
Immune Evasion Strategies during Chronic Hepatitis B and C Virus Infection

Abstract
1. Introduction
2. Host Responses to HBV and HCV Infection
2.1. Cell-Intrinsic Innate Immune Responses
2.2. Innate Immune Responses
2.2.1. NK Cell Responses in Chronic Viral Hepatitis
2.2.2. NKT Cell Responses in Chronic Viral Hepatitis
2.2.3. Kupffer Cell Responses in Chronic Viral Hepatitis
2.2.4. The Role of Myeloid-Derived Suppressor Cells in Chronic Viral Hepatitis
2.2.5. Dendritic Cells in Chronic Viral Hepatitis
2.2.6. Extracellular Vesicles in HBV and HCV Innate Immune Responses
2.3. Adaptive Immune Responses
2.3.1. T Cell Responses to HBV and HCV
2.3.2. B Cell Responses in HBV and HCV
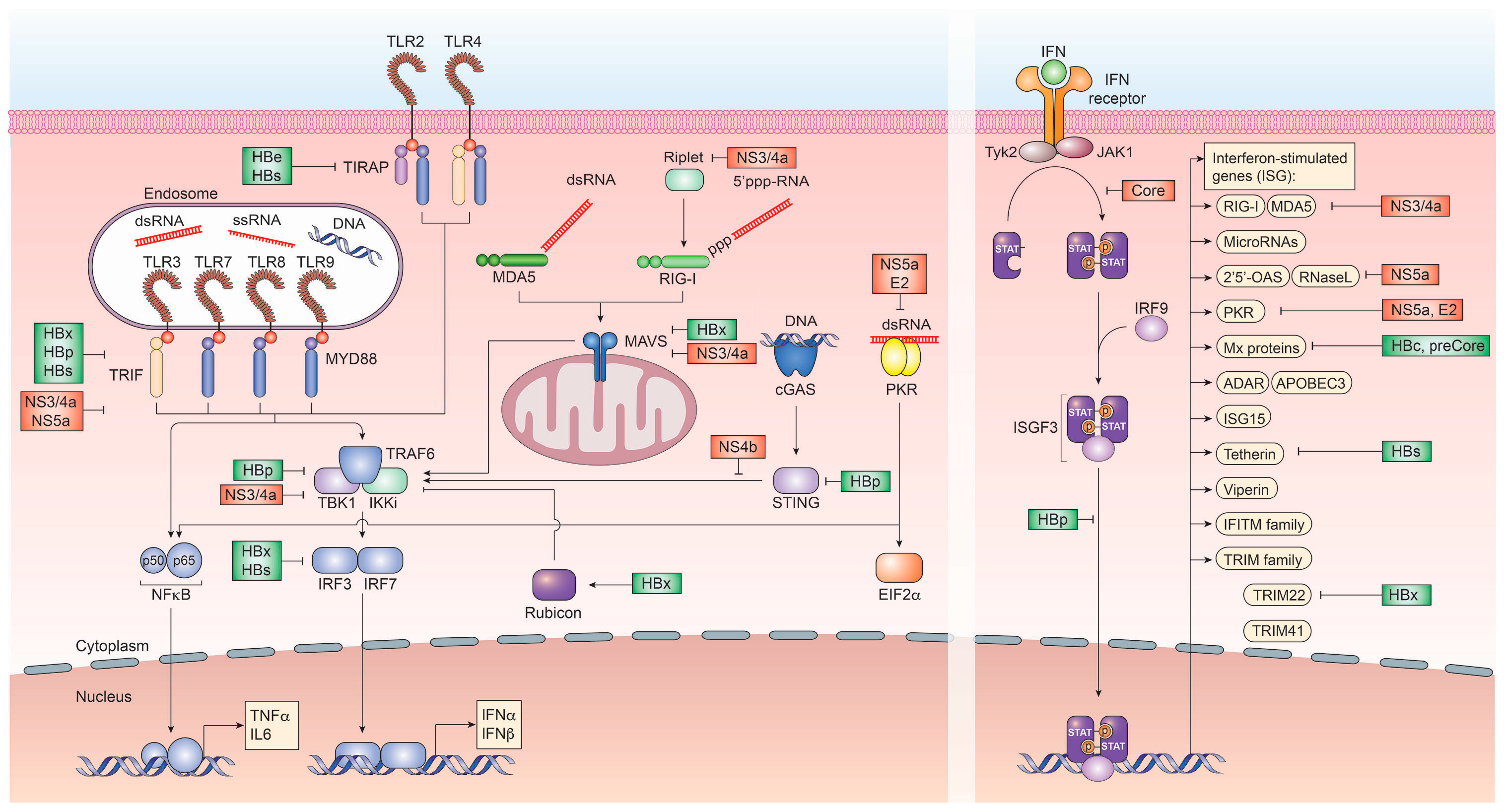
3. Immune Evasion Strategies of HCV
3.1. Viral Proteins and Their Role in Immune Evasion
3.2. Viral Variability and Immune Evasion
3.3. Evasion of Humoral Immune Responses
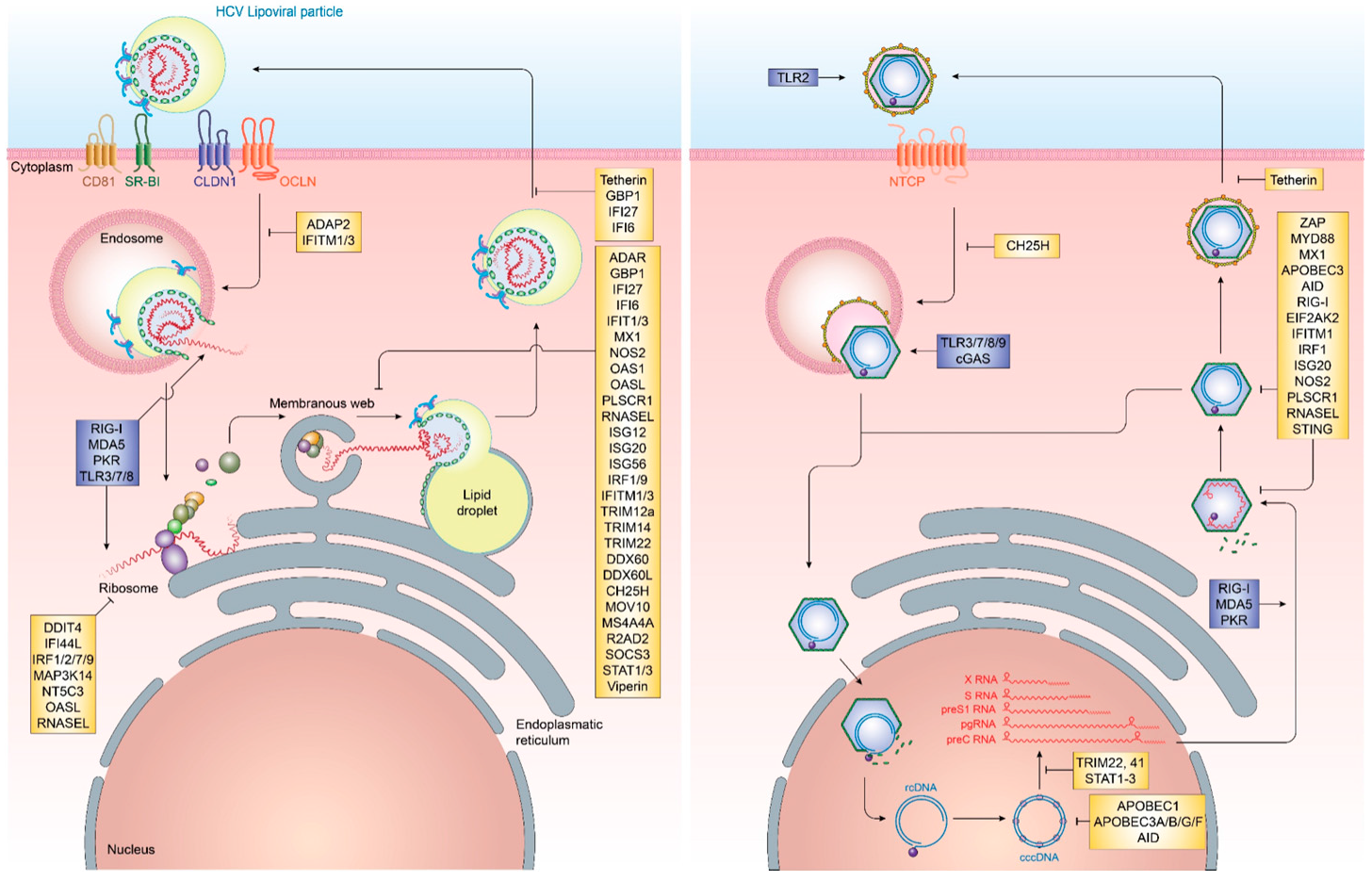
3.4. Viral Mimicry of Cellular Lipids
4. Immune Evasion Strategies of HBV
4.1. Viral Proteins and Their Role in Immune Evasion
4.2. Viral Mimicry of HBV cccDNA as Minichromosome
4.3. Evasion of Innate and Adaptive Immune Responses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis, G.; Zoulim, F.; Tacke, F.; European Association for the Study of the Liver. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cacciapuoti, C. Hepatitis C virus (HCV) genotypes distribution: An epidemiological up-date in europe. Infect. Agent. Cancer 2016, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.V.; King, L.Y.; Chung, R.T. Hepatitis C virus-associated cancer. Annu. Rev. Pathol. 2015, 10, 345–370. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.A.; Feld, J.J. Acute hepatitis C: Management in the rapidly evolving world of HCV. Curr. Gastroenterol. Rep. 2014, 16, 371. [Google Scholar] [CrossRef] [PubMed]
- Dustin, L.B.; Rice, C.M. Flying under the radar: The immunobiology of hepatitis C. Annu. Rev. Immunol. 2007, 25, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Kumar, M.; Sarin, S.K. Management of acute hepatitis B and reactivation of hepatitis B. Liver Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Bortolotti, F.; Donato, F. Natural history of chronic hepatitis B: Special emphasis on disease progression and prognostic factors. J. Hepatol. 2008, 48, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L.; Thio, C.L.; Martin, M.P.; Qi, Y.; Ge, D.; O’Huigin, C.; Kidd, J.; Kidd, K.; Khakoo, S.I.; Alexander, G.; et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009, 461, 798–801. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.J. The natural history of chronic hepatitis B virus infection. Hepatology 2009, 49, S45–S55. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.J. Natural history of chronic hepatitis B. Clin. Liver Dis. 2010, 14, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Papatheodoridis, G.V. Hepatitis B e antigen-negative chronic hepatitis B: Natural history and treatment. Semin. Liver Dis. 2006, 26, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.F.; Chang, M.H. Natural history of chronic hepatitis B virus infection from infancy to adult life—The mechanism of inflammation triggering and long-term impacts. J. Biomed. Sci. 2015, 22, 92. [Google Scholar] [CrossRef] [PubMed]
- Hakim, M.S.; Spaan, M.; Janssen, H.L.; Boonstra, A. Inhibitory receptor molecules in chronic hepatitis B and C infections: Novel targets for immunotherapy? Rev. Med. Virol. 2014, 24, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No love lost between viruses and interferons. Annu. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef] [PubMed]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I interferon in chronic virus infection and cancer. Trends Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-lambda: Immune functions at barrier surfaces and beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Metz, P.; Reuter, A.; Bender, S.; Bartenschlager, R. Interferon-stimulated genes and their role in controlling hepatitis C virus. J. Hepatol. 2013, 59, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Shu, Q.; Lennemann, N.J.; Sarkar, S.N.; Sadovsky, Y.; Coyne, C.B. Adap2 is an interferon stimulated gene that restricts RNA virus entry. PLoS Pathog. 2015, 11, e1005150. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Kitamura, K.; Simadu, M.; Koura, M.; Muramatsu, M. Concerted action of activation-induced cytidine deaminase and uracil-DNA glycosylase reduces covalently closed circular DNA of duck hepatitis B virus. FEBS Lett. 2013, 587, 3148–3152. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Huang, Y.; Chen, Y.; Tu, Z.; Hu, J.; Tavis, J.E.; Huang, A.; Hu, Y. Association of hepatitis B virus covalently closed circular DNA and human APOBEC3b in hepatitis B virus-related hepatocellular carcinoma. PLoS ONE 2016, 11, e0157708. [Google Scholar] [CrossRef] [PubMed]
- Renard, M.; Henry, M.; Guetard, D.; Vartanian, J.P.; Wain-Hobson, S. APOBEC1 and APOBEC3 cytidine deaminases as restriction factors for hepadnaviral genomes in non-humans in vivo. J. Mol. Biol. 2010, 400, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.C.; Suspene, R.; Henry, M.; Guetard, D.; Wain-Hobson, S.; Vartanian, J.P. Human APOBEC1 cytidine deaminase edits HBV DNA. Retrovirology 2009, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Guetard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8321–8326. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Han, X.; Guan, G.; Wu, N.; Sun, J.; Pak, V.; Liang, G. TGF-beta triggers HBV cccDNA degradation through AID-dependent deamination. FEBS Lett. 2016, 590, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Que, L.; Liu, G.; Kitamura, K.; Wakae, K.; Li, Y.; Nishitsuji, H.; Ujino, S.; Shimotohno, K.; Muramatsu, M. Molecular characterization of AID-mediated reduction of hepatitis B virus transcripts. Virology 2017, 510, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Kitamura, K.; Wang, Z.; Liu, G.; Chowdhury, S.; Fu, W.; Koura, M.; Wakae, K.; Honjo, T.; Muramatsu, M. RNA editing of hepatitis B virus transcripts by activation-induced cytidine deaminase. Proc. Natl. Acad. Sci. USA 2013, 110, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Watashi, K.; Tsukuda, S.; Aly, H.H.; Fukasawa, M.; Fujimoto, A.; Suzuki, R.; Aizaki, H.; Ito, T.; Koiwai, O.; et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 2014, 443, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, S.; Yi, Z.; Tian, H.; Aliyari, R.; Li, Y.; Chen, G.; Liu, P.; Zhong, J.; Chen, X.; et al. Interferon-inducible cholesterol-25-hydroxylase inhibits hepatitis C virus replication via distinct mechanisms. Sci. Rep. 2014, 4, 7242. [Google Scholar] [CrossRef] [PubMed]
