
Advances in Oncolytic Viral Therapy in Melanoma: A Comprehensive Review

 , ,
, ,  ,
,
Abstract
1. Introduction
2. A Brief History of Oncolytic Viral Therapy
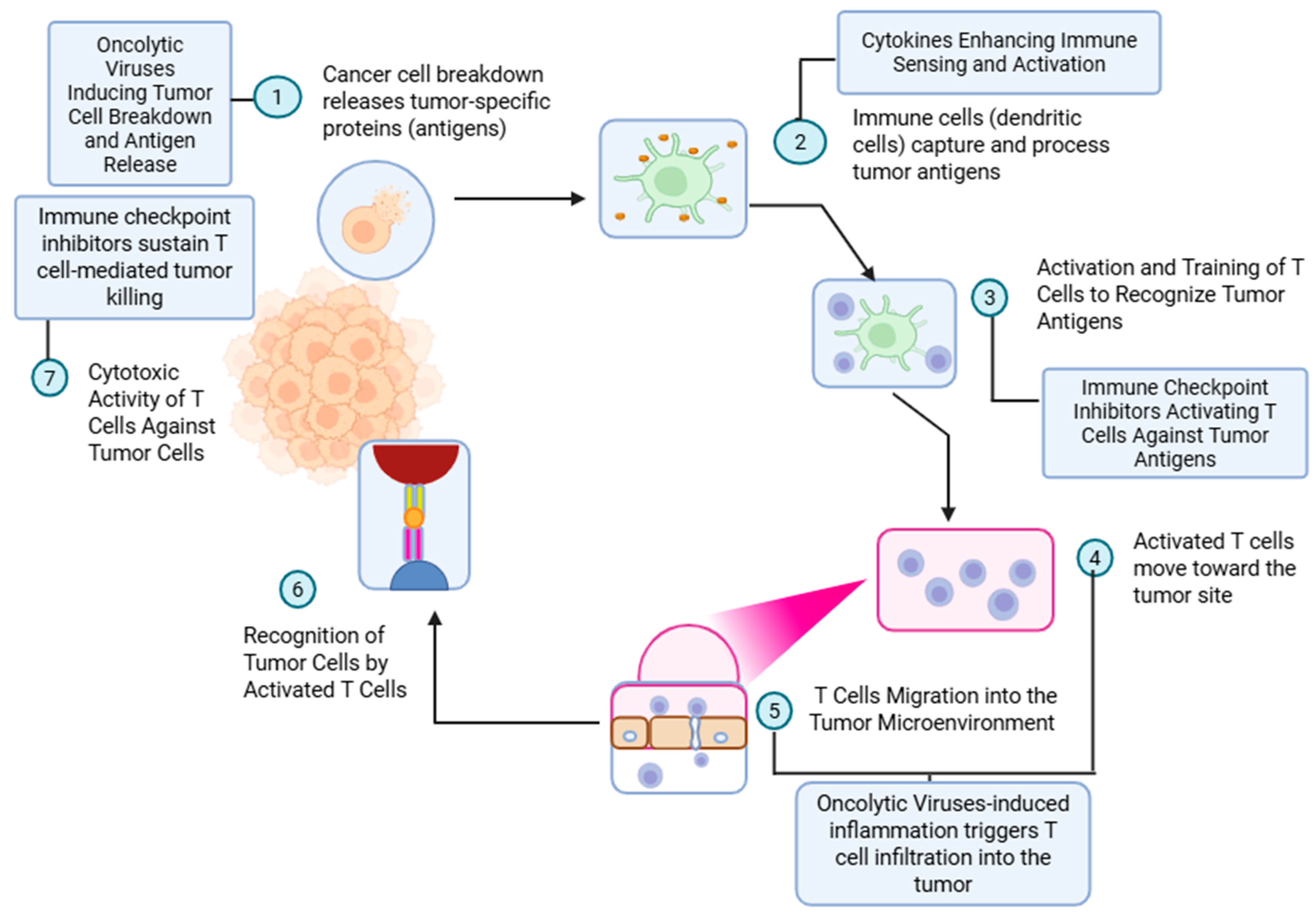
3. Mechanism of Oncolytic Virus Therapy
3.1. Selective Infection and Replication in Tumor Cells
3.2. Stimulating the Immune System to Cause Cell Death
3.3. Delivering Therapeutic Genes and Altering Tumor Neovasculature
4. Talimogene Laherparepvec: The First Oncolytic Viral Therapy
4.1. T-VEC as Monotherapy
4.2. T-VEC with Pembrolizumab in Treatment-Naïve Advanced Melanoma
4.3. T-VEC with Ipilimumab in Treatment-Naïve Advanced Melanoma
4.4. Neoadjuvant T-VEC with Nivolumab
4.5. T-VEC in Combination with Pembrolizumab in Advanced Melanoma Refractory to Anti-PD1 Therapy
5. Vusolimogene Oderparepvec: The Novel Drug
6. Oncolytic Viruses in Clinical Development for Patients with Melanoma
6.1. DNA Viruses
6.1.1. Herpesvirus
6.1.2. Adenovirus
6.1.3. Vaccinia Virus
6.1.4. Parvovirus
6.2. RNA Viruses
6.2.1. Reovirus
6.2.2. Poliovirus
6.2.3. Coxsackievirus
6.2.4. Seneca Valley Virus
6.2.5. Negative-Sense ssRNA Viruses
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | Antigen-presenting cell |
| CAR | Coxsackie and Adenovirus Receptor |
| CD | Cluster of differentiation |
| CI | Confidence interval |
| CR | Complete response |
| CRR | Complete response rate |
| DAF | Decay-accelerating factor |
| DAMP | Damage-associated molecular pattern |
| DC | Dendritic cell |
| DCR | Disease control rate |
| DNA | Deoxyribonucleic acid |
| DoR | Duration of response |
| DRR | Durable response rate |
| dsDNA | Double-stranded DNA |
| EFS | Event-free survival |
| FDA | Food and Drug Administration |
| GALV-GP-R | Glycoprotein gibbon ape leukemia virus surface glycoprotein with the R-sequence deleted |
| GBM | Glioblastoma multiforme |
| H-1PV | H-1 parvovirus |
| HR | Hazard ratio |
| HSV | Herpesvirus |
| HVEM | Herpes Virus Entry Mediator |
| ICAM-1 | Intracellular adhesion molecule 1 |
| ICD | Immunogenic cell death |
| ICI | Immune checkpoint inhibitor |
| ICP | Infected cell protein |
| IFN | Interferon |
| IM | Intramuscular |
| IQR | Interquartile range |
| IRES | Internal ribosome entry site |
| IV | Intravenous |
| MeV | Measles virus |
| NDV | Newcastle disease virus |
| OR | Odds ratio |
| ORR | Overall response rate |
| OS | Overall survival |
| OV | Oncolytic virus |
| PD-1 | Programmed cell death protein-1 |
| PFS | Progression-free survival |
| PFU | Plaque-forming unit |
| PR | Partial response |
| REV | Russian encephalitis virus |
| RFS | Recurrence-free survival |
| RNA | Ribonucleic acid |
| RoPV1 | Rodent protoparvovirus 1 |
| SD | Stable disease |
| ssRNA | Single-stranded RNA |
| SVV | Seneca Valley virus |
| TIL | Tumor-infiltrating lymphocyte |
| TLR | Toll-like receptor |
| TRAE | Treatment-related adverse event |
| TME | Tumor microenvironment |
| T-VEC | Talimogene laherparepvec |
| VSV | Vesicular stomatitis virus |
| VuSO | Vusolimogene oderparepvec |
| WNV | West Nile virus |
References
- Coley, W.B., II. Contribution to the Knowledge of Sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Middleton, M.R.; Hoeller, C.; Michielin, O.; Robert, C.; Caramella, C.; Öhrling, K.; Hauschild, A. Intratumoural immunotherapies for unresectable and metastatic melanoma: Current status and future perspectives. Br. J. Cancer 2020, 123, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic virotherapy: Basic principles, recent advances and future directions. Signal Transduct. Target. Ther. 2023, 8, 156. [Google Scholar] [CrossRef]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra137. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Lecoq, H. Discovery of the first virus, the tobacco mosaic virus: 1892 or 1898? C R. Acad. Sci. III 2001, 324, 929–933. [Google Scholar] [CrossRef]
- Dock, G. The influence of complicating diseases upon leukaemia. Am. J. Med. Sci. (1827-1924) 1904, 127, 563. [Google Scholar] [CrossRef]
- Pasquinucci, G. Possible effect of measles on leukaemia. Lancet 1971, 1, 136. [Google Scholar] [CrossRef] [PubMed]
