
The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
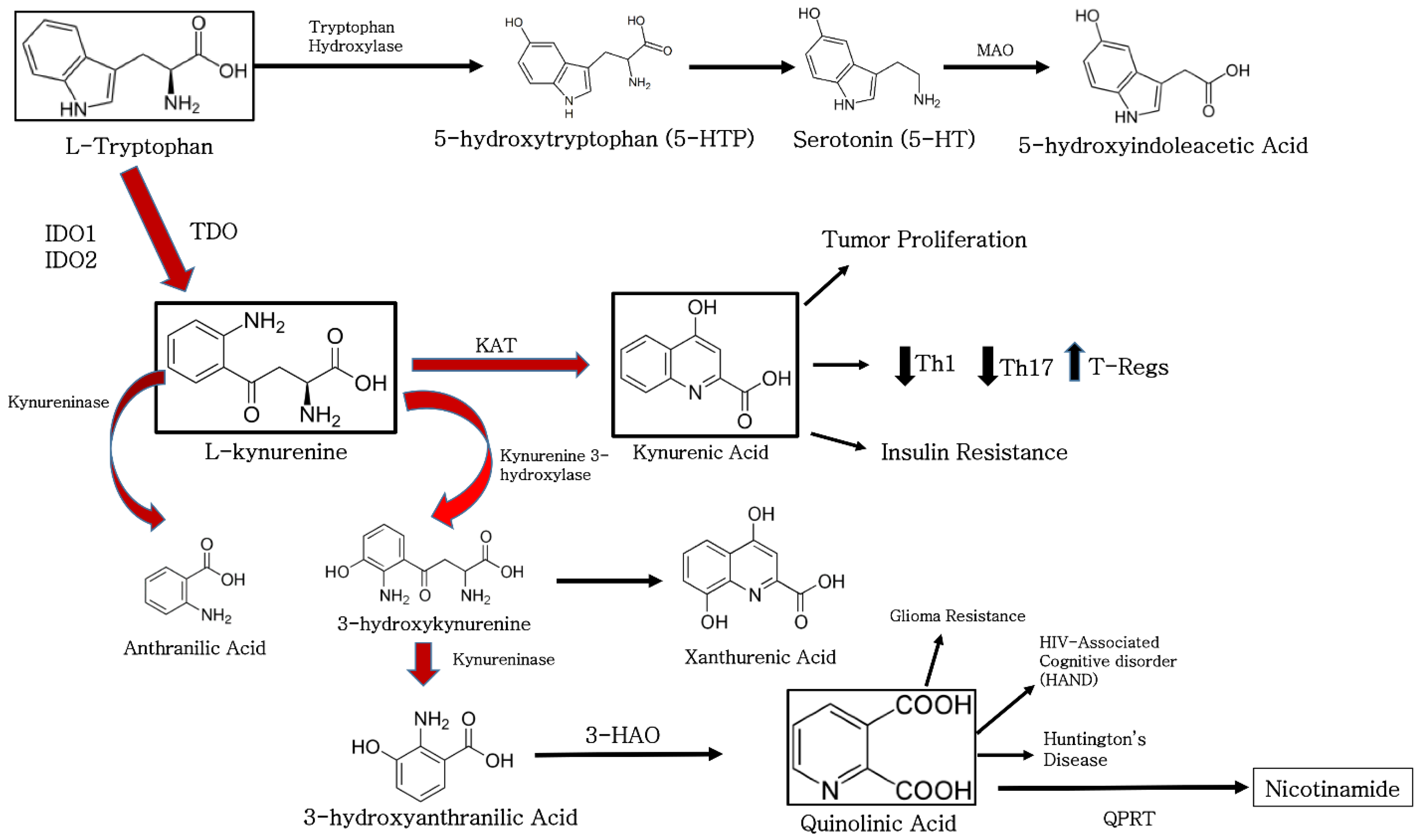
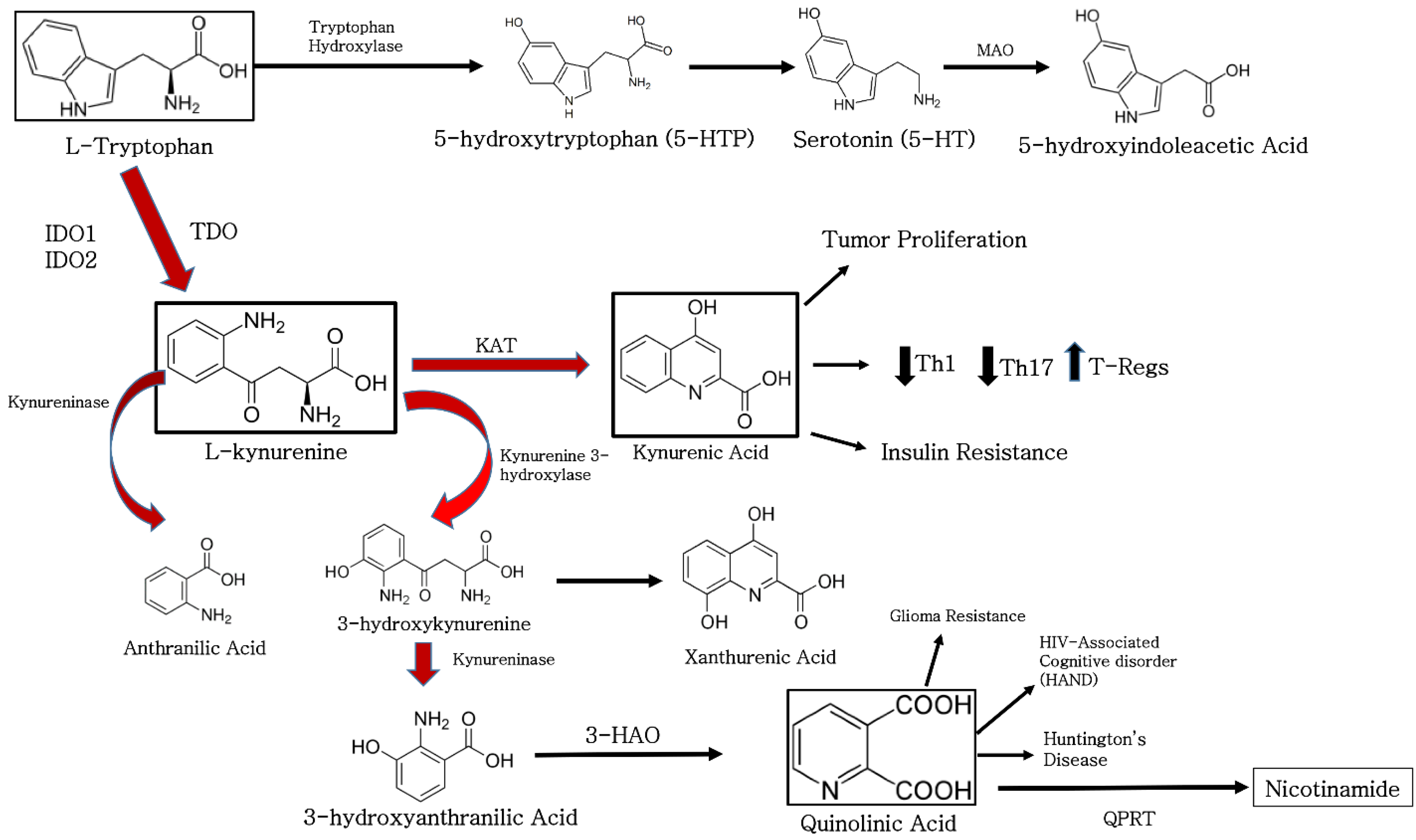
2. The Function of Indoleamine 2, 3-Dioxygenase in Biological Systems
2.1. IDO Function in Stem Cells
2.2. The Function of IDO in Cells of the Nervous System
3. Mechanisms of IDO1 Induction and Function
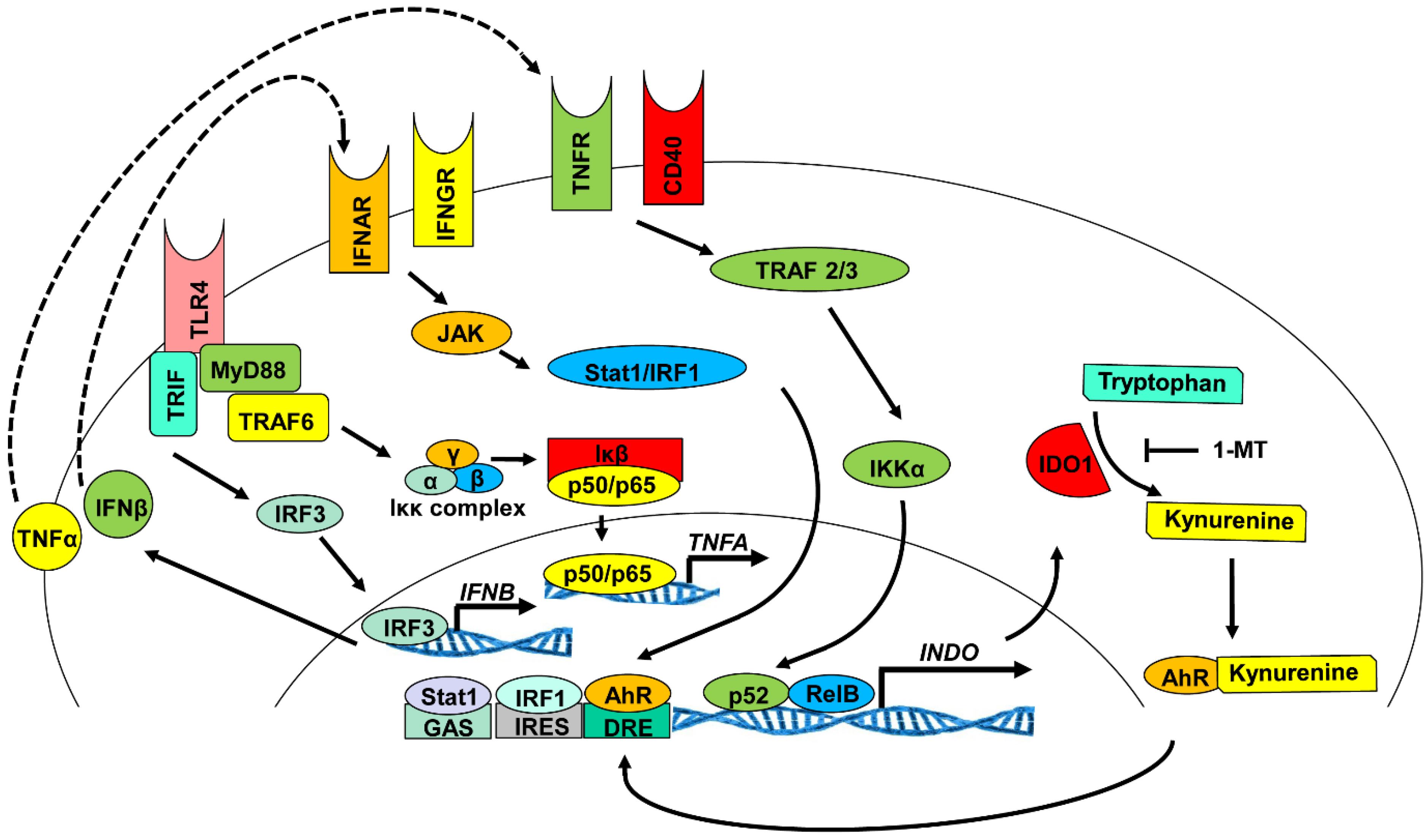
3.1. Signaling Pathways Responsible for the Induction of IDO1 Expression
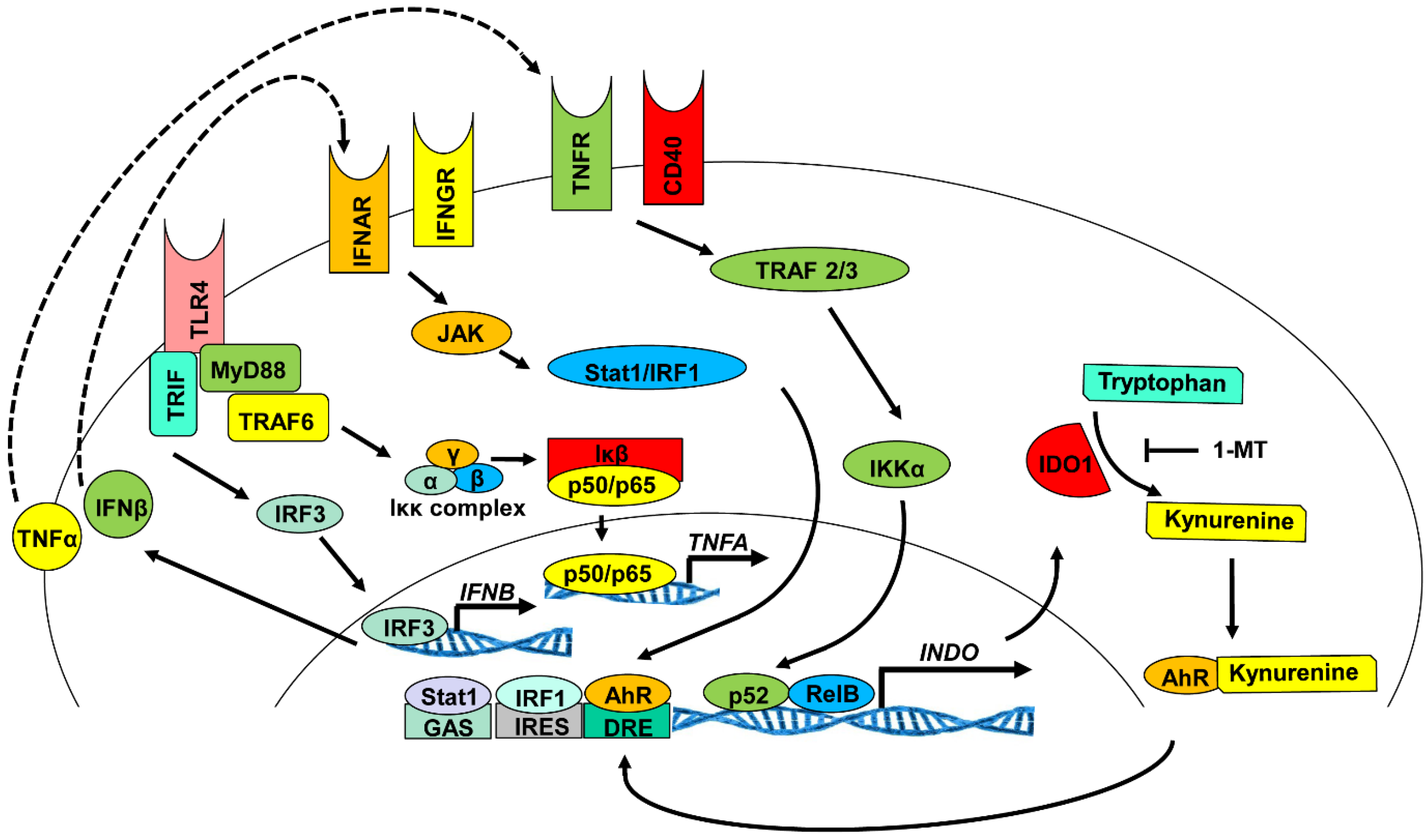
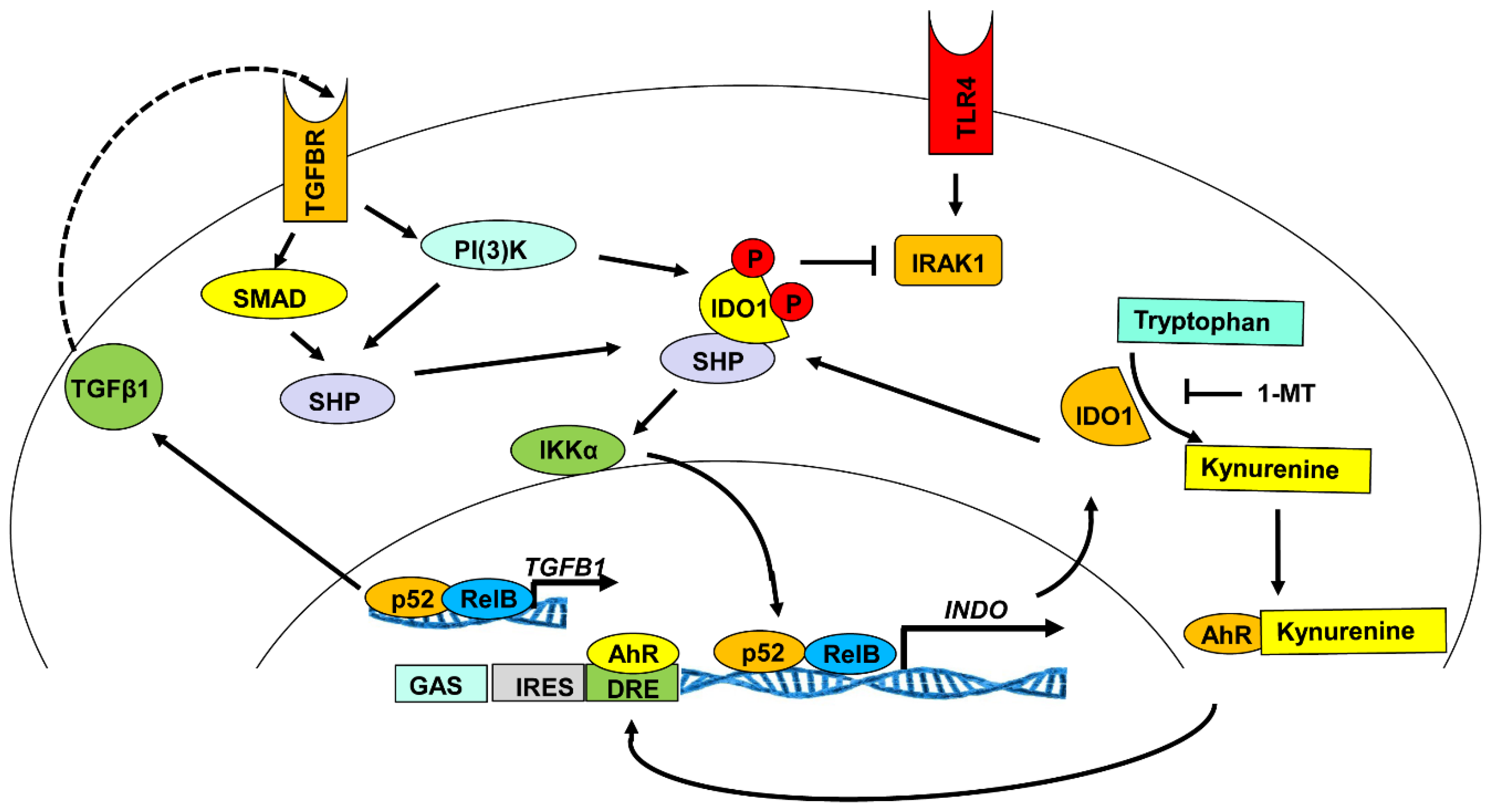
3.2. Enzymatic Activity of IDO1
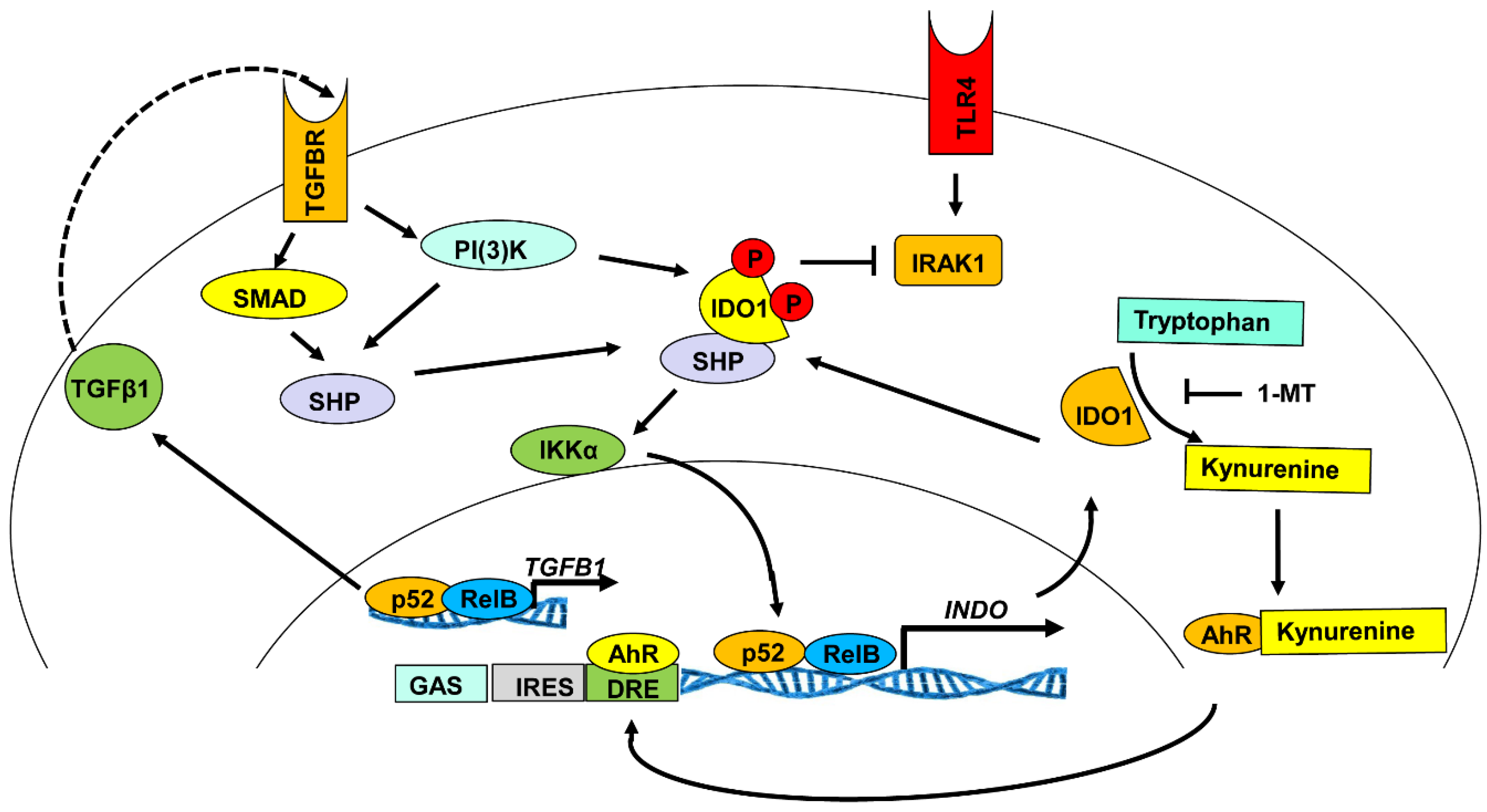
3.3. Indoleamine 2, 3-Dioxygenase 1 Signaling Activity
4. The Role of IDO in Immune Suppression
4.1. The Function of IDO in Organ and Tissue Graft Survival
4.2. The Function of Indoleamine 2, 3-Dioxygenase in Viral Infection
4.3. The Role of Indoleamine 2, 3-Dioxygenase in the Promotion of Cancer Cell Survival
4.4. The Role of Indoleamine 2, 3-Dioxygenase in Tissue-Specific Autoimmunity
5. Immune Suppressive Vaccines: The Case for CTB-Autoantigens and Their Relationship to IDO1
5.1. Immunosuppressive Vaccine Induction of Indoleamine 2, 3-Dioxygenase
6. Conclusions
Acknowledgement
Author Contributions
Conflicts of Interest
References
- Sugimoto, H.; Oda, S.; Otsuki, T.; Hino, T.; Yoshida, T.; Shiro, Y. Crystal structure of human indoleamine 2, 3-dioxygenase: Catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. USA 2006, 103, 2611–2616. [Google Scholar] [CrossRef] [PubMed]
- Heitger, A. Regulation of expression and function of IDO in human dendritic cells. Curr. Med. Chem. 2011, 18, 2222–2233. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, H.J.; Ball, H.J.; Ho, Y.F.; Austin, C.J.; Whittington, C.M.; Belov, K.; Maghzal, G.J.; Jermiin, L.S.; Hunt, N.H. Characterization and evolution of vertebrate indoleamine 2, 3-dioxygenases IDOs from monotremes and marsupials. Comp. Biochem. Physiol. 2009, 153, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.J.; Sanchez-Perez, A.; Weiser, S.; Austin, C.J.D.; Astelbauer, F.; Miu, J.; McQuillan, J.A.; Stocker, R.; Jermiin, L.S.; Hunt, N.H. Characterization of an indoleamine 2, 3-dioxygenase-like protein found in humans and mice. Gene 2007, 396, 203–213. [Google Scholar] [CrossRef]
- Zhang, Y.; Kang, S.A.; Mukherjee, T.; Bale, S.; Crane, B.R.; Begley, T.P.; Ealick, S.E. Crystal structure and mechanism of tryptophan 2, 3-dioxygenase, a heme enzyme involved in tryptophan catabolism and in quinolinate biosynthesis. Biochemistry 2007, 46, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Panozzo, C.; Nawara, M.; Suski, C.; Kucharczyka, R.; Skoneczny, M.; Bécam, A.-M.; Rytka, J.; Herbert, C.J. Aerobic and anaerobic NAD+ metabolism in Saccharomyces cerevisiae. FEBS Lett. 2002, 517, 97–102. [Google Scholar] [CrossRef]
- Metz, R.; Duhadaway, J.B.; Kamasani, U.; Laury-Kleintop, L.; Muller, A.J.; Prendergast, G.C. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2, 3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007, 67, 7082–7087. [Google Scholar] [CrossRef] [PubMed]
- Lob, S.; Königsrainer, A.; Zieker, D.; Brücher, B.L.; Rammensee, H.G.; Opelz, G.; Terness, P. IDO1 and IDO2 are expressed in human tumors: Levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol. Immunother. 2009, 58, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Metz, R.; Muller, A.J.; Merlo, L.M.; Mandik-Nayak, L. IDO2 in immunomodulation and autoimmune disease. Front. Immunol. 2014. [Google Scholar] [CrossRef]
- Najfeld, V.; Menninger, J.; Muhleman, D.; Comings, D.E.; Gupta, S.L. Localization of indoleamine 2, 3-dioxygenase gene (INDO) to chromosome 8p12 ≥ p11 by fluorescent in situ hybridization. Cytogenet. Cell Genet. 1993, 64, 231–232. [Google Scholar] [CrossRef]
- Belladonna, M.L.; Puccetti, P.; Orabona, C.; Fallarino, F.; Vacca, C.; Volpi, C.; Gizzi, S.; Pallotta, M.T.; Fioretti, M.C.; Grohmann, U. Immunosuppression via tryptophan catabolism: The role of kynurenine pathway enzymes. Transplantation 2007, 84, S17–S20. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Chandler, P.; Lee, G.K.; Johnson, T.; Keskin, D.B.; Lee, J.; Munn, D.H. Indoleamine 2, 3-dioxygenase, immunosuppression and pregnancy. J. Reprod. Immunol. 2002, 57, 143–150. [Google Scholar] [CrossRef]