- Anggakusuma; Romero-Brey, I.; Berger, C.; Colpitts, C.C.; Boldanova, T.; Engelmann, M.; Todt, D.; Perin, P.M.; Behrendt, P.; Vondran, F.W.; et al. Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 2015, 62, 702–714. [Google Scholar]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular rna helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, Y.; Yan, F.; Tu, J.; Yang, X.; Xing, Y.; Chen, Z. 5′-triphosphate-siRNA activates RIG-I-dependent type I interferon production and enhances inhibition of hepatitis B virus replication in HepG2.2.15 cells. Eur. J. Pharmacol. 2013, 721, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. Ddx60, a dexd/h box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell Biol. 2011, 31, 3802–3819. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Okamoto, M.; Morioka, Y.; Okabe, M.; Matsumoto, M.; Seya, T. Ddx60 is involved in RIG-I-dependent and independent antiviral responses, and its function is attenuated by virus-induced EGFR activation. Cell Rep. 2015, 11, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Grunvogel, O.; Esser-Nobis, K.; Reustle, A.; Schult, P.; Muller, B.; Metz, P.; Trippler, M.; Windisch, M.P.; Frese, M.; Binder, M.; et al. Ddx60l is an interferon-stimulated gene product restricting hepatitis C virus replication in cell culture. J. Virol. 2015, 89, 10548–10568. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.I.; Kwon, S.N.; Park, S.H.; Kim, Y.K.; Choi, S.Y.; Kim, J.P.; Ahn, B.Y. PKR protein kinase is activated by hepatitis C virus and inhibits viral replication through translational control. Virus Res. 2009, 142, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Guo, H.; Xu, C.; Chang, J.; Gu, B.; Wang, L.; Block, T.M.; Guo, J.T. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 2008, 82, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Baek, K.W.; Cho, E.Y.; Ahn, B.Y. PKR-dependent mechanisms of interferon-alpha for inhibiting hepatitis B virus replication. Mol. Cells 2011, 32, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Morris, A.; Mendez, H.; Koch, R.; Silverman, R.H.; Williams, B.R.; Chisari, F.V. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J. Virol. 2002, 76, 2617–2621. [Google Scholar] [CrossRef] [PubMed]
- Itsui, Y.; Sakamoto, N.; Kurosaki, M.; Kanazawa, N.; Tanabe, Y.; Koyama, T.; Takeda, Y.; Nakagawa, M.; Kakinuma, S.; Sekine, Y.; et al. Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J. Viral. Hepat. 2006, 13, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Kwon, Y.C.; Liu, S.; Hagedorn, C.H.; Ray, R.B.; Ray, R. Interferon-alpha inducible protein 6 impairs EGFR activation by CD81 and inhibits hepatitis C virus infection. Sci. Rep. 2015, 5, 9012. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Michal, J.J.; Zhang, L.; Ding, B.; Lunney, J.K.; Liu, B.; Jiang, Z. Interferon induced IFIT family genes in host antiviral defense. Int. J. Biol. Sci. 2013, 9, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Metz, P.; Dazert, E.; Ruggieri, A.; Mazur, J.; Kaderali, L.; Kaul, A.; Zeuge, U.; Windisch, M.P.; Trippler, M.; Lohmann, V.; et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology 2012, 56, 2082–2093. [Google Scholar] [CrossRef] [PubMed]
- Raychoudhuri, A.; Shrivastava, S.; Steele, R.; Kim, H.; Ray, R.; Ray, R.B. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J. Virol. 2011, 85, 12881–12889. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Woodward, J.; Lau, D.T.; Barnes, A.; Joyce, M.; McFarlane, N.; McKeating, J.A.; Tyrrell, D.L.; Gale, M., Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 2013, 57, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Qin, B.; Zhang, X.; Zhu, W.; Kemper, T.; Ma, Z.; Trippler, M.; Schlaak, J.; Chen, X.; Lu, M. Interferon-induced proteins with tetratricopeptide repeats 1 and 2 are cellular factors that limit hepatitis B virus replication. J. Innate. Immun. 2014, 6, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Dong, H.; Zhu, H.; Nelson, D.; Liu, C.; Lambiase, L.; Li, X. Identification of the IFITM3 gene as an inhibitor of hepatitis C viral translation in a stable stat1 cell line. J. Viral. Hepat. 2011, 18, e523–e529. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, N.; Kurosaki, M.; Sakamoto, N.; Enomoto, N.; Itsui, Y.; Yamashiro, T.; Tanabe, Y.; Maekawa, S.; Nakagawa, M.; Chen, C.H.; et al. Regulation of hepatitis C virus replication by interferon regulatory factor 1. J. Virol. 2004, 78, 9713–9720. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.; Hopkins, S.; Baugh, J.; Chatterji, U.; Hernandez, F.; Hiscott, J.; Sluder, A.; Lin, K.; Gallay, P.A. HCV NS5a and IRF9 compete for cypa binding. J. Hepatol. 2013, 58, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nie, H.; Mao, R.; Mitra, B.; Cai, D.; Yan, R.; Guo, J.T.; Block, T.M.; Mechti, N.; Guo, H. Interferon-inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem-loop structure of viral RNA. PLoS Pathog. 2017, 13, e1006296. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.R.; Funami, K.; Oshiumi, H.; Mengao, D.; Takaki, H.; Matsumoto, M.; Aly, H.H.; Watashi, K.; Chayama, K.; Seya, T. Interferon-stimulated gene of 20 kda protein (ISG20) degrades RNA of hepatitis B virus to impede the replication of HBV in vitro and in vivo. Oncotarget 2016, 7, 68179–68193. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Pan, T.; Xu, J.; Zhang, Y.; Song, W.; Yi, Z.; Yuan, Z. Hepatitis C virus replicative double-stranded RNA is a potent interferon inducer that triggers interferon production through MDA5. J. Gen. Virol. 2016, 97, 2868–2882. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, L.; Chen, L.; Feng, W.; Xu, Y.; Chen, F.; Liu, X.; Chen, Z.; Liu, W. Mxa inhibits hepatitis B virus replication by interaction with hepatitis B core antigen. Hepatology 2012, 56, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Jiao, B.; Chen, Y.; Li, S.; Chen, L. Mxa is a positive regulator of type I ifn signaling in HCV infection. J. Med. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, S.; Chen, Q.; Peng, L.; Zhai, J.; Liu, Y.; Yuan, Z. Inhibition of hepatitis B virus replication by MYD88 involves accelerated degradation of pregenomic RNA and nuclear retention of pre-s/s RNAs. J. Virol. 2010, 84, 6387–6399. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.W.; Su, I.J.; Lai, M.D.; Chang, W.T.; Huang, W.; Lei, H.Y. The role of inducible nitric oxide synthase in a murine acute hepatitis B virus (HBV) infection model induced by hydrodynamics-based in vivo transfection of HBV-DNA. J. Hepatol. 2003, 39, 834–842. [Google Scholar] [CrossRef]
