/mL) (Sigma, St. Louis, MO, USA). The viability of cells was checked by trypan blue dye exclusion assay. The cell number was adjusted to be at 1 × 107 cells/mL. One hundred microliters of medium with cells were added in triplicates to the cell culture plates and stimulated with recombinant VP2 protein at a concentration of 5 µg/mL, and virulent NDV at a concentration of 10 µg/mL; ConA (10 µg/mL) was used as positive control along with non-stimulated control. The plates were incubated at 37 °C in 5% CO2 atmosphere with 95% relative humidity. After 60 h, 20 µL of MTT dye (5 mg/mL) was added to all the wells and incubated for further 4 h. One hundred microliters of DMSO was added to all the wells and mixed to dissolve the formazan crystals. Spectrophotometric reading was taken at 570 nm and stimulation index (S.I) was calculated as the ratio between mean OD value of stimulated cells and the mean OD value of the non-stimulated control.
2.10.2. Flow Cytometric Analysis
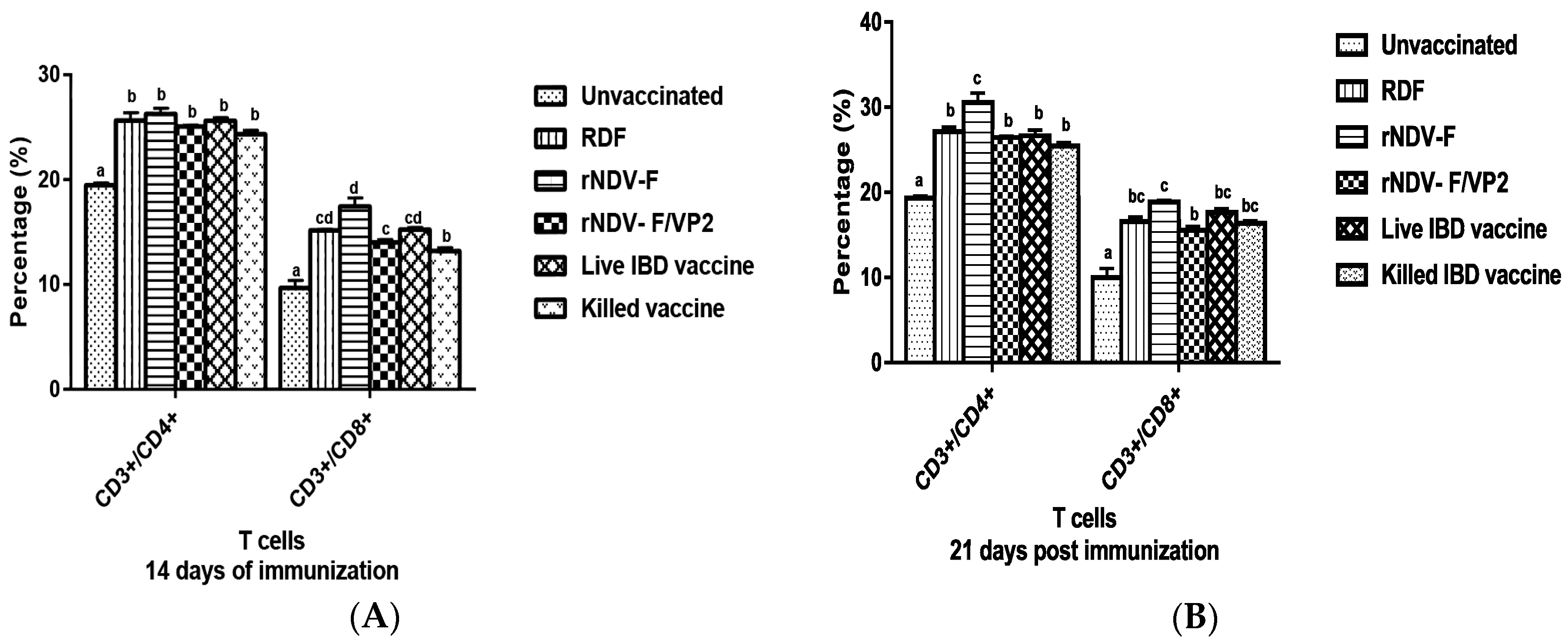
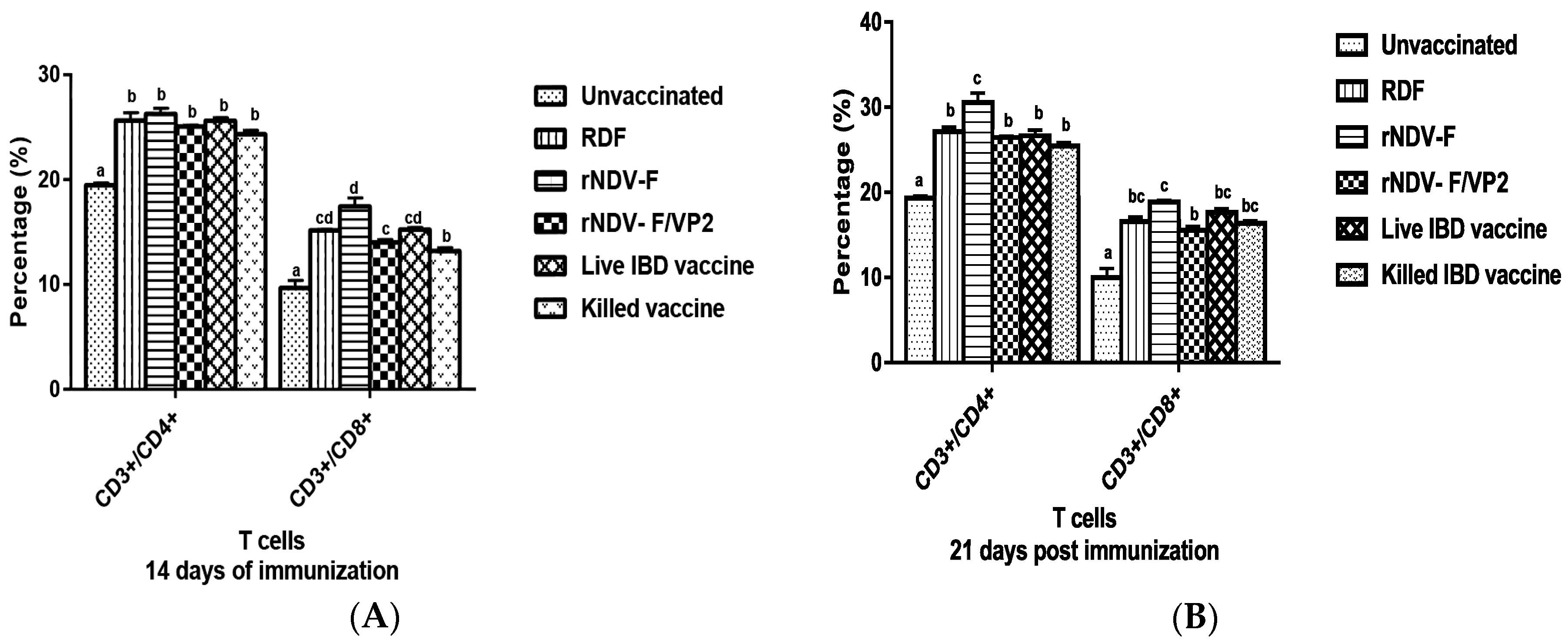
At 14 and 21 dpi, PBMCs were collected from the vaccinated and control groups (n = 6) and analyzed by flow cytometry. Briefly, 1 × 105 cells were taken in a microcentrifuge tube to which 1:10 dilution of anti-chicken CD3, CD4 and CD8 monoclonal antibodies (mAb) (Serotec, Oxford, UK) were added. The tubes were incubated in the dark at room temperature for 30 min. The cells were then washed twice with PBS to remove the unbound mAb molecules and analyzed in a FACS Calibur Flow Cytometer (BD Biosciences, San Jose, CA, USA) to estimate the percentage of CD4 and CD8 cells in the sample.