- Mellor, A. Indoleamine 2, 3-dioxygenase and regulation of T cell immunity. Biochem. Biophys. Res. Commun. 2005, 338, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2, 3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Riminucci, M.; Gronthos, S.; Robey, P.G. Bone marrow stromal stem cells: Nature, biology, and potential applications. Stem Cells 2001, 19, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Chen, X.; Huang, Y.; Li, W.; Li, J.; Cao, K.; Cao, G.; Zhang, L.; Li, F.; Roberts, A.I. Phylogenetic distinction of iNOS and IDO function in mesenchymal stem cell-mediated immunosuppression in mammalian species. Cell Death Differ. 2014, 21, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.F.; Su, J.J.; Roberts, A.I.; Shou, P.; Rabson, A.B.; Ren, G.W. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 2012, 33, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Meisel, R.; Laryea, M.; Göbel, U.; Däubener, W.; Dilloo, D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2, 3-dioxygenase-mediated tryptophan degradation. Blood 2004, 103, 4619–4621. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.W.; Su, J.; Zhang, L.; Zhao, X.; Ling, W.; L’huillie, A.; Zhang, J.; Lu, Y.; Roberts, A.I.; Ji, W.; et al. Species variation in the mechanisms of mesenchymal stem cell-mediated Immunosuppression. Stem Cells 2009, 27, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.; Rüssing, D.; Adams, O.; Ailyati, A.; Sik Kim, K.; Schroten, H.; Däubener, W. Role of human brain microvascular endothelial cells during central nervous system infection. Significance of indoleamine 2, 3-dioxygenase in antimicrobial defence and immunoregulation. Thromb. Haemost. 2005, 94, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Kwidzinski, E.; Bechmann, I. IDO expression in the brain: A double-edged sword. J. Mol. Med. (Berl) 2007, 85, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.S.; Zhao, M.L.; Rivieccio, M.; Choi, S.; Connolly, E.; Zhao, Y.; Takikawa, O.; Brosnan, C.F.; Lee, S.C. Astrocyte indoleamine 2, 3-dioxygenase is induced by the TLR3 ligand poly(I:C): Mechanism of induction and role in antiviral response. J. Virol. 2007, 81, 9838–9850. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, P.A.; Begolka, W.S.; Olson, J.K.; Elhofy, A.; Karpus, W.J.; Miller, S.D. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 2005, 49, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Krumbholz, M.; Giese, T.; Hartmann, G.; Aloisi, F.; Meinl, E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J. Neuroimmunol. 2005, 159, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Meli, E.; Moroni, F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J. Neurochem. 2001, 77, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. USA 1996, 93, 12553–12558. [Google Scholar] [CrossRef] [PubMed]
- Heine, W.; Radke, M.; Wutzke, K.D. The significance of tryptophan in human nutrition. Amino Acids 1995, 9, 91–205. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Ohta, Y.; Shinohara, R.; Ishiguro, I. Contribution of serum albumin to the transport of orally administered L-tryptophan into liver of rats with L-tryptophan depletion. Amino Acids 1999, 16, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Mbongue, J.C.; Nicholas, D.A.; Zhang, K.; Kim, N.S.; Hamilton, B.N.; Larios, M.; Zhang, G.; Umezawa, K.; Firek, A.F.; Langridge, W.H. Induction of indoleamine 2, 3-dioxygenase in human dendritic cells by a cholera toxin B subunit-proinsulin vaccine. PLoS ONE 2015, 10, e0118562. [Google Scholar] [CrossRef] [PubMed]
- Tas, S.W.; Vervoordeldonk, M.J.; Hajji, N.; Schuitemake, J.H.N.; van der Sluijs, K.F.; May, M.J.; Ghosh, S.; Kapsenberg, M.L.; Tak, P.P.; de Jong, E.C. Noncanonical NF-kappaB signaling in dendritic cells is required for indoleamine 2, 3-dioxygenase (IDO) induction and immune regulation. Blood 2007, 110, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Du, M.X.; Sotero-Esteva, W.D.; Taylor, M.W. Analysis of transcription factors regulating induction of indoleamine 2, 3-dioxygenase by IFN-gamma. J. Interferon Cytokine Res. 2000, 20, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Courtney, J.M.; Falkenhagen, D.; Ivanovich, P.; Schuett, W. Adsorption technologies and blood purification procedures: Honoring horst klinkmann. Artif. Organs 2002, 26, 84–208. [Google Scholar]