- Zygiert, Z. Hodgkin’s disease: Remissions after measles. Lancet 1971, 1, 593. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.W. Effects of glandular fever infection in acute leukaemia. Br. Med. J. 1953, 1, 589–593. [Google Scholar] [CrossRef]
- Hoster, H.A.; Zanes, R.P., Jr.; Von Haam, E. Studies in Hodgkin’s syndrome; the association of viral hepatitis and Hodgkin’s disease; a preliminary report. Cancer Res. 1949, 9, 473–480. [Google Scholar] [PubMed]
- Moore, A.E. The destructive effect of the virus of Russian Far East encephalitis on the transplantable mouse sarcoma 180. Cancer 1949, 2, 525–534. [Google Scholar] [CrossRef]
- Moore, A.E. Inhibition of growth of five transplantable mouse tumors by the virus of Russian Far East encephalitis. Cancer 1951, 4, 375–382. [Google Scholar] [CrossRef]
- Southam, C.M.; Moore, A.E. Clinical studies of viruses as antineoplastic agents with particular reference to Egypt 101 virus. Cancer 1952, 5, 1025–1034. [Google Scholar] [CrossRef]
- Huebner, R.J.; Rowe, W.P.; Schatten, W.E.; Smith, R.R.; Thomas, L.B. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer 1956, 9, 1211–1218. [Google Scholar] [CrossRef]
- Asada, T. Treatment of human cancer with mumps virus. Cancer 1974, 34, 1907–1928. [Google Scholar] [CrossRef]
- Newman, W.; Southam, C.M. Virus treatment in advanced cancer; a pathological study of fifty-seven cases. Cancer 1954, 7, 106–118. [Google Scholar] [CrossRef]
- Hammon, W.M.; Yohn, D.S.; Casto, B.C.; Atchison, R.W. Oncolytic potentials of nonhuman viruses for human cancer. I. Effects of twenty-four viruses on human cancer cell lines. J. Natl. Cancer Inst. 1963, 31, 329–345. [Google Scholar] [PubMed]
- Yohn, D.S.; Hammon, W.M.; Atchison, R.W.; Casto, B.C. Oncolytic potentials of nonhuman viruses for human cancer. II. Effects of five viruses on heterotransplantable human tumors. J. Natl. Cancer Inst. 1968, 41, 523–529. [Google Scholar] [PubMed]
- Molomut, N.; Padnos, M. Inhibition of transplantable and spontaneous murine tumours by the M-P virus. Nature 1965, 208, 948–950. [Google Scholar] [CrossRef]
- Southam, C.M.; Moore, A.E. West Nile, Ilheus, and Bunyamwera virus infections in man. Am. J. Trop. Med. Hyg. 1951, 31, 724–741. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Kawaoka, Y. The origins of new pandemic viruses: The acquisition of new host ranges by canine parvovirus and influenza A viruses. Annu. Rev. Microbiol. 2005, 59, 553–586. [Google Scholar] [CrossRef]
- Murray, D.R.; Cassel, W.A.; Torbin, A.H.; Olkowski, Z.L.; Moore, M.E. Viral oncolysate in the management of malignant melanoma. II. Clinical studies. Cancer 1977, 40, 680–686. [Google Scholar] [CrossRef]
- Cassel, W.A.; Murray, D.R.; Phillips, H.S. A phase II study on the postsurgical management of Stage II malignant melanoma with a Newcastle disease virus oncolysate. Cancer 1983, 52, 856–860. [Google Scholar] [CrossRef]
- Cassel, W.A.; Murray, D.R. A ten-year follow-up on stage II malignant melanoma patients treated postsurgically with Newcastle disease virus oncolysate. Med. Oncol. Tumor Pharmacother. 1992, 9, 169–171. [Google Scholar] [CrossRef]
- Batliwalla, F.M.; Bateman, B.A.; Serrano, D.; Murray, D.; Macphail, S.; Maino, V.C.; Ansel, J.C.; Gregersen, P.K.; Armstrong, C.A. A 15-year follow-up of AJCC stage III malignant melanoma patients treated postsurgically with Newcastle disease virus (NDV) oncolysate and determination of alterations in the CD8 T cell repertoire. Mol. Med. 1998, 4, 783–794. [Google Scholar] [CrossRef]
- Schirrmacher, V. Fifty Years of Clinical Application of Newcastle Disease Virus: Time to Celebrate! Biomedicines 2016, 4, 16. [Google Scholar] [CrossRef]
- Hastie, E.; Grdzelishvili, V.Z. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J. Gen. Virol. 2012, 93, 2529–2545. [Google Scholar] [CrossRef] [PubMed]
- Felt, S.A.; Grdzelishvili, V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: A 5-year update. J. Gen. Virol. 2017, 98, 2895–2911. [Google Scholar] [CrossRef] [PubMed]