- Naganuma, A.; Nozaki, A.; Tanaka, T.; Sugiyama, K.; Takagi, H.; Mori, M.; Shimotohno, K.; Kato, N. Activation of the interferon-inducible 2′-5′-oligoadenylate synthetase gene by hepatitis C virus core protein. J. Virol. 2000, 74, 8744–8750. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Wakita, T.; Esumi, M. 2′,5′-oligoadenylate synthetase-like gene highly induced by hepatitis C virus infection in human liver is inhibitory to viral replication in vitro. Biochem. Biophys. Res. Commun. 2010, 392, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, X.; Liu, J.; Ding, X.; Han, M.; Hu, W.; Wang, X.; Zhou, Z.; Wang, S. Inhibition of hepatitis B virus replication by phospholipid scramblase 1 in vitro and in vivo. Antiviral. Res. 2012, 94, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Dansako, H.; Ueda, Y.; Okumura, N.; Satoh, S.; Sugiyama, M.; Mizokami, M.; Ikeda, M.; Kato, N. The cyclic GMP-AMP synthetase-sting signaling pathway is required for both the innate immune response against HBV and the suppression of HBV assembly. FEBS J. 2016, 283, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.S.; Ahmed, S.; Napoletano, S.; Schroeder, M.; Johnston, J.A.; Hegarty, J.E.; O'Farrelly, C.; Stevenson, N.J. Hepatitis C virus (HCV)-induced suppressor of cytokine signaling (SOCS) 3 regulates proinflammatory TNF-alpha responses. J. Leukoc. Biol. 2014, 96, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.X.; Zhang, L.; Peng, L.F.; Sun, E.; Chung, W.J.; Jang, J.Y.; Tsai, W.L.; Hyppolite, G.; Chung, R.T. Suppressor of cytokine signaling 3 suppresses hepatitis C virus replication in an mTOR-dependent manner. J. Virol. 2010, 84, 6060–6069. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Investig. 2012, 122, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Turkson, J.; Hassanein, T.; Siddiqui, A. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: Role of STAT-3 in HCV replication. J. Virol. 2005, 79, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; Takeuchi, K.; Chihara, K.; Honjoh, C.; Kato, Y.; Yoshiki, H.; Hotta, H.; Sada, K. STAT1 is essential for the inhibition of hepatitis C virus replication by interferon-lambda but not by interferon-alpha. Sci. Rep. 2016, 6, 38336. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhao, X.; Cai, D.; Liu, Y.; Block, T.M.; Guo, J.T.; Guo, H. The interferon-inducible protein tetherin inhibits hepatitis B virus virion secretion. J. Virol. 2015, 89, 9200–9212. [Google Scholar] [CrossRef] [PubMed]
- Amet, T.; Byrd, D.; Hu, N.; Sun, Q.; Li, F.; Zhao, Y.; Hu, S.; Grantham, A.; Yu, Q. Bst-2 expression in human hepatocytes is inducible by all three types of interferons and restricts production of hepatitis C virus. Curr. Mol. Med. 2014, 14, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Dafa-Berger, A.; Kuzmina, A.; Fassler, M.; Yitzhak-Asraf, H.; Shemer-Avni, Y.; Taube, R. Modulation of hepatitis C virus release by the interferon-induced protein BST-2/tetherin. Virology 2012, 428, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Trippler, M.; Pei, R.; Lu, M.; Broering, R.; Gerken, G.; Schlaak, J.F. Toll-like receptor activated human and murine hepatic stellate cells are potent regulators of hepatitis C virus replication. J. Hepatol. 2009, 51, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Gonzalez, V.D.; Li, Q.; Modi, A.A.; Chen, W.; Noureddin, M.; Rotman, Y.; Liang, T.J. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 2012, 142, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, S.; Zhao, X.; Chen, M.; Lin, Y.; Xia, Y.; Sun, C.; Yang, X.; Wang, J.; Guo, Y.; et al. Poly(I:C) treatment leads to interferon-dependent clearance of hepatitis B virus in a hydrodynamic injection mouse model. J. Virol. 2014, 88, 10421–10431. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Tian, Y.; Chan, S.T.; Kim, J.Y.; Cho, C.; Ou, J.H. TNF-alpha induced by hepatitis C virus via TLR7 and TLR8 in hepatocytes supports interferon signaling via an autocrine mechanism. PLoS Pathog. 2015, 11, e1004937. [Google Scholar] [CrossRef] [PubMed]
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.V. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 2005, 79, 7269–7272. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Y.; Li, C.; Wu, Y.; Guo, L.; Peng, C.; Huang, Y.; Cheng, G.; Qin, F.X. TRIM14 inhibits hepatitis C virus infection by spry domain-dependent targeted degradation of the viral NS5a protein. Sci. Rep. 2016, 6, 32336. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Duan, Z.; Xu, W.; Xiong, S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located ring domain. Hepatology 2009, 50, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhao, X.; Sun, D.; Yang, L.; Chong, C.; Pan, Y.; Chi, X.; Gao, Y.; Wang, M.; Shi, X.; et al. Interferon alpha (IFNalpha)-induced TRIM22 interrupts HCV replication by ubiquitinating NS5a. Cell Mol. Immunol. 2016, 13, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Guo, J.T.; Wu, J.Z.; Yang, G. Identification and characterization of multiple TRIM proteins that inhibit hepatitis B virus transcription. PLoS ONE 2013, 8, e70001. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Eyre, N.S.; Yip, E.; Narayana, S.; Li, K.; Fiches, G.; McCartney, E.M.; Jangra, R.K.; Lemon, S.M.; Beard, M.R. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5a. Hepatology 2011, 54, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Nie, H.; Cai, D.; Zhang, J.; Liu, H.; Yan, R.; Cuconati, A.; Block, T.M.; Guo, J.T.; Guo, H. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog. 2013, 9, e1003494. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Q.; Dai, J.; Bai, L.; Tang, H. The efficacy of zinc finger antiviral protein against hepatitis B virus transcription and replication in tansgenic mouse model. Virol. J. 2015, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [PubMed]