- Chon, S.Y.; Hassanain, H.H.; Pine, R.; Gupta, S.L. Involvement of two regulatory elements in interferon-gamma-regulated expression of human indoleamine 2, 3-dioxygenase gene. J. Interferon Cytokine Res. 1995, 15, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Hale, P.T.; Carlin, J.M. The role of IFN-gamma and TNF-alpha-responsive regulatory elements in the synergistic induction of indoleamine dioxygenase. J. Interferon Cytokine Res. 2005, 25, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Konan, K.V.; Taylor, M.W. Importance of the two interferon-stimulated response element (ISRE) sequences in the regulation of the human indoleamine 2, 3-dioxygenase gene. J. Biol. Chem. 1996, 271, 19140–19145. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Bebien, M.; Otero, D.C.; Johnson-Vroom, K.E.; Cao, Y.; Vu, D.; Jegga, A.G.; Aronow, B.J.; Ghosh, G.; Rickert, R.C.; et al. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, P.; Grohmann, U. IDO and regulatory T cells: A role for reverse signalling and non-canonical NF-kappaB activation. Nat. Rev. Immunol. 2007, 7, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Fernandez, M.V.; Plumas, J.; Chaperot, L.; Bhardwaj, N. Activation of the noncanonical NF-kappaB pathway by HIV controls a dendritic cell immunoregulatory phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 14122–14127. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Hale, P.T.; Carlin, J.M. NF-kappa B activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN-gamma and tumor necrosis factor-alpha. Cytokine 2006, 35, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Katz, B.; Spinola, S.M. Haemophilus ducreyi lipooligosaccharides induce expression of the immunosuppressive enzyme indoleamine 2, 3-dioxygenase via type I interferons and tumor necrosis factor alpha in human dendritic cells. Infect. Immun. 2011, 79, 3338–3347. [Google Scholar] [CrossRef] [PubMed]
- Fujigaki, S.; Saito, K.; Sekikawa, K.; Tone, S.; Takikawa, O.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Lipopolysaccharide induction of indoleamine 2, 3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur. J. Immunol. 2001, 31, 2313–2318. [Google Scholar] [PubMed]
- Horvath, C.M. The Jak-STAT pathway stimulated by interferon gamma. Sci. STKE 2004. [Google Scholar] [CrossRef]
- Jurgens, B.; Hainz, U.; Fuchs, D.; Felzmann, T.; Heitger, A. Interferon-gamma-triggered indoleamine 2, 3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood 2009, 114, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2, 3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Wu, D.; Goth, S.R.; Baek, J.; Lollies, A.; Domhardt, R.; Grindel, A.; Pessah, I.N. Aryl hydrocarbon receptor signaling regulates NF-kappaB RelB activation during dendritic-cell differentiation. Immunol. Cell Biol. 2013, 91, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Mizuno, S.; Ishikawa, E.; Tanemura, A.; Murata, Y.; Kuriyama, N.; Azumi, Y.; Kishiwada, M.; Usui, M.; Sakurai, H.; et al. Negative prognostic impact of renal replacement therapy in adult living-donor liver transplant recipients: Preoperative recipient condition and donor factors. Transplant. Proc. 2014, 46, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [PubMed]
- Zaher, S.S.; Germain, C.; Fu, H.; Larkin, D.F.; George, A.J. 3-Hydroxykynurenine suppresses CD4+ T-cell proliferation, induces T-regulatory-cell development, and prolongs corneal allograft survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2640–2648. [Google Scholar] [CrossRef] [PubMed]
- Pop, S.M.; Wong, C.P.; He, Q.; Wang, Y.; Wallet, M.A.; Goudy, K.S.; Tisch, R. The type and frequency of immunoregulatory CD4+ T-cells govern the efficacy of antigen-specific immunotherapy in nonobese diabetic mice. Diabetes 2007, 56, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Egilmez, N.K. Indoleamine 2, 3-dioxygenase and dendritic cell tolerogenicity. Immunol. Investig. 2012, 41, 738–764. [Google Scholar] [CrossRef] [PubMed]