- Nagalo, B.M.; Breton, C.A.; Zhou, Y.; Arora, M.; Bogenberger, J.M.; Barro, O.; Steele, M.B.; Jenks, N.J.; Baker, A.T.; Duda, D.G.; et al. Oncolytic Virus with Attributes of Vesicular Stomatitis Virus and Measles Virus in Hepatobiliary and Pancreatic Cancers. Mol. Ther. Oncolytics 2020, 18, 546–555. [Google Scholar] [CrossRef]
- Levaditi, C.; Nicolau, S. Sur le culture du virus vaccinal dans les neoplasmes epithelieux. CR Soc. Biol. 1922, 86, 928. [Google Scholar]
- Pack, G.T. Note on the experimental use of rabies vaccine for melanomatosis. AMA Arch. Dermatol. Syphilol. 1950, 62, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.E. Viruses with oncolytic properties and their adaptation to tumors. Ann. N. Y. Acad. Sci. 1952, 54, 945–952. [Google Scholar] [CrossRef]
- Southam, C.M. Present status of oncolytic virus studies. Trans. N. Y. Acad. Sci. 1960, 22, 657–673. [Google Scholar] [CrossRef]
- Speir, R.W.; Southam, C.M. Interference of Newcastle disease virus with neuropathogenicity of oncolytic viruses in mice. Ann. N. Y. Acad. Sci. 1960, 83, 551–563. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef]
- Badrinath, N.; Heo, J.; Yoo, S.Y. Viruses as nanomedicine for cancer. Int. J. Nanomed. 2016, 11, 4835–4847. [Google Scholar]
- Farassati, F.; Yang, A.-D.; Lee, P.W. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat. Cell Biol. 2001, 3, 745–750. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef]
- Vile, R.G.; Hart, I.R. Targeting of cytokine gene expression to malignant melanoma cells using tissue specific promoter sequences. Ann. Oncol. 1994, 5 (Suppl. S4), 59–65. [Google Scholar] [CrossRef] [PubMed]
- Savontaus, M.; Sauter, B.; Huang, T.; Woo, S. Transcriptional targeting of conditionally replicating adenovirus to dividing endothelial cells. Gene Ther. 2002, 9, 972–979. [Google Scholar] [CrossRef]
- Deng, L.; Fan, J.; Ding, Y.; Zhang, J.; Zhou, B.; Zhang, Y.; Huang, B. Oncolytic efficacy of thymidine kinase-deleted vaccinia virus strain Guang9. Oncotarget 2017, 8, 40533. [Google Scholar] [CrossRef]
- Liu, B.; Robinson, M.; Han, Z.; Branston, R.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.; Thornton, M.; Bullock, P. ICP34. 5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef]
- Potts, K.G.; Hitt, M.M.; Moore, R.B. Oncolytic viruses in the treatment of bladder cancer. Adv. Urol. 2012, 2012, 404581. [Google Scholar] [CrossRef]
- Chung, R.Y.; Saeki, Y.; Chiocca, E.A. B-myb promoter retargeting of herpes simplex virus γ34. 5 gene-mediated virulence toward tumor and cycling cells. J. Virol. 1999, 73, 7556–7564. [Google Scholar] [CrossRef]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed]
- Haag, F.; Adriouch, S.; Braß, A.; Jung, C.; Möller, S.; Scheuplein, F.; Bannas, P.; Seman, M.; Koch-Nolte, F. Extracellular NAD and ATP: Partners in immune cell modulation. Purinergic Signal. 2007, 3, 71–81. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Nino-Castro, A.C.; Schultze, J.L. Regulatory dendritic cells: There is more than just immune activation. Front. Immunol. 2012, 3, 274. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Johnson, P.; Knipe, D.; Chiocca, E. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther. 2003, 10, 983–990. [Google Scholar] [CrossRef]
- Verweij, M.C.; Horst, D.; Griffin, B.D.; Luteijn, R.D.; Davison, A.J.; Ressing, M.E.; Wiertz, E.J. Viral inhibition of the transporter associated with antigen processing (TAP): A striking example of functional convergent evolution. PLoS Pathog. 2015, 11, e1004743. [Google Scholar] [CrossRef]
- Goldsmith, K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected cell protein (ICP) 47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J. Exp. Med. 1998, 187, 341–348. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Budnick, I.; Singh, M.; Thiruppathi, M.; Alharshawi, K.; Elshabrawy, H.; Holterman, M.J.; Prabhakar, B.S. Dual role of GM-CSF as a pro-inflammatory and a regulatory cytokine: Implications for immune therapy. J. Interferon Cytokine Res. 2015, 35, 585–599. [Google Scholar] [CrossRef]