- Lebosse, F.; Testoni, B.; Fresquet, J.; Facchetti, F.; Galmozzi, E.; Fournier, M.; Hervieu, V.; Berthillon, P.; Berby, F.; Bordes, I.; et al. Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J. Hepatol. 2017, 66, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M.; et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Gordien, E.; Rosmorduc, O.; Peltekian, C.; Garreau, F.; Brechot, C.; Kremsdorf, D. Inhibition of hepatitis B virus replication by the interferon-inducible MXA protein. J. Virol. 2001, 75, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, E.; Vigano, M.; Aghemo, A.; Lampertico, P.; Colombo, M. PEGIFN-alpha2a for the treatment of chronic hepatitis B and C: A 10-year history. Expert Rev. Anti-Infect. Ther. 2013, 11, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Trepo, C. A brief history of hepatitis milestones. Liver Int. 2014, 34 (Suppl. S1), 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yoshio, S.; Kanto, T.; Kuroda, S.; Matsubara, T.; Higashitani, K.; Kakita, N.; Ishida, H.; Hiramatsu, N.; Nagano, H.; Sugiyama, M.; et al. Human blood dendritic cell antigen 3 (BDCA3)(+) dendritic cells are a potent producer of interferon-lambda in response to hepatitis C virus. Hepatology 2013, 57, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; et al. Il28b is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nishida, N.; Sugiyama, M.; Kurosaki, M.; Matsuura, K.; Sakamoto, N.; Nakagawa, M.; Korenaga, M.; Hino, K.; Hige, S.; et al. Genome-wide association of IL28b with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009, 41, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28b predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28b) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8(+) t cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Parham, P.; Moffett, A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat. Rev. Immunol. 2013, 13, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NK cell receptors. Annu. Rev. Immunol. 1998, 16, 359–393. [Google Scholar] [CrossRef] [PubMed]
- Lysakova-Devine, T.; O’Farrelly, C. Tissue-specific NK cell populations and their origin. J. Leukoc. Biol. 2014, 96, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Bost, A.; Glass, J.I.; Tan, S.L. Cytokine-activated natural killer cells exert direct killing of hepatoma cells harboring hepatitis C virus replicons. J. Interferon Cytokine Res. 2006, 26, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.M.; Yoon, J.C.; Park, J.H.; Lee, J.M. Hepatitis C virus impairs natural killer cell activity via viral serine protease NS3. PLoS ONE 2017, 12, e0175793. [Google Scholar] [CrossRef] [PubMed]
- Kafrouni, M.I.; Brown, G.R.; Thiele, D.L. Virally infected hepatocytes are resistant to perforin-dependent CTL effector mechanisms. J. Immunol. 2001, 167, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Holder, K.A.; Russell, R.S.; Grant, M.D. Natural killer cell function and dysfunction in hepatitis C virus infection. Biomed. Res. Int. 2014, 2014, 903764. [Google Scholar] [CrossRef] [PubMed]
- Kakimi, K.; Guidotti, L.G.; Koezuka, Y.; Chisari, F.V. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med. 2000, 192, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.; Brunetto, M.; Reynolds, G.; Christophides, T.; Kennedy, P.T.; Lampertico, P.; Das, A.; Lopes, A.R.; Borrow, P.; Williams, K.; et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J. Exp. Med. 2007, 204, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Peppa, D.; Gill, U.S.; Reynolds, G.; Easom, N.J.; Pallett, L.J.; Schurich, A.; Micco, L.; Nebbia, G.; Singh, H.D.; Adams, D.H.; et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J. Exp. Med. 2013, 210, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Peppa, D. NK cells: A double-edged sword in chronic hepatitis B virus infection. Front. Immunol. 2013, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhu, Y.Y.; Chen, J.; Ye, Y.B.; Li, J.Y.; Liu, Y.R.; Hu, M.L.; Zheng, Y.C.; Jiang, J.J. Activated natural killer cells accelerate liver damage in patients with chronic hepatitis B virus infection. Clin. Exp. Immunol. 2015, 180, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Mizuhara, H.; O’Neill, E.; Seki, N.; Ogawa, T.; Kusunoki, C.; Otsuka, K.; Satoh, S.; Niwa, M.; Senoh, H.; Fujiwara, H. T cell activation-associated hepatic injury: Mediation by tumor necrosis factors and protection by interleukin 6. J. Exp. Med. 1994, 179, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Biermer, M.; Puro, R.; Schneider, R.J. Tumor necrosis factor alpha inhibition of hepatitis B virus replication involves disruption of capsid integrity through activation of NF-kappab. J. Virol. 2003, 77, 4033–4042. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; MacDonald, H.R.; Kronenberg, M.; Smyth, M.J.; Van Kaer, L. NKT cells: What’s in a name? Nat. Rev. Immunol. 2004, 4, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Murata, K.; Sweet, L.; Publicover, J.; Hu, Z.; Kaser, A.; Bosse, E.; Iqbal, J.; Hussain, M.M.; Balschun, K.; et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat. Med. 2012, 18, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Boltjes, A.; Movita, D.; Boonstra, A.; Woltman, A.M. The role of kupffer cells in hepatitis B and hepatitis C virus infections. J. Hepatol. 2014, 61, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Burgio, V.L.; Ballardini, G.; Artini, M.; Caratozzolo, M.; Bianchi, F.B.; Levrero, M. Expression of co-stimulatory molecules by kupffer cells in chronic hepatitis of hepatitis C virus etiology. Hepatology 1998, 27, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimkhani, M.R.; Mohar, I.; Crispe, I.N. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 2011, 54, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, R.; Xu, L.; Yin, W.; Chen, Y.; Zheng, X.; Lian, Z.; Wei, H.; Tian, Z. Kupffer cells support hepatitis B virus-mediated CD8+ T cell exhaustion via hepatitis b core antigen-TLR2 interactions in mice. J. Immunol. 