- Mbongue, J.; Nicholas, D.; Firek, A.; Langridge, W. The role of dendritic cells in tissue-specific autoimmunity. J. Immunol. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Shinde, R.; Liu, H.; Gnana-Prakasam, J.P.; Veeranan-Karmegam, R.; Huang, L.; Ravishankar, B.; Bradley, J.; Kvirkvelia, N.; McMenamin, M.; et al. Amino acid metabolism inhibits antibody-driven kidney injury by inducing autophagy. J. Immunol. 2015, 194, 5713–5724. [Google Scholar] [CrossRef] [PubMed]
- Belladonna, M.L.; Orabona, C.; Grohmann, U.; Puccetti, P. TGF-beta and kynurenines as the key to infectious tolerance. Trends Mol. Med. 2009, 15, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Terness, P.; Bauer, T.M.; Röse, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2, 3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2, 3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, M.T.; Orabona, C.; Bianchi, R.; Vacca, C.; Fallarino, F.; Belladonna, M.L.; Volpi, C.; Mondanelli, G.; Gargaro, M.; Allegrucci, M.; et al. Forced IDO1 expression in dendritic cells restores immunoregulatory signalling in autoimmune diabetes. J. Cell. Mol. Med. 2014, 18, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Belladonna, M.L.; Volpi, C.; Bianchi, R.; Vacca, C.; Orabona, C.; Pallotta, M.T.; Boon, L.; Gizzi, S.; Fioretti, M.C.; Grohmann, U.; et al. Cutting edge: Autocrine TGF-beta sustains default tolerogenesis by IDO-competent dendritic cells. J. Immunol. 2008, 181, 5194–5198. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, J.; Weeratna, R.; Payette, P.; Jurk, M.; Schetter, C.; Laucht, M.; Wader, T.; Tluk, S.; Liu, M.; Davis, H.L.; et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 2004, 34, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Baban, B.; Chandler, P.R.; Manlapat, A.; Kahler, D.J.; Munn, D.H. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2, 3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J. Immunol. 2005, 175, 5601–5605. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A., 3rd; Kahler, D.J.; Baban, B.; Chandler, P.R.; Kang, B.; Shimoda, M.; Koni, P.A.; Pihkala, J.; Vilagos, B.; Busslinger, M.; et al. B-lymphoid cells with attributes of dendritic cells regulate T cells via indoleamine 2, 3-dioxygenase. Proc. Natl. Acad. Sci. USA 2010, 107, 10644–10648. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Volpi, C.; Zelante, T.; Vacca, C.; Calvitti, M.; Fioretti, M.C.; Puccetti, P.; Romani, L.; Grohmann, U. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J. Immunol. 2009, 183, 6303–6312. [Google Scholar] [CrossRef] [PubMed]
- Lind, E.F.; Ahonen, C.L.; Wasiuk, A.; Kosaka, Y.; Becher, B.; Bennett, K.A.; Noelle, R.J. Dendritic cells require the NF-kappaB2 pathway for cross-presentation of soluble antigens. J. Immunol. 2008, 181, 354–363. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Hou, J.; Zhou, J.; Zhao, W.; Xu, H.; Zheng, Y.; Yu, Y.; Liu, S.; Cao, X. Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat. Immunol. 2008, 9, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Tabata, M.; Taguri, M.; Manabe, S.; Morita, S.; Takanashi, S. Extensive reconstruction of the left anterior descending coronary artery with an internal thoracic artery graft. Ann. Thorac. Surg. 2011, 91, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Sucher, R.; Fischler, K.; Oberhuber, R.; Kronberger, I.; Margreiter, C.; Ollinger, R.; Schneeberger, S.; Fuchs, D.; Werner, E.R.; Watschinger, K.; et al. IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated Ag-4 Ig-mediated long-term solid organ allograft survival. J. Immunol. 2012, 188, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.; Rössner, S.; Onderka, J.; Lechmann, M.; Pallotta, M.T.; Fallarino, F.; Boon, L.; Nicolette, C.; DeBenedette, M.A.; Tcherepanova, I.Y.; et al. Topical application of soluble CD83 induces IDO-mediated immune modulation, increases Foxp3+ T cells, and prolongs allogeneic corneal graft survival. J. Immunol. 2013, 191, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Brandacher, G.; Cakar, F.; Winkler, C.; Schneeberger, S.; Obrist, P.; Bösmüller, C.; Werner-Felmayer, G.; Werner, E.R.; Bonatti, H.; Margreiter, R.; et al. Non-invasive monitoring of kidney allograft rejection through IDO metabolism evaluation. Kidney Int. 2007, 71, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Z.; Liu, L.; Liu, K.; Bizargity, P.; Hancock, W.W.; Visner, G.A. Reduced cytotoxic function of effector CD8+ T cells is responsible for indoleamine 2, 3-dioxygenase-dependent immune suppression. J. Immunol. 