- Uchida, H.; Hamada, H.; Nakano, K.; Kwon, H.; Tahara, H.; Cohen, J.B.; Glorioso, J.C. Oncolytic herpes simplex virus vectors fully retargeted to tumor-associated antigens. Curr. Cancer Drug Targets 2018, 18, 162–170. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef]
- Breitbach, C.J.; De Silva, N.S.; Falls, T.J.; Aladl, U.; Evgin, L.; Paterson, J.; Sun, Y.Y.; Roy, D.G.; Rintoul, J.L.; Daneshmand, M. Targeting tumor vasculature with an oncolytic virus. Mol. Ther. 2011, 19, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Khushalani, N.I.; Harrington, K.J.; Melcher, A.; Bommareddy, P.K.; Zamarin, D. Breaking the barriers in cancer care: The next generation of herpes simplex virus-based oncolytic immunotherapies for cancer treatment. Mol. Ther. Oncolytics 2023, 31, 100729. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Thorne, S.H.; Bartlett, D.L. Oncolytic virotherapy: Molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim. Biophys. Acta 2008, 1785, 217–231. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Hamid, O.; Hoffner, B.; Gasal, E.; Hong, J.; Carvajal, R.D. Oncolytic immunotherapy: Unlocking the potential of viruses to help target cancer. Cancer Immunol. Immunother. 2017, 66, 1249–1264. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Rutkowski, P.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Queirolo, P.; Dummer, R.; Butler, M.O.; Hill, A.G.; et al. Final, 10-Year Outcomes with Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2025, 392, 11–22. [Google Scholar] [CrossRef]
- Gastman, B.; Robert, C.; Gogas, H.; Rutkowski, P.; Long, G.V.; Chaney, M.F.; Joshi, H.; Lin, Y.-L.; Snyder, W.; Chesney, J.A. Primary analysis of a phase 2, open-label, multicenter trial of talimogene laherparepvec (T-VEC) plus pembrolizumab (pembro) for the treatment (Tx) of patients (pts) with advanced melanoma (MEL) who progressed on prior anti–PD-1 therapy: MASTERKEY-115. J. Clin. Oncol. 2022, 40, 9518. [Google Scholar] [CrossRef]
- Chesney, J.A.; Puzanov, I.; Collichio, F.A.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone for advanced melanoma: 5-year final analysis of a multicenter, randomized, open-label, phase II trial. J. Immunother. Cancer 2023, 11, e006270. [Google Scholar] [CrossRef]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A Phase I Study of OncoVEXGM-CSF, a Second-Generation Oncolytic Herpes Simplex Virus Expressing Granulocyte Macrophage Colony-Stimulating Factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor–Encoding, Second-Generation Oncolytic Herpesvirus in Patients With Unresectable Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Ӧhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III–IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Stahlie, E.H.A.; Mulder, E.; Reijers, S.; Balduzzi, S.; Zuur, C.L.; Klop, W.M.C.; van der Hiel, B.; Van de Wiel, B.A.; Wouters, M.; Schrage, Y.M.; et al. Single agent Talimogene Laherparepvec for stage IIIB-IVM1c melanoma patients: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2022, 175, 103705. [Google Scholar] [CrossRef]
- Franke, V.; Berger, D.M.S.; Klop, W.M.C.; van der Hiel, B.; van de Wiel, B.A.; ter Meulen, S.; Wouters, M.W.J.M.; van Houdt, W.J.; van Akkooi, A.C.J. High response rates for T-VEC in early metastatic melanoma (stage IIIB/C-IVM1a). Int. J. Cancer 2019, 145, 974–978. [Google Scholar] [CrossRef]
- Perez, M.C.; Zager, S.J.; Amatruda, T.; Conry, R.; Ariyan, C.; Desai, C.; Kirkwood, J.M.; Treichel, S.; Cohan, D.; Raskin, L. Observational Study of Talimogene Laherparepvec use for Melanoma in Clinical Practice in the United States (COSMUS-1). Melanoma Manag. 2019, 6, MMT19. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Gastman, B.R.; McCahon, L.; Buchbinder, E.I.; Puzanov, I.; Nanni, M.; Lewis, J.M.; Carvajal, R.D.; Singh-Kandah, S.; Desai, A.M.; et al. Observational Study Of Talimogene Laherparepvec Use in the Anti-PD-1 era for Melanoma in the US (COSMUS-2). Melanoma Manag. 2020, 7, 5–8. [Google Scholar] [CrossRef]
- Franke, V.; Stahlie, E.H.A.; van der Hiel, B.; van de Wiel, B.A.; Wouters, M.W.J.M.; van Houdt, W.J.; van Akkooi, A.C.J. Re-introduction of T-VEC Monotherapy in Recurrent Melanoma is Effective. J. Immunother. 2022, 45, 263–266. [Google Scholar] [CrossRef]