2015, 195, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.; et al. Regulation of toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2007, 45, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Guo, M.; Li, G.; Yu, D.; Zhang, X.; Lan, K.; Deng, Q. Hepatitis B virus core protein sensitizes hepatocytes to tumor necrosis factor-induced apoptosis by suppression of the phosphorylation of mitogen-activated protein kinase kinase 7. J. Virol. 2015, 89, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.C.; Roggerson, K.M.; Lee, H.C.; Golden-Mason, L.; Rosen, H.R.; Hahn, Y.S. Hepatitis C virus-induced myeloid-derived suppressor cells suppress NK cell IFN-gamma production by altering cellular metabolism via arginase-1. J. Immunol. 2016, 196, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Gill, U.S.; Quaglia, A.; Sinclair, L.V.; Jover-Cobos, M.; Schurich, A.; Singh, K.P.; Thomas, N.; Das, A.; Chen, A.; et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat. Med. 2015, 21, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Heath, W.R.; Carbone, F.R. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 2009, 10, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N. The divergence and interplay between PDC and MDC in humans. Front. Biosci. 2009, 14, 808–817. [Google Scholar] [CrossRef]
- Schildberg, F.A.; Hegenbarth, S.I.; Schumak, B.; Scholz, K.; Limmer, A.; Knolle, P.A. Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur. J. Immunol. 2008, 38, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, V.M.; Hon, H.; Ibegbu, C.; Knechtle, S.J.; Kirk, A.D.; Grakoui, A. Hepatic enrichment and activation of myeloid dendritic cells during chronic hepatitis C virus infection. Hepatology 2012, 56, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.H.; Lin, Y.L.; Chen, C.J.; Lin, T.Y.; Hsu, Y.C.; Chen, L.K.; Chiang, B.L. Hepatitis B virus surface antigen can activate human monocyte-derived dendritic cells by nuclear factor kappa b and p38 mitogen-activated protein kinase mediated signaling. Microbiol. Immunol. 2012, 56, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Di Tacchio, M.; Berardi, S.; Santantonio, T.; Fasano, M.; Ferrone, S.; Vacca, A.; Dammacco, F.; Racanelli, V. Dendritic cell maturation in HCV infection: Altered regulation of MHC class I antigen processing-presenting machinery. J. Hepatol. 2014, 61, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lukes, Y.; Anderson, J.; Fileta, B.; Reinhardt, B.; Sjogren, M. Hepatitis C virus E2 envelope protein induces dendritic cell maturation. J. Viral. Hepat. 2007, 14, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Sarobe, P.; Lasarte, J.J.; Zabaleta, A.; Arribillaga, L.; Arina, A.; Melero, I.; Borras-Cuesta, F.; Prieto, J. Hepatitis C virus structural proteins impair dendritic cell maturation and inhibit in vivo induction of cellular immune responses. J. Virol. 2003, 77, 10862–10871. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Ait-Goughoulte, M.; Truscott, S.M.; Meyer, K.; Blazevic, A.; Abate, G.; Ray, R.B.; Hoft, D.F.; Ray, R. Hepatitis C virus inhibits cell surface expression of HLA-DR, prevents dendritic cell maturation, and induces interleukin-10 production. J. Virol. 2008, 82, 3320–3328. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.E.; Zannetti, C.; Lucifora, J.; Norder, H.; Protzer, U.; Hainaut, P.; Zoulim, F.; Tommasino, M.; Trepo, C.; Hasan, U.; et al. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS ONE 2011, 6, e26315. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Decembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe. 2012, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, K.; Liu, Y.; Xu, Y.; Zhang, F.; Yang, H.; Liu, J.; Pan, T.; Chen, J.; Wu, M.; et al. Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat. Immunol. 2013, 14, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; He, Z.; Yuan, J.; Wen, W.; Huang, X.; Hu, Y.; Lin, C.; Pan, J.; Li, R.; Deng, H.; et al. IFITM3-containing exosome as a novel mediator for anti-viral response in dengue virus infection. Cell Microbiol. 2015, 17, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, Q.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell Mol. Immunol. 2017, 14, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Boyd, B.; Chisari, F.V. Virion-independent transfer of replication-competent hepatitis C virus RNA between permissive cells. J. Virol. 2015, 89, 2956–2961. [Google Scholar] [CrossRef] [PubMed]
- Cosset, F.L.; Dreux, M. HCV transmission by hepatic exosomes establishes a productive infection. J. Hepatol. 2014, 60, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.; Sun, L.; Zhou, L.; Ma, T.C.; Song, L.; Wu, J.G.; Li, J.L.; Ho, W.Z. Toll-like receptor 3-activated macrophages confer anti-HCV activity to hepatocytes through exosomes. FASEB J. 2016, 30, 4132–4140. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, A.; Maelfait, J.; Davenne, T.; Partridge, T.; Peng, Y.; Mayer, A.; Dong, T.; Kaever, V.; Borrow, P.; Rehwinkel, J. Viruses transfer the antiviral second messenger cgamp between cells. Science 2015, 349, 1228–1232. [Google Scholar] [CrossRef] [PubMed]
- McKinney, E.F.; Smith, K.G. T cell exhaustion and immune-mediated disease-the potential for therapeutic exhaustion. Curr. Opin. Immunol. 2016, 43, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Martin, B.; Thimme, R. Restoration of HBV-specific CD8+ T cell function by PD-1 blockade in inactive carrier patients is linked to T cell differentiation. J. Hepatol. 2014, 61, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, H.T.; Tsai, H.F.; Liao, H.J.; Lin, Y.J.; Chen, L.; Chen, P.J.; Hsu, P.N. PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PLoS ONE 2012, 7, e39179. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology 2010. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.; Isogawa, M.; Freeman, G.J.; Chisari, F.V. PD-1:PD-l1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J. Immunol. 2007, 178, 2714–2720. [Google Scholar] [CrossRef] [PubMed]
- Franzese, O.; Kennedy, P.T.; Gehring, A.J.; Gotto, J.; Williams, R.; Maini, M.K.; Bertoletti, A. Modulation of the CD8+-T-cell response by CD4+ CD25+ regulatory T cells in patients with hepatitis B virus infection. J. Virol. 2005, 79, 3322–3328. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kobayashi, K.; Ueno, Y.; Shiina, M.; Niitsuma, H.; Kanno, N.; Kobayashi, T.; Shimosegawa, T. Mechanism of T cell hyporesponsiveness to hbcag is associated with regulatory T cells in chronic hepatitis B. World J. Gastroenterol. 2006, 12, 4310–4317. [Google Scholar] [CrossRef] [PubMed]