2.11. Statistical Analysis
SPSS 20.0 statistical software was used to analyze the results. One-way ANOVA was employed to evaluate the differences in the mean values of various parameters and Waller-Duncan as post-hoc test.
4. Discussion
In this study, we have generated a recombinant NDV-F virus (rNDV- F) with an incorporated IBDV VP2 gene from a very virulent IBDV strain into it (rNDV-F/VP2), using a low virulent NDV-F strain to be used as a vaccine vector. The rNDV-F/VP2 was evaluated as a bivalent vaccine candidate against ND and IBD in SPF chickens. Currently, lentogenic NDV strains Hitchner B1, Lasota, F and mesogenic R2B strains are widely used as live vaccines in India [
10]. The strain F vaccine is usually used in young chickens but is also suitable for use as a vaccine in chickens of all ages. As a viral vector, NDV strain LaSota has been widely used to express glycoproteins of avian metapneumovirus subgroup C [
20], glycoprotein gB and gD genes of infectious laryngotracheitis [
21,
22], HA of H5 avian influenza [
23], VP2 gene of IBDV [
11,
12]. Besides poultry viral diseases, LaSota as a viral vector has been used to express rabies glycoprotein [
24], haemagglutinin (H) or fusion protein (F) of canine distemper [
25]. In this study, we have reported for the first time generation of a recombinant NDV ‘F’ strain viral vector expressing the VP2 capsid gene of vvIBDV, which has the ability to induce both the arms of immune responses. It has been reported earlier that the LaSota strain often causes post-vaccination respiratory signs and it could be used as a booster vaccine in flocks vaccinated with F or B1 [
26]. However, the recombinant virus generated with NDV strain F with IBDV VP2 was found to be safe, stable and able to show protective efficacy against virulent NDV and very virulent IBDV challenges. In most of the earlier reports, researchers have utilized glycoproteins as foreign antigens to be expressed from NDV viral vector. VP2 protein of IBDV is a non-glycosylated capsid protein of IBDV. The absence of appropriate signal in VP2 protein to direct them to golgi apparatus prompted us to utilize the HN signal from NDV to be fused with the VP2 gene for it to be incorporated into the virion of rNDV-F/VP2 virus. Hence, to obtain the actual virus titer, a real time PCR assay was performed by cloning a portion of F gene into a T/A cloning vector and used as a standard. This incorporation of VP2 was evidenced by an induction of sufficient immune responses in SPF chickens. The rNDV-F/VP2 virus was slightly attenuated, as compared to the rNDV-F virus, may be due to the increase in size of the full-length NDV F genome after insertion of the foreign protein VP2 gene, as reported earlier by several groups [
27,
28]. The results showed that the rNDV-F or rNDV-F/VP2 were safe and no abnormal clinical signs and death were observed in chickens even at 10-fold higher than the recommended dose. This vaccine would be more acceptable for vaccination in young chicks considering the earlier reports that the lentogenic LaSota produces mild respiratory symptoms in young chickens.