- Long, G.; Dummer, R.; Johnson, D.; Michielin, O.; Martin-Algarra, S.; Treichel, S.; Chan, E.; Diede, S.; Ribas, A. 429 Long-term analysis of MASTERKEY-265 phase 1b trial of talimogene laherparepvec (T-VEC) plus pembrolizumab in patients with unresectable stage IIIB-IVM1c melanoma. BMJ Spec. J. 2020, 170, 1109–1119. [Google Scholar]
- Chesney, J.A.; Ribas, A.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined With Pembrolizumab for Advanced Melanoma. J. Clin. Oncol. 2023, 41, 528–540. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Zijlker, L.P.; Houdt, W.J.v.; Stahlie, E.H.A.; Franke, V.; Rohaan, M.W.; Delatzakis, A.; Zuur, C.; Klop, W.M.C.; Wiel, B.A.v.d.; Kuijpers, A.; et al. Neoadjuvant T-VEC + nivolumab combination therapy for resectable early metastatic (stage IIIB/C/D-IV M1a) melanoma with injectable disease: NIVEC trial. J. Clin. Oncol. 2023, 41, 9546. [Google Scholar] [CrossRef]
- Dummer, R.; Gyorki, D.; Hyngstrom, J.; Berger, A.; Conry, R.; Demidov, L.; Sharma, A.; Treichel, S.; Radcliffe, H.; Gorski, K. Neoadjuvant talimogene laherparepvec plus surgery versus surgery alone for resectable stage IIIB–IVM1a melanoma: A randomized, open-label, phase 2 trial. Nat. Med. 2021, 27, 1789–1796. [Google Scholar] [CrossRef]
- Dummer, R.; Gyorki, D.E.; Hyngstrom, J.R.; Ning, M.; Lawrence, T.; Ross, M.I. Final 5-Year Follow-Up Results Evaluating Neoadjuvant Talimogene Laherparepvec Plus Surgery in Advanced Melanoma: A Randomized Clinical Trial. JAMA Oncol. 2023, 9, 1457–1459. [Google Scholar] [CrossRef]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer 2019, 7, 214. [Google Scholar] [CrossRef]
- Emamekhoo, H.; Patel, S.; Rodriguez, E.; Riaz, M.K.; Giaccone, G.; Furqan, M.; Sacco, J.J.; Bommareddy, P.; Raza, S.; He, S.; et al. IGNYTE: A Phase 1/2 Multi-Cohort Clinical Trial of RP1 ± Nivolumab in Patients with Non-Small Cell Lung Cancer and Other Solid Tumors. Int. J. Radiat. Oncol. Biol. Phys. 2022, 112, e12–e13. [Google Scholar] [CrossRef]
- Guedan, S.; Grases, D.; Rojas, J.J.; Gros, A.; Vilardell, F.; Vile, R.; Mercade, E.; Cascallo, M.; Alemany, R. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012, 19, 1048–1057. [Google Scholar] [CrossRef]
- Wong, M.K.K.; Sacco, J.J.; Robert, C.; Michels, J.; Bowles, T.L.; In, G.K.; Tsai, K.K.; Lebbe, C.; Gaudy-Marqueste, C.; Couselo, E.M.; et al. Efficacy and safety of RP1 combined with nivolumab in patients with anti–PD-1–failed melanoma from the IGNYTE clinical trial. J. Clin. Oncol. 2024, 42, 9517. [Google Scholar] [CrossRef]
- Luke, J.J.; Kong, G.; Gullo, G.; Robert, C. A randomized, controlled, multicenter, phase 3 study of vusolimogene oderparepvec (VO) combined with nivolumab vs treatment of physician’s choice in patients with advanced melanoma that has progressed on anti–PD-1 and anti–CTLA-4 therapy (IGNYTE-3). J. Clin. Oncol. 2024, 42, TPS9604. [Google Scholar] [CrossRef]
- Harrington, K.J.; Sacco, J.J.; Olsson-Brown, A.C.; Chan, T.Y.; Nenclares, P.; Leslie, I.; Saleem, I.; Bommareddy, P.; Ahlers, C.M.; Coffin, R.S.; et al. A phase 1 trial of RP2, a first-in-class, enhanced potency oncolytic HSV expressing an anti-CTLA-4 antibody as a single agent and combined with nivolumab in patients with advanced solid tumors. J. Clin. Oncol. 2022, 40, TPS2704. [Google Scholar] [CrossRef]
- Harrington, K.J.; Sacco, J.J.; Olsson-Brown, A.; Chan, T.; Nenclares, P.; Leslie, I.; Bommareddy, P.; Ahlers, C.; Wolff, J.; Middleton, M.R. 827P An open-label, multicenter, phase I study of RP2 as a single agent and in combination with nivolumab in patients with solid tumors: Safety, efficacy, and biomarker results. Ann. Oncol. 2022, 33, S926–S927. [Google Scholar] [CrossRef]
- Sacco, J.J.; Harrington, K.J.; Olsson-Brown, A.; Chan, T.Y.; Nenclares, P.; Leslie, I.; Bommareddy, P.; Kalbasi, A.; Xie, B.; Mishal, M.; et al. Safety, efficacy, and biomarker results from an open-label, multicenter, phase 1 study of RP2 alone or combined with nivolumab in a cohort of patients with uveal melanoma. J. Clin. Oncol. 2024, 42, 9511. [Google Scholar] [CrossRef]