2009, 183, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.L.; Lin, Y.H.; Xiao, H.; Xing, S.; Chen, H.; Chi, P.D.; Zhang, G. Epstein-Barr virus infection induces indoleamine 2, 3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-kappaB pathways: Impairment in T cell functions. J. Virol. 2014, 88, 6660–6671. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.S.; Naif, H.; Thuruthyil, S.J.; Nasr, N.; Littlejohn, T.; Takikawa, O.; Kapoor, V. Induction of indolamine 2, 3-dioxygenase in primary human macrophages by human immunodeficiency virus type 1 is strain dependent. J. Virol. 2000, 74, 4110–4115. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Herbeuval, J.P.; Hardy, A.W.; Anderson, S.A.; Dolan, M.J.; Fuchs, D.; Shearer, G.M. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2, 3-dioxygenase in plasmacytoid dendritic cells. Blood 2007, 109, 3351–3359. [Google Scholar] [CrossRef] [PubMed]
- Douek, D.C.; Roederer, M.; Koup, R.A. Emerging concepts in the immunopathogenesis of AIDS. Annu. Rev. Med. 2009, 60, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Donaghy, H.; Harman, A.N.; Kim, M.; Turville, S.G. Manipulation of dendritic cell function by viruses. Curr. Opin. Microbiol. 2010, 13, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Pantaleo, G.; Stanley, S.; Weissman, D. Immunopathogenic mechanisms of HIV infection. Ann. Intern. Med. 1996, 124, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, D.; Forsman, A.; Hagberg, L.; Larsson, M.; Norkrans, G.; Reibnegger, G.; Werner, E.R.; Wachter, H. Immune activation and decreased tryptophan in patients with HIV-1 infection. J. Interferon Res. 1990, 10, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Linde, A. Diagnosis of Epstein-Barr virus-related diseases. Scand. J. Infect. Dis. Suppl. 1996, 100, 83–88. [Google Scholar] [PubMed]
- Maeda, E.; Akahane, M.; Kiryu, S.; Kato, N.; Yoshikawa, T.; Hayashi, N.; Aoki, S.; Minami, M.; Uozaki, H.; Fukayama, M.; et al. Spectrum of Epstein-Barr virus-related diseases: A pictorial review. Jpn. J .Radiol. 2009, 27, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.; Herrera, R.; Veluppillai, P.; Greenspan, J.; Greenspan, D.; Palefsky, J.M. Epstein-Barr virus (EBV)-infected monocytes facilitate dissemination of EBV within the oral mucosal epithelium. J. Virol. 2007, 81, 5484–5496. [Google Scholar] [CrossRef] [PubMed]
- Walling, D.M.; Ray, A.J.; Nichols, J.E.; Flaitz, C.M.; Nichols, C.M. Epstein-Barr virus infection of Langerhans cell precursors as a mechanism of oral epithelial entry, persistence, and reactivation. J. Virol. 2007, 81, 7249–7268. [Google Scholar] [CrossRef] [PubMed]
- Savard, M.; Bélanger, C.; Tremblay, M.J.; Dumais, N.; Flamand, L.; Borgeat, P.; Gosselin, J. EBV suppresses prostaglandin E2 biosynthesis in human monocytes. J. Immunol. 2000, 164, 6467–6473. [Google Scholar] [CrossRef] [PubMed]
- Savard, M.; Bélanger, C.; Tardif, M.; Gourde, P.; Flamand, L.; Gosselin, J. Infection of primary human monocytes by Epstein-Barr virus. J. Virol. 2000, 74, 2612–2619. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; Prendergast, G.C. Marrying immunotherapy with chemotherapy: Why say IDO? Cancer Res. 2005, 65, 8065–8068. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Lob, S.; Konigsrainer, A.; Schafer, R.; Rammensee, H.G.; Opelz, G.; Terness, P. Levo- but not dextro-1-methyl tryptophan abrogates the IDO activity of human dendritic cells. Blood 2008, 111, 2152–2154. [Google Scholar] [CrossRef] [PubMed]