- Stoop, J.N.; van der Molen, R.G.; Baan, C.C.; van der Laan, L.J.; Kuipers, E.J.; Kusters, J.G.; Janssen, H.L. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology 2005, 41, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Stoop, J.N.; van der Molen, R.G.; Kuipers, E.J.; Kusters, J.G.; Janssen, H.L. Inhibition of viral replication reduces regulatory T cells and enhances the antiviral immune response in chronic hepatitis B. Virology 2007, 361, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Ueno, Y.; Kobayashi, K.; Kakazu, E.; Shiina, M.; Inoue, J.; Tamai, K.; Wakui, Y.; Tanaka, Y.; Ninomiya, M.; et al. Hepatitis B virus replication could enhance regulatory T cell activity by producing soluble heat shock protein 60 from hepatocytes. J. Infect. Dis. 2010, 202, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ikeda, F.; Stadanlick, J.; Nunes, F.A.; Alter, H.J.; Chang, K.M. Suppression of HCV-specific t cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 2003, 38, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.; Hansi, N.; Easom, N.J.W.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef] [PubMed]
- Giang, E.; Dorner, M.; Prentoe, J.C.; Dreux, M.; Evans, M.J.; Bukh, J.; Rice, C.M.; Ploss, A.; Burton, D.R.; Law, M. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA. 2012, 109, 6205–6210. [Google Scholar] [CrossRef] [PubMed]
- De Jong, Y.P.; Dorner, M.; Mommersteeg, M.C.; Xiao, J.W.; Balazs, A.B.; Robbins, J.B.; Winer, B.Y.; Gerges, S.; Vega, K.; Labitt, R.N.; et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.T.; Goldstein, S.T.; Tufa, J.; Tauillii, S.; Margolis, H.S.; Mahoney, F.J. Long term antibody response to hepatitis B vaccination beginning at birth and to subsequent booster vaccination. Pediatr. Infect. Dis. J. 2003, 22, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Hudu, S.A.; Malik, Y.A.; Niazlin, M.T.; Harmal, N.S.; Adnan, A.; Alshrari, A.S.; Sekawi, Z. Antibody and immune memory persistence post infant hepatitis B vaccination. Patient Preference Adherence 2013, 7, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Ciupe, S.M.; Ribeiro, R.M.; Perelson, A.S. Antibody responses during hepatitis B viral infection. PLoS Comput. Biol. 2014, 10, e1003730. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Wang, X.; Zhou, Y.; Zhang, T.; Ho, W. HCV dsRNA-activated macrophages inhibit HCV replication in hepatocytes. Hepat. Mon. 2015, 15, e29282. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Ding, Q.; Lu, J.; Tao, W.; Huang, B.; Zhao, Y.; Niu, J.; Liu, Y.J.; Zhong, J. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 2015, 62, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4a protease-mediated cleavage of the toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4a cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Oshiumi, H.; Azuma, M.; Kato, N.; Matsumoto, M.; Seya, T. IPS-1 is essential for type III IFN production by hepatocytes and dendritic cells in response to hepatitis C virus infection. J. Immunol. 2014, 192, 2770–2777. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.Q.; Waggoner, S.N.; Cruise, M.W.; Hall, C.; Xie, X.; Oldach, D.W.; Hahn, Y.S. SOCS1 and SOCS3 are targeted by hepatitis C virus core/GC1qr ligation to inhibit T-cell function. J. Virol. 2005, 79, 15417–15429. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.J.; Kato, N.; Shu, H.B.; Zhong, J. Hepatitis C virus NS4b blocks the interaction of sting and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Joyce, M.A.; Van Buuren, N.; Levin, A.; Kirkegaard, K.; Gale, M., Jr.; Tyrrell, D.L.; Wozniak, R.W. The hepatitis C virus-induced membranous web and associated nuclear transport machinery limit access of pattern recognition receptors to viral replication sites. PLoS Pathog. 2016, 12, e1005428. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Q.; Barton, D.J. Activation and evasion of the antiviral 2′-5′ oligoadenylate synthetase/ribonuclease l pathway by hepatitis C virus mRNA. RNA 2002, 8, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, T.; Okushin, K.; Enooku, K.; Fujinaga, H.; Moriya, K.; Yotsuyanagi, H.; Aizaki, H.; Suzuki, T.; Matsuura, Y.; Koike, K. Nonstructural 5a protein of hepatitis C virus interferes with toll-like receptor signaling and suppresses the interferon response in mouse liver. PLoS ONE 2017, 12, e0170461. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Shi, S.T.; Romano, P.R.; Barber, G.N.; Lai, M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 1999, 285, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Keck, Z.Y.; Foung, S.K. Viral evasion and challenges of hepatitis C virus vaccine development. Curr. Opin. Virol. 2016, 20, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, N.; Moratorio, G.; Cristina, J.; Moreno, P. Hepatitis C virus genetic variability and evolution. World J. Hepatol. 2015, 7, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Sagnelli, E.; Argentini, C.; Genovese, D.; Pisaturo, M.; Coppola, N.; Taffon, S.; Sagnelli, C.; Rapicetta, M. Virological and epitope evolution of HCV infection from acute hepatitis C to subsequent episodes of HCV-related acute liver cell necrosis. Infection 2009, 37, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Neveu, B.; Debeaupuis, E.; Echasserieau, K.; le Moullac-Vaidye, B.; Gassin, M.; Jegou, L.; Decalf, J.; Albert, M.; Ferry, N.; Gournay, J.; et al. Selection of high-avidity CD8 T cells correlates with control of hepatitis C virus infection. Hepatology 2008, 48, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Moriya, O.; Yoshimoto, T.; Hayashi, H.; Akatsuka, T.; Matsui, M. Immunogenic variation between multiple HLA-A*0201-restricted, hepatitis C virus-derived epitopes for cytotoxic T lymphocytes. Viral. Immunol. 2006, 19, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Gondar, V.; Molina-Jimenez, F.; Hishiki, T.; Garcia-Buey, L.; Koutsoudakis, G.; Shimotohno, K.; Benedicto, I.; Majano, P.L. Apolipoprotein E, but not apolipoprotein B, is essential for efficient cell-to-cell transmission of hepatitis C virus. J. Virol. 2015, 89, 9962–9973. [Google Scholar] [CrossRef] [PubMed]