- Shepard, D.R.; Ahmed, M.; Bekaii-Saab, T.S.; Wolff, J. An open-label clinical trial of RP2 and RP3 oncolytic immunotherapy in combination with atezolizumab and bevacizumab for the treatment of patients with advanced colorectal carcinoma. J. Clin. Oncol. 2023, 41, TPS3628. [Google Scholar] [CrossRef]
- Harrington, K.J.; Rullan, A.; Deighton, L.; Barata, J.; Castro, H.; Ahlers, C.M.; McRae, J.; Bommareddy, P.; Coffin, R.S.; Middleton, M.R. An open-label, multicenter, phase 1 study of RP3 as a single agent and in combination with nivolumab in patients (pts) with solid tumors. J. Clin. Oncol. 2022, 40, TPS2705. [Google Scholar] [CrossRef]
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Trends Microbiol. 2000, 8, 410–418. [Google Scholar] [CrossRef]
- Mastrangelo, M.J.; Maguire, H.C., Jr.; Eisenlohr, L.C.; Laughlin, C.E.; Monken, C.E.; McCue, P.A.; Kovatich, A.J.; Lattime, E.C. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999, 6, 409–422. [Google Scholar] [CrossRef]
- Lutzky, J.; Sullivan, R.J.; Cohen, J.V.; Ren, Y.; Li, A.; Haq, R. Phase 1b study of intravenous coxsackievirus A21 (V937) and ipilimumab for patients with metastatic uveal melanoma. J. Cancer Res. Clin. Oncol. 2023, 149, 6059–6066. [Google Scholar] [CrossRef]
- Curti, B.D.; Richards, J.; Hyngstrom, J.R.; Daniels, G.A.; Faries, M.; Feun, L.; Margolin, K.A.; Hallmeyer, S.; Grose, M.; Zhang, Y.; et al. Intratumoral oncolytic virus V937 plus ipilimumab in patients with advanced melanoma: The phase 1b MITCI study. J. Immunother. Cancer 2022, 10, e005224. [Google Scholar] [CrossRef]
- Silk, A.W.; O’Day, S.J.; Kaufman, H.L.; Bryan, J.; Norrell, J.T.; Imbergamo, C.; Portal, D.; Zambrano-Acosta, E.; Palmeri, M.; Fein, S.; et al. A phase 1b single-arm trial of intratumoral oncolytic virus V937 in combination with pembrolizumab in patients with advanced melanoma: Results from the CAPRA study. Cancer Immunol. Immunother. 2023, 72, 1405–1415. [Google Scholar] [CrossRef]
- Smith, K.E.R.; Peng, K.W.; Pulido, J.S.; Weisbrod, A.J.; Strand, C.A.; Allred, J.B.; Newsom, A.N.; Zhang, L.; Packiriswamy, N.; Kottke, T.; et al. A phase I oncolytic virus trial with vesicular stomatitis virus expressing human interferon beta and tyrosinase related protein 1 administered intratumorally and intravenously in uveal melanoma: Safety, efficacy, and T cell responses. Front. Immunol. 2023, 14, 1279387. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Ross, M.I.; Agarwala, S.S.; Taylor, M.H.; Vetto, J.T.; Neves, R.I.; Daud, A.; Khong, H.T.; Ungerleider, R.S.; Tanaka, M.; et al. Final results of a phase II multicenter trial of HF10, a replication-competent HSV-1 oncolytic virus, and ipilimumab combination treatment in patients with stage IIIB-IV unresectable or metastatic melanoma. J. Clin. Oncol. 2017, 35, 9510. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Curti, B.; Daniels, G.A.; Hallmeyer, S.; Whitman, E.D.; Lutzky, J.; Spitler, L.E.; Zhou, K.; Bommareddy, P.K.; Grose, M.; et al. Clinical Responses of Oncolytic Coxsackievirus A21 (V937) in Patients With Unresectable Melanoma. J. Clin. Oncol. 2021, 39, 3829–3838. [Google Scholar] [CrossRef] [PubMed]
- Manservigi, R.; Argnani, R.; Marconi, P. HSV recombinant vectors for gene therapy. Open Virol. J. 2010, 4, 123. [Google Scholar] [CrossRef]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef]
- Weed, D.J.; Nicola, A.V. Herpes simplex virus membrane fusion. Cell Biol. Herpes Viruses 2017, 223, 29–47. [Google Scholar]
- Maruzuru, Y.; Shindo, K.; Liu, Z.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kato, A.; Kawaguchi, Y. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J. Virol. 2014, 88, 7445–7454. [Google Scholar] [CrossRef]
- Shen, Y.; Nemunaitis, J. Herpes simplex virus 1 (HSV-1) for cancer treatment. Cancer Gene Ther. 2006, 13, 975–992. [Google Scholar] [CrossRef]
- Johnson, D.B.; Puzanov, I.; Kelley, M.C. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy 2015, 7, 611–619. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Benkő, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Chu, R.L.; Post, D.E.; Khuri, F.R.; Van Meir, E.G. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin. Cancer Res. 2004, 10, 5299–5312. [Google Scholar] [CrossRef]