- Thaker, A.I.; Rao, M.S.; Bishnupuri, K.S.; Kerr, T.A.; Foster, L.; Marinshaw, J.M.; Newberry, R.D.; Stenson, W.F.; Ciorba, M.A. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology 2013, 145, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Ino, K.; Yamamoto, E.; Shibata, K.; Kajiyama, H.; Yoshida, N.; Terauchi, M.; Nawa, A.; Nagasaka, T.; Takikawa, O.; Kikkawa, F. Inverse correlation between tumoral indoleamine 2, 3-dioxygenase expression and tumor-infiltrating lymphocytes in endometrial cancer: Its association with disease progression and survival. Clin. Cancer Res. 2008, 14, 2310–2317. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.F.; Idema, A.J.; Bol, K.F.; Grotenhuis, J.A.; de Vries, I.J.; Wesseling, P.; Adema, G.J. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J. Neuroimmunol. 2010, 225, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Bolduc, A.R.; Hoda, M.N.; Gamble, D.N.; Dolisca, S.-B.; Bolduc, A.K.; oang, K.; Ashley, C.; McCall, D.; Rojiani, A.M. The indoleamine 2, 3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J. Immunother. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Li, C.-F.; Kuo, C.-C.; Tsai, K.K.; Hou, M.-F.; Hung, W.-C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2, 3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.; Rawal, B.; Fulp, J.; Lee, J.H.; Lopez, A.; Bui, M.M.; Khalil, F.; Antonia, S.; Yfantis, H.G.; Lee, D.H.; Dorsey, T.H.; Ambs, S. Analysis of indoleamine 2–3 dioxygenase (IDO1) expression in breast cancer tissue by immunohistochemistry. Cancer Immunol. Immunother. 2013, 62, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Boyland, E.; Williams, D.C. Metabolism of tryptophan. 2. Metabolism of tryptophan in patients suffering from cancer of the bladder. Biochem. J. 1956, 64, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Friberg, M.; Jennings, R.; Alsarraj, M.; Dessureault, S.; Cantor, A.; Extermann, M.; Mellor, A.L.; Munn, D.H.; Antonia, S.J. Indoleamine 2, 3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 2002, 101, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2, 3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Ueno, H.; Fay, J.; Banchereau, J. Dendritic cells: A critical player in cancer therapy? J. Immunother. 2008, 31, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J.; Mellman, I. Designing vaccines based on biology of human dendritic cell subsets. Immunity 2010, 33, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Ueno, H.; Banchereau, J. Recent developments in cancer vaccines. J. Immunol. 2011, 186, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, H.; Sprent, J. A defect in central tolerance in NOD mice. Nat. Immunol. 2001, 2, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Bianchi, R.; Orabona, C.; Vacca, C.; Belladonna, M.L.; Fioretti, M.C.; Serreze, D.V.; Grohmann, U.; Puccetti, P. CTLA-4-Ig activates forkhead transcription factors and protects dendritic cells from oxidative stress in nonobese diabetic mice. J. Exp. Med. 2004, 200, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Martino, G.; Hartung, H.P. Immunopathogenesis of multiple sclerosis: The role of T cells. Curr. Opin. Neurol. 1999, 12, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Opitz, C.A.; Ho, P.P.; Agrawal, A.; Lutz, C.; Weller, M.; Mellor, A.L.; Steinman, L.; Wick, W.; Platten, M. Mouse mesenchymal stem cells suppress antigen-specific TH cell immunity independent of indoleamine 2,3-dioxygenase 1 (IDO1). Stem Cells Dev. 2010, 19, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, K.; Zou, J.P.; Tschetter, J.R.; Ward, J.M.; Shearer, G.M. Effect of indoleamine 2, 3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 129, 186–196. [Google Scholar] [CrossRef]
- Kwidzinski, E.; Bunse, J.; Kovac, A.D.; Ullrich, O.; Zipp, F.; Nitsch, R.; Bechmann, I. IDO (indolamine 2, 3-dioxygenase) expression and function in the CNS. Adv. Exp. Med. Biol. 2003, 527, 113–118. [Google Scholar] [PubMed]
- Lemos, H.; Huang, L.; Chandler, P.R.; Mohamed, E.; Souza, G.R.; Li, L.; Pacholczyk, G.; Barber, G.N.; Hayakawa, Y.; Munn, D.H.; Mellor, A.L. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J. Immunol. 2014, 192, 5571–5578. [Google Scholar] [CrossRef] [PubMed]
- Cernea, S.; Dobreanu, M.; Raz, I. Prevention of type 1 diabetes: Today and tomorrow. Diabetes Metab. Res. Rev. 2010, 26, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K. Vaccines for type 1 diabetes in the late stage of clinical development. Indian J. Pharmacol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.C. The prospect of vaccination to prevent type 1 diabetes. Hum. Vaccin. 2005, 1, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Odumosu, O.; Nicholas, D.; Yano, H.; Langridge, W. AB toxins: A paradigm switch from deadly to desirable. Toxins (Basel) 2010, 2, 1612–1645. [Google Scholar] [CrossRef] [PubMed]