IBDV is very resistant in the environment and the commonly used sanitizers are not sufficient to prevent the infection. Thus, vaccination plays an important role in the prevention of the disease. Different modified live vaccines (MLVs) have been developed and classified as mild intermediate, intermediate plus IBD vaccines depending upon their ability to break through the interference of maternally derived antibodies. However, MLVs are not fully efficacious against IBDV field challenges and induce moderate to severe bursal lesions and immunosuppression. The major capsid protein VP2 has proved to be an important target for generating cellular and humoral immune responses capable of conferring active and passive protection against IBDV infection [
29,
30]. There are several reports using recombinant live viral vectors including herpes virus of turkey [
31,
32], NDV [
11], fowl pox virus [
33], Marek’s disease virus [
34] and avian adenovirus [
35] as expression vectors incorporating VP2 with the aim of protecting birds against IBD. Humoral immunity plays an important role in the clearance of IBD following vaccination with rapid infiltration of T cells into the bursa and upregulation of CMI related genes following infection with very virulent IBD led to the hypothesis that CMI responses are also crucial for protection against IBDV [
36]. Similarly, CMI plays an important role in protection of chickens vaccinated against NDV and contributes to viral clearance [
37]. Although humoral antibody remains the primary mechanism in affording protection against virulent NDV or IBDV, CMI responses whose quantification are labor intensive is considered equally important in the field challenges [
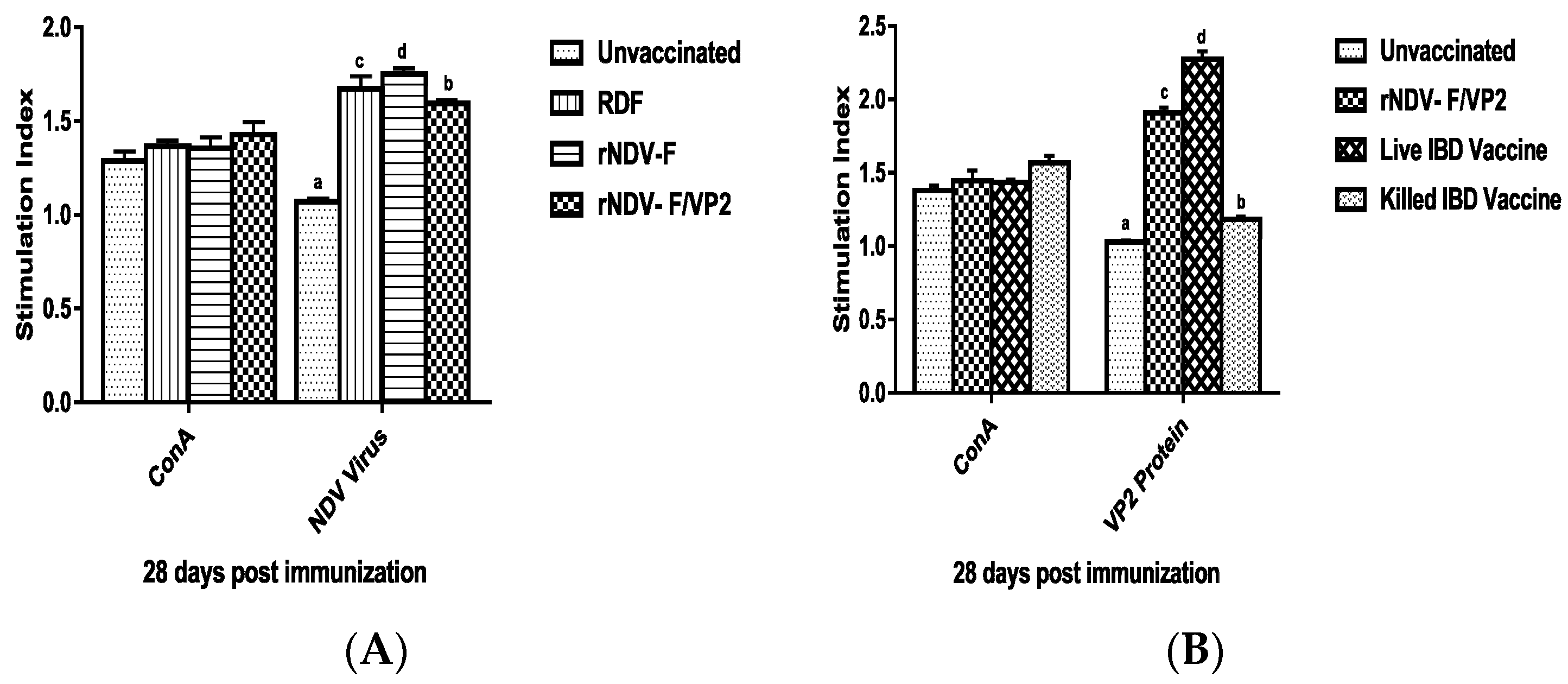
38]. The present study reports the induction of both humoral and cell mediated immune responses by the recombinant virus rNDV-F/VP2 against NDV and IBDV. The subsets of T lymphocytes, including cytokine-secreting CD4
+ T helper cells (MHC class II) and CD8
+ cytotoxic T lymphocytes (MHC class I) as presented in the host post vaccination are the principal cells of the CMI response. The birds vaccinated with the recombinant viruses showed a significantly higher CD4
+ and CD8
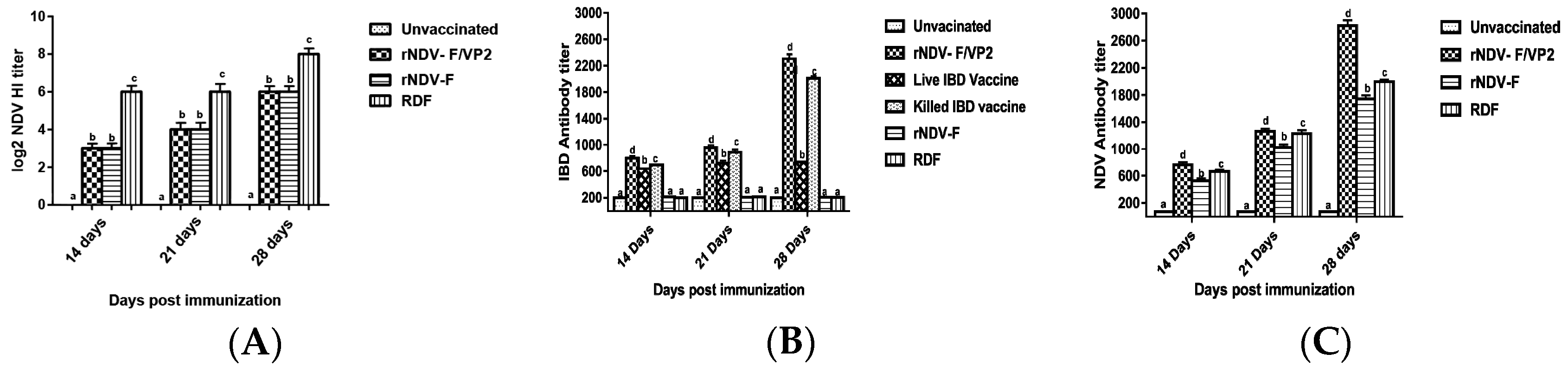