- Pantua, H.; Diao, J.; Ultsch, M.; Hazen, M.; Mathieu, M.; McCutcheon, K.; Takeda, K.; Date, S.; Cheung, T.K.; Phung, Q.; et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J. Mol. Biol. 2013, 425, 1899–1914. [Google Scholar] [CrossRef] [PubMed]
- Bankwitz, D.; Doepke, M.; Hueging, K.; Weller, R.; Bruening, J.; Behrendt, P.; Lee, J.Y.; Vondran, F.W.R.; Manns, M.P.; Bartenschlager, R.; et al. Maturation of secreted HCV particles by incorporation of secreted apoe protects from antibodies by enhancing infectivity. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Hepatitis B virus-induced parkin-dependent recruitment of linear ubiquitin assembly complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 2016, 12, e1005693. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Z.; Hu, C.; Qian, F.; Cheng, Y.; Wu, M.; Shi, B.; Chen, J.; Hu, Y.; Yuan, Z. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J. Immunol. 2013, 190, 5142–5151. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, M.; Zhang, X.; Zhang, W.; Zhang, Z.; Chen, L.; He, J.; Zheng, Y.; Chen, C.; Wang, F.; et al. Hepatitis B virus polymerase impairs interferon-alpha-induced STAT activation through inhibition of importin-alpha5 and protein kinase C-delta. Hepatology 2013, 57, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Peng, N.; Xie, J.; Hao, Q.; Zhang, M.; Zhang, Y.; Xia, Z.; Xu, G.; Zhao, F.; Wang, Q.; et al. Human hepatitis B virus surface and E antigens inhibit major vault protein signaling in interferon induction pathways. J. Hepatol. 2015, 62, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, K.; Matsunaga, S.; Watashi, K.; Sugiyama, M.; Kimura, H.; Yamamoto, N.; Mizokami, M.; Wakita, T.; Ryo, A. Molecular dissection of HBV evasion from restriction factor tetherin: A new perspective for antiviral cell therapy. Oncotarget 2015, 6, 21840–21852. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ryu, W.S. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: Implications for immune evasion. PLoS Pathog. 2010, 6, e1000986. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B virus polymerase disrupts k63-linked ubiquitination of sting to block innate cytosolic DNA-sensing pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, A.; Cui, L.; Hao, R.; Wang, Y.; He, J.; Guo, D. Hepatitis B virus polymerase suppresses nf-kappab signaling by inhibiting the activity of IKKs via interaction with HSP90beta. PLoS ONE 2014, 9, e91658. [Google Scholar]
- Wan, Y.; Cao, W.; Han, T.; Ren, S.; Feng, J.; Chen, T.; Wang, J.; Broering, R.; Lu, M.; Zhu, Y. Inducible rubicon facilitates viral replication by antagonizing interferon production. Cell Mol. Immunol. 2017, 14, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, H. Mechanism of inhibiting type i interferon induction by hepatitis B virus x protein. Protein Cell 2010, 1, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The SMC5/6 complex restricts hbv when localized to ND10 without inducing an innate immune response and is counteracted by the HBV x protein shortly after infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Park, E.S.; Kim, D.H.; Cho, K.C.; Kim, K.P.; Park, Y.K.; Ahn, S.H.; Park, S.H.; Kim, K.H.; Kim, C.W.; et al. Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5′-UTR of TRIM22. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Quiroga, J.A.; Carreno, V. Hepatitis B virus downregulates the human interferon-inducible mxa promoter through direct interaction of precore/core proteins. J. Gen. Virol. 2003, 84, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schranz, P.; Schroder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Locarnini, S.; Zoulim, F. Molecular genetics of HBV infection. Antivir. Ther. 2010, 15 (Suppl. S3), 3–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Li, Y.N.; Zhao, J.R.; Zhang, J.; Yan, Z. HBC binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics 2011, 6, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBX binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Riviere, L.; Gerossier, L.; Ducroux, A.; Dion, S.; Deng, Q.; Michel, M.L.; Buendia, M.A.; Hantz, O.; Neuveut, C. Hbx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccdna involving setdb1 histone methyltransferase. J. Hepatol. 2015, 63, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.A.; Suslov, A.; Ketterer, S.; Heim, M.H.; Wieland, S.F. Hepatitis B virus covalently closed circular DNA homeostasis is independent of the lymphotoxin pathway during chronic HBV infection. J. Viral. Hepat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bouzidi, M.S.; Caval, V.; Suspene, R.; Hallez, C.; Pineau, P.; Wain-Hobson, S.; Vartanian, J.P. APOBEC3de antagonizes hepatitis B virus restriction factors APOBEC3f and APOBEC3g. J. Mol. Biol. 2016, 428, 3514–3528. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Henry, M.; Marchio, A.; Suspene, R.; Aynaud, M.M.; Guetard, D.; Cervantes-Gonzalez, M.; Battiston, C.; Mazzaferro, V.; Pineau, P.; et al. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 2010, 6, e1000928. [Google Scholar] [CrossRef] [PubMed]
- Kock, J.; Blum, H.E. Hypermutation of hepatitis B virus genomes by APOBEC3g, APOBEC3c and APOBEC3h. J. Gen. Virol. 2008, 89, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Rosler, C.; Malim, M.H.; von Weizsacker, F. Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3c. Hepatology 2007, 46, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3g. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Tal, G.; Lider, O.; Shaul, Y. Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J. Immuno.l 2005, 175, 3165–3176. [Google Scholar] [CrossRef]
- Purvina, M.; Hoste, A.; Rossignol, J.M.; Lagaudriere-Gesbert, C. Human hepatitis B viral e antigen and its precursor p20 inhibit T lymphocyte proliferation. Biochem. Biophys. Res. Commun. 2012, 417, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Kuo, C.F.; Akbari, O.; Ou, J.H. Maternal-derived hepatitis B virus E antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity 2016, 44, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Peppa, D.; Micco, L.; Javaid, A.; Kennedy, P.T.; Schurich, A.; Dunn, C.; Pallant, C.; Ellis, G.; Khanna, P.; Dusheiko, G.; et al. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLoS Pathog. 2010, 6, e1001227. [Google Scholar] [CrossRef] [PubMed]