- Denes, B.; Krausova, V.; Fodor, N.; Timiryasova, T.; Henderson, D.; Hough, J.; Yu, J.; Fodor, I.; Langridge, W.H. Protection of NOD mice from type 1 diabetes after oral inoculation with vaccinia viruses expressing adjuvanted islet autoantigens. J. Immunother. 2005, 28, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Bergerot, I.; Ploix, C.; Petersen, J.; Moulin, V.; Rask, C.; Fabien, N.; Lindblad, M.; Mayer, A.; Czerkinsky, C.; Holmgren, J.; Thivolet, C. A cholera toxoid-insulin conjugate as an oral vaccine against spontaneous autoimmune diabetes. Proc. Natl. Acad. Sci. USA 1997, 94, 4610–4614. [Google Scholar] [CrossRef] [PubMed]
- George-Chandy, A.; Eriksson, K.; Lebens, M.; Nordström, I.; Schön, E.; Holmgren, J. Cholera toxin B subunit as a carrier molecule promotes antigen presentation and increases CD40 and CD86 expression on antigen-presenting cells. Infect. Immun. 2001, 69, 5716–5725. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Chong, D.K.; Langridge, W.H. Efficacy of a food plant-based oral cholera toxin B subunit vaccine. Nat. Biotechnol. 1998, 16, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Odumosu, O.; Nicholas, D.; Payne, K.; Langridge, W. Cholera toxin B subunit linked to glutamic acid decarboxylase suppresses dendritic cell maturation and function. Vaccine 2011, 29, 8451–8458. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, A.; Colucci, M.; Pugliese, O.; Quintieri, F.; Boirivant, M. Cholera toxin B subunit promotes the induction of regulatory T cells by preventing human dendritic cell maturation. J. Leukoc. Biol. 2008, 84, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Suzuki, N.; Yamashita, N.; Nagafuchi, H.; Nakajima, T.; Wakisaka, S.; Yamamoto, S.; Sakane, T. Characterization of T cells specific for an epitope of human 60-kD heat shock protein (HSP) in patients with Behcet’s disease (BD) in Japan. Clin. Exp. Immunol. 1997, 108, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Pickering, M.C.; Haskard, D.O. Behcet’s syndrome. J. R. Coll. Physicians Lond. 2000, 34, 169–177. [Google Scholar] [PubMed]
- Phipps, P.A.; Stanford, M.R.; Sun, J.B.; Xiao, B.G.; Holmgren, J.; Shinnick, T.; Hasan, A.; Mizushima, Y.; Lehner, T. Prevention of mucosally induced uveitis with a HSP60-derived peptide linked to cholera toxin B subunit. Eur. J. Immunol. 2003, 33, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.; Whittall, T.; Bergmeier, L.A.; Lindblad, M.; Lundin, S.; Shinnick, T.; Mizushima, Y.; Holmgren, J.; Lehner, T. Oral tolerization with peptide 336–351 linked to cholera toxin B subunit in preventing relapses of uveitis in Behcet’s disease. Clin. Exp. Immunol. 2004, 137, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.S.; Reid, H.H.; Beddoe, T.; Tynan, F.E.; Perugini, M.A.; Johns, T.G.; Bernard, C.C.; Rossjohn, J. The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 11059–11064. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Rodriguez, M. Multiple sclerosis, seizures, and antiepileptics: Role of IL-18, IDO, and melatonin. Eur. J. Neurol. 2011, 18, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.B.; Czerkinsky, C.; Holmgren, J. B Lymphocytes treated in vitro with antigen coupled to cholera toxin B subunit induce antigen-specific Foxp3(+) regulatory T cells and protect against experimental autoimmune encephalomyelitis. J. Immunol. 2012, 188, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Tisch, R.; McDevitt, H. Insulin-dependent diabetes mellitus. Cell 1996, 85, 291–297. [Google Scholar] [CrossRef]
- Eisenbarth, G.S. Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med. 1986, 314, 1360–1368. [Google Scholar] [PubMed]
- Melendez-Ramirez, L.Y.; Richards, R.J.; Cefalu, W.T. Complications of type 1 diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Ploix, C.; Bergerot, I.; Durand, A.; Czerkinsky, C.; Holmgren, J.; Thivolet, C. Oral administration of cholera toxin B-insulin conjugates protects NOD mice from autoimmune diabetes by inducing CD4+ regulatory T-cells. Diabetes 1999, 48, 2150–2156. [Google Scholar] [CrossRef] [PubMed]