- Charman, M.; Herrmann, C.; Weitzman, M.D. Viral and cellular interactions during adenovirus DNA replication. FEBS Lett. 2019, 593, 3531–3550. [Google Scholar] [CrossRef]
- Schaack, J.; Bennett, M.L.; Colbert, J.D.; Torres, A.V.; Clayton, G.H.; Ornelles, D.; Moorhead, J. E1A and E1B proteins inhibit inflammation induced by adenovirus. Proc. Natl. Acad. Sci. USA 2004, 101, 3124–3129. [Google Scholar] [CrossRef]
- Abudoureyimu, M.; Lai, Y.; Tian, C.; Wang, T.; Wang, R.; Chu, X. Oncolytic adenovirus—A nova for gene-targeted oncolytic viral therapy in HCC. Front. Oncol. 2019, 9, 1182. [Google Scholar] [CrossRef]
- Li, S.; Ou, M.; Wang, G.; Tang, L. Application of conditionally replicating adenoviruses in tumor early diagnosis technology, gene-radiation therapy and chemotherapy. Appl. Microbiol. Biotechnol. 2016, 100, 8325–8335. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zheng, S.; Li, X.-F.; Huang, J.-J.; Zheng, X.; Li, Z. Intra-tumor injection of H101, a recombinant adenovirus, in combination with chemotherapy in patients with advanced cancers: A pilot phase II clinical trial. World J. Gastroenterol. WJG 2004, 10, 3634. [Google Scholar] [CrossRef]
- Makower, D.; Rozenblit, A.; Kaufman, H.; Edelman, M.; Lane, M.E.; Zwiebel, J.; Haynes, H.; Wadler, S. Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin. Cancer Res. 2003, 9, 693–702. [Google Scholar]
- Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J. Urol. 2012, 188, 2391–2397. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 1–21. [Google Scholar] [CrossRef]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.-S.; Falls, T.; Burns, J.; Garcia, V. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef]
- Park, B.-H.; Hwang, T.; Liu, T.-C.; Sze, D.Y.; Kim, J.-S.; Kwon, H.-C.; Oh, S.Y.; Han, S.-Y.; Yoon, J.-H.; Hong, S.-H. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: A phase I trial. Lancet Oncol. 2008, 9, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, C.; Marchini, A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses 2019, 11, 562. [Google Scholar] [CrossRef]
- Hajda, J.; Lehmann, M.; Krebs, O.; Kieser, M.; Geletneky, K.; Jäger, D.; Dahm, M.; Huber, B.; Schöning, T.; Sedlaczek, O.; et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer 2017, 17, 576. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef]
- Antar, A.A.; Konopka, J.L.; Campbell, J.A.; Henry, R.A.; Perdigoto, A.L.; Carter, B.D.; Pozzi, A.; Abel, T.W.; Dermody, T.S. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 2009, 5, 59–71. [Google Scholar] [CrossRef]
- McSherry, E.A.; McGee, S.F.; Jirstrom, K.; Doyle, E.M.; Brennan, D.J.; Landberg, G.; Dervan, P.A.; Hopkins, A.M.; Gallagher, W.M. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int. J. Cancer 2009, 125, 1343–1351. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, W.; Huang, B.; Liu, Z.; Sun, L.; Zhang, Q.; Qiu, X.; Xu, K.; Wang, E. Overexpression of JAM-A in non-small cell lung cancer correlates with tumor progression. PLoS ONE 2013, 8, e79173. [Google Scholar] [CrossRef]
- Xu, P.-P.; Sun, Y.-F.; Fang, Y.; Song, Q.; Yan, Z.-X.; Chen, Y.; Jiang, X.-F.; Fei, X.-C.; Zhao, Y.; Leboeuf, C. JAM-A overexpression is related to disease progression in diffuse large B-cell lymphoma and downregulated by lenalidomide. Sci. Rep. 2017, 7, 7433. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wendlandt, E.; Tricot, G.; Zhan, F.; Carew, J.S.; Nawrocki, S.T. Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 2015, 6, 41275. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R.G. Structure and Function of the Protein Kinase R. In Interferon: The 50th Anniversary; Pitha, P.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 253–292. [Google Scholar] [CrossRef]