- Tjwa, E.T.; van Oord, G.W.; Hegmans, J.P.; Janssen, H.L.; Woltman, A.M. Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis B. J. Hepatol. 2011, 54, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M.U. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 2009. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Hou, N.; Meng, J.; Wang, X.; Zhang, X.; Zhao, D.; Liu, Y.; Zhu, F.; Zhang, L.; Sun, W.; et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (TIM-3) mediates natural killer cell suppression in chronic hepatitis B. J. Hepatol. 2010, 52, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Bonorino, P.; Ramzan, M.; Camous, X.; Dufeu-Duchesne, T.; Thelu, M.A.; Sturm, N.; Dariz, A.; Guillermet, C.; Pernollet, M.; Zarski, J.P.; et al. Fine characterization of intrahepatic NK cells expressing natural killer receptors in chronic hepatitis B and C. J. Hepatol. 2009, 51, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Li, J.; Wands, J.R.; Wen, Y.M. Hepatitis B virus genetic variants: Biological properties and clinical implications. Emerg. Microbes. Infect. 2013, 2, e10. [Google Scholar] [CrossRef] [PubMed]
- Shouval, D. Hepatitis B vaccines. J. Hepatol. 2003, 39 (Suppl. 1), S70–S76. [Google Scholar] [CrossRef]
- Poovorawan, Y.; Theamboonlers, A.; Sanpavat, S.; Chumdermpadetsuk, S.; Safary, A.; Vandepapeliere, P. Long-term antibody persistence after booster vaccination with combined tetravalent diphtheria tetanus, whole-cell bordetella pertussis and hepatitis B vaccine in healthy infants. Ann. Trop. Paediatr. 1997, 17, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Iino, S.; Shiraki, K.; Akahane, Y.; Okamoto, H.; Domoto, K.; Mishiro, S. Safety and efficacy of a recombinant yeast-derived pre-S2 + S-containing hepatitis B vaccine (TGP-943): Phase 1, 2 and 3 clinical testing. Vaccine 1994, 12, 1090–1096. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Interferon-Stimulated Gene | ISG Class | Effect on HCV | Effect on HBV | Reference |
|---|---|---|---|---|
| ADAP2 | Direct antiviral | Inhibits entry | - | [20] |
| ADAR | Direct antiviral | RNA editing | - | [21] |
| APOBEC1, 3A, 3B, 3G, 3F | Direct antiviral | - | cccDNA/HBV DNA editing | [22,23,24,25,26,27] |
| AID | Direct antiviral | - | cccDNA editing | [28,29,30] |
| CH25H | Unknown | Inhibits replication | Inhibits entry | [31,32,33,34] |
| DDIT4 | Unknown | Inhibits infection | - | [21] |
| DDX58 (RIG-I) | RNA sensor | Enhanced viral sensing | Inhibits replication | [21,35,36,37] |
| DDX60 | RIG-I enhancer | Inhibits infection | - | [21,38,39] |
| DDX60L | Unknown | Inhibits replication | - | [40] |
| EIF2AK2 (PKR) | RNA sensor | Inhibition of translation | Inhibition of replication | [41,42,43,44] |
| GBP1 | Unknown | Inhibits infection | - | [45] |
| IFI44L | Unknown | Inhibits infection | - | [21] |
| IFI6 | Unknown | Inhibits HCV entry | - | [45,46] |
| IFI27 | Unknown | Inhibits infection | - | [21] |
| IFIT1 | Direct antiviral | Sequesters viral nucleic acids | - | [45,47] |
| IFIT3 | MAVS/TBK1 enhancer | Inhibits infection | - | [48] |
| IFITM1 | Direct antiviral | Inhibits entry and replication | Inhibition of replication | [48,49,50,51] |
| IFITM3 | Direct antiviral | Inhibits translation | - | [52] |
| IRF1 | ISG inducer | Inhibits infection | Inhibition of replication | [21,44,53] |
| IRF2 | Unknown | Inhibits infection | - | [21] |
| IRF7 | IFN inducer | Inhibits infection | - | [21] |
| IRF9 | IFN inducer | Inhibits infection | - | [54] |
| ISG12 | Ubiquitin-dependent degradation | Viral protein degradation | - | [45] |
| ISG20 | Direct antiviral | Cleaves RNA genome | Inhibits encapsidation, Cleaves RNA genome | [42,55,56] |
| ISG56 | Unknown | Inhibits replication | - | [49] |
| MAP3K14 | Unknown | Inhibits infection | - | [21] |
| MDA5 | RNA sensor | Inhibits infection | - | [21,57] |
| MOV10 | Unknown | Inhibits infection | - | [21] |
| MS4A4A | Unknown | Inhibits infection | - | [21] |
| MX1 | ISG inducer | Inhibits infection | Inhibits replication | [45,58,59] |
| MYD88 | IFN inducer | - | RNA degradation | [60] |
| NOS2 | Unknown | - | Inhibits replication | [48,61] |
| NT5C3 | Unknown | Inhibits infection | - | [21] |
| OAS1 | RNase I activator | Inhibits infection | - | [45,62] |
| OASL | Unknown | Inhibits replication | - | [21,63] |
| PLSCR1 | Unknown | Inhibits infection | Inhibits replication | [48,64] |
| RNASEL | Direct antiviral | Cleaves RNA genome | Inhibition of replication | [44,48] |
| R2AD2 | Direct antiviral | Inhibits replication | - | [48] |
| STING | IFN inducer | - | Inhibits HBV assembly | [65] |
| SOCS3 | IFN negative regulator | Inhibits replication | - | [66,67] |
| STAT1/3 | IFN inducer | Inhibits replication | Inhibits transcription | [68,69,70] |
| BST2 (Tetherin) | Direct antiviral | Inhibits HCV secretion | Inhibits HBV secretion | [71,72,73] |
| TLR3 | RNA sensor | Inhibits infection | Inhibits replication | [74,75,76] |
| TLR7 | RNA sensor | Inhibits infection | Inhibits replication | [77,78,79] |
| TLR8 | RNA sensor | Inhibits infection | Inhibits replication | [78,79] |
| TRIM14 | Unknown | Degradation of NS5a | - | [48,80] |
| TRIM12a | Unknown | Inhibits infection | - | [21] |
| TRIM22 | Unknown | Ubiquitination of NS5a | Inhibits transcription | [81,82] |
| TRIM41 | E3 ubiquitin ligase activator | - | Inhibits transcription | [83] |
| Viperin | Direct antiviral | Inhibits replication | - | [84] |
| ZAP | Unknown | - | Inhibits replication | [85,86] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega-Prieto, A.M.; Dorner, M. Immune Evasion Strategies during Chronic Hepatitis B and C Virus Infection. Vaccines 2017, 5, 24. https://doi.org/10.3390/vaccines5030024
Ortega-Prieto AM, Dorner M. Immune Evasion Strategies during Chronic Hepatitis B and C Virus Infection. Vaccines. 2017; 5(3):24. https://doi.org/10.3390/vaccines5030024
Chicago/Turabian StyleOrtega-Prieto, Ana Maria, and Marcus Dorner. 2017. "Immune Evasion Strategies during Chronic Hepatitis B and C Virus Infection" Vaccines 5, no. 3: 24. https://doi.org/10.3390/vaccines5030024
APA StyleOrtega-Prieto, A. M., & Dorner, M. (2017). Immune Evasion Strategies during Chronic Hepatitis B and C Virus Infection. Vaccines, 5(3), 24. https://doi.org/10.3390/vaccines5030024