- Denes, B.; Yu, J.; Fodor, N.; Takátsy, Z.; Fodor, I.; Langridge, W.H. Suppression of hyperglycemia in NOD mice after inoculation with recombinant vaccinia viruses. Mol. Biotechnol. 2006, 34, 317–327. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, W.; Shi, X.; Jin, Y.; Zhang, Y. Protection against autoimmune diabetes by silkworm-produced GFP-tagged CTB-insulin fusion protein. Clin. Dev. Immunol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Stroehla, B.C. The epidemiology of autoimmune diseases. Autoimmun. Rev. 2003, 2, 119–125. [Google Scholar] [CrossRef]
- Jacobson, D.L.; Gange, S.J.; Rose, N.R.; Graham, N.M. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 1997, 84, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.J.; Rau, L.M. Autoimmune diseases: A leading cause of death among young and middle-aged women in the United States. Am. J. Public Health 2000, 90, 1463–1466. [Google Scholar] [PubMed]
- Ganguly, D.; Haak, S.; Sisirak, V.; Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013, 13, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.N.; Lew, A.M.; Wu, L. The potential role of dendritic cells in the therapy of Type 1 diabetes. Immunotherapy 2013, 5, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.L.; Reizis, B. Dendritic cells: Arbiters of immunity and immunological tolerance. Cold Spring Harb. Perspect. Biol. 2012, 4, a007401. [Google Scholar] [CrossRef] [PubMed]
- Garbi, N.; Kreutzberg, T. Dendritic cells enhance the antigen sensitivity of T cells. Front. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Itano, A.A.; McSorley, S.J.; Reinhardt, R.L.; Ehst, B.D.; Ingulli, E.; Rudensky, A.Y.; Jenkins, M.K. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity 2003, 19, 47–57. [Google Scholar] [CrossRef]
- Wallet, M.A.; Flores, R.R.; Wanga, Y.; Yi, Z.; Kroger, C.J.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Wang, B.; Tisch, R. MerTK regulates thymic selection of autoreactive T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 4810–4815. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, G.X.; Gran, B.; Fallarino, F.; Yu, S.; Li, H.; Cullimore, M.L.; Rostami, A.; Xu, H. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J. Immunol. 2010, 185, 5953–5961. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Thivolet, C. Nasal administration of CTB-insulin induces active tolerance against autoimmune diabetes in non-obese diabetic (NOD) mice. Clin. Exp. Immunol. 2002, 130, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Petrovsky, N.; Silva, D.; Schatz, D.A. Vaccine therapies for the prevention of type 1 diabetes mellitus. Paediatr. Drugs 2003, 5, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Odumosu, O.; Payne, K.; Baez, I.; Jutzy, J.; Wall, N.; Langridge, W. Suppression of dendritic cell activation by diabetes autoantigens linked to the cholera toxin B subunit. Immunobiology 2011, 216, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, K.; Hirose, K.; Oabe, Y.; Fukunaga, Y.; Takahashi, A.; Shiroishi, M.; Kajikawa, M.; Tabata, S.; Nakamura, S.; Takai, T. The long-term immunosuppressive effects of disulfide-linked HLA-G dimer in mice with collagen-induced arthritis. Hum. Immunol. 2013, 74, 433–438. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mbongue, J.C.; Nicholas, D.A.; Torrez, T.W.; Kim, N.-S.; Firek, A.F.; Langridge, W.H.R. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines 2015, 3, 703-729. https://doi.org/10.3390/vaccines3030703
Mbongue JC, Nicholas DA, Torrez TW, Kim N-S, Firek AF, Langridge WHR. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines. 2015; 3(3):703-729. https://doi.org/10.3390/vaccines3030703
Chicago/Turabian StyleMbongue, Jacques C., Dequina A. Nicholas, Timothy W. Torrez, Nan-Sun Kim, Anthony F. Firek, and William H.R. Langridge. 2015. "The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity" Vaccines 3, no. 3: 703-729. https://doi.org/10.3390/vaccines3030703
APA StyleMbongue, J. C., Nicholas, D. A., Torrez, T. W., Kim, N.-S., Firek, A. F., & Langridge, W. H. R. (2015). The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines, 3(3), 703-729. https://doi.org/10.3390/vaccines3030703