+ T lymphocytes as compared to the commercial vaccines against IBDV and NDV in Flow cytometric analysis. The results were supported by the lymphocyte transformation test, wherein the rNDV-F and rNDV-F/VP2 induced antigen specific lymphocyte proliferative response when stimulated with virulent NDV virus or VP2 protein respectively. The recombinant viruses were extremely effective in eliciting antibody mediated immune reactions beside cell mediated reactions, as indicated by production of high titers of antigen specific antibodies. In this study, the HI antibody titers could attain 64 with the rNDV-F or rNDV-F/VP2 against NDV which was 2 log2 titer less than the parental vaccine RDF at 28 days post vaccination. HI titers of 32 or higher are protective against NDV infection [
39]. However, the recombinant virus could provide only 80% protection against NDV challenge. In a similar study, recombinant fowl pox virus expressing NDV fusion and haemagglutinin genes and ILTV gB gene induced detectable ELISA antibody titres against NDV and ILTV but failed to elicit significant HI titers against NDV [
40].
As the number of vaccine candidates developed using NDV reverse genetics are increasing every year, researchers are able to decipher the differences in the immune responses accorded by these recombinant viruses. It has been noted that all the recombinant NDV vaccines are not equal in their ability to replicate in chickens. Hence, each vaccine that is created should be evaluated for its ability to replicate in chickens, and to induce a protective immune response against a virulent NDV challenge [
41]. However, significantly higher levels of antibody titers were noticed in NDV and IBDV ELISA of the rNDV-F/VP2 group reflecting the presence of higher levels of neutralizing antibodies against both NDV and IBDV, proving that the recombinant virus could elicit a strong humoral response. Further on vvIBDV challenge, none of the vaccinated birds showed clinical signs or mortality and the control unvaccinated birds started showing symptoms from 3rd day post challenge. All the dead birds showed gross bursal lesions, haemorrhages in the thigh muscles typical of IBD. The vaccinated groups showed significantly less bursal lesions with mild depletion of lymphocytes and intact follicular structure.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}