- Müller, L.; Berkeley, R.; Barr, T.; Ilett, E.; Errington-Mais, F. Past, Present and Future of Oncolytic Reovirus. Cancers 2020, 12, 3219. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, C.L.; Wimmer, E.; Racaniello, V.R. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 1989, 56, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Bossert, B.; Arita, M.; Nomoto, A.; Wimmer, E. Dual stem loops within the poliovirus internal ribosomal entry site control neurovirulence. J. Virol. 1999, 73, 958–964. [Google Scholar] [CrossRef]
- Sloan, A.E.; Buerki, R.A.; Murphy, C.; Kelly, A.T.; Ambady, P.; Brown, M.; Butowski, N.A.; Cavaliere, R.; Curry, W.T.; Desjardins, A.; et al. LUMINOS-101: Phase 2 study of PVSRIPO with pembrolizumab in recurrent glioblastoma. J. Clin. Oncol. 2021, 39, TPS2065. [Google Scholar] [CrossRef]
- Beasley, G.M.; Nair, S.K.; Farrow, N.E.; Landa, K.; Selim, M.A.; Wiggs, C.A.; Jung, S.H.; Bigner, D.D.; True Kelly, A.; Gromeier, M.; et al. Phase I trial of intratumoral PVSRIPO in patients with unresectable, treatment-refractory melanoma. J. Immunother. Cancer 2021, 9, e002203. [Google Scholar] [CrossRef]
- Inman, B.A.; Balar, A.V.; Milowsky, M.I.; Pruthi, R.S.; Polasek, M.J.; Morris, S.R.; Mixson, L.; Orr, K.; Woodson, E.M.; Kelly, A.T.; et al. Abstract CT242: LUMINOS-103: A basket trial evaluating the safety and efficacy of PVSRIPO in patients with advanced solid tumors. Cancer Res. 2021, 81, CT242. [Google Scholar] [CrossRef]
- Jahan, N.; Wimmer, E.; Mueller, S. A host-specific, temperature-sensitive translation defect determines the attenuation phenotype of a human rhinovirus/poliovirus chimera, PV1(RIPO). J. Virol. 2011, 85, 7225–7235. [Google Scholar] [CrossRef] [PubMed]
- Shafren, D.R.; Dorahy, D.J.; Ingham, R.A.; Burns, G.F.; Barry, R.D. Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J. Virol. 1997, 71, 4736–4743. [Google Scholar] [CrossRef]
- Shafren, D.; Quah, M.; Wong, Y.; Andtbacka, R.H.I.; Kaufman, H.L.; Au, G.G. Combination of a novel oncolytic immunotherapeutic agent, CAVATAK (coxsackievirus A21) and immune-checkpoint blockade significantly reduces tumor growth and improves survival in an immune competent mouse melanoma model. J. Immunother. Cancer 2014, 2, P125. [Google Scholar] [CrossRef]
- Reddy, P.S.; Burroughs, K.D.; Hales, L.M.; Ganesh, S.; Jones, B.H.; Idamakanti, N.; Hay, C.; Li, S.S.; Skele, K.L.; Vasko, A.J.; et al. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J. Natl. Cancer Inst. 2007, 99, 1623–1633. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Senzer, N.N.; Stephenson, J., Jr.; Loesch, D.; Burroughs, K.D.; Reddy, P.S.; Hann, C.L.; Hallenbeck, P.L. Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin. Cancer Res. 2011, 17, 888–895. [Google Scholar] [CrossRef]
- Duke, T.; Mgone, C.S. Measles: Not just another viral exanthem. Lancet 2003, 361, 763–773. [Google Scholar] [CrossRef]
- Blechacz, B.; Russell, S.J. Measles virus as an oncolytic vector platform. Curr. Gene Ther. 2008, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Iankov, I.D.; Dispenzieri, A.; Galanis, E. Attenuated oncolytic measles virus strains as cancer therapeutics. Curr. Pharm. Biotechnol. 2012, 13, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Vigil, A.; Kelly, K.; García-Sastre, A.; Fong, Y. Genetically engineered Newcastle disease virus for malignant melanoma therapy. Gene Ther. 2009, 16, 796–804. [Google Scholar] [CrossRef]
- Barber, G.N. VSV-tumor selective replication and protein translation. Oncogene 2005, 24, 7710–7719. [Google Scholar] [CrossRef]
- Zhang, Y.; Nagalo, B.M. Immunovirotherapy Based on Recombinant Vesicular Stomatitis Virus: Where Are We? Front. Immunol. 2022, 13, 898631. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Othus, M.; Chen, Y.; Wright, G.P.; Yost, K.J.; Hyngstrom, J.R.; Hu-Lieskovan, S.; Lao, C.D.; Fecher, L.A.; Truong, T.-G.; et al. Neoadjuvant–Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N. Engl. J. Med. 2023, 388, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Lucas, M.W.; Scolyer, R.A.; Wiel, B.A.v.d.; Menzies, A.M.; Lopez-Yurda, M.; Hoeijmakers, L.L.; Saw, R.P.M.; Lijnsvelt, J.M.; Maher, N.G.; et al. Neoadjuvant Nivolumab and Ipilimumab in Resectable Stage III Melanoma. N. Engl. J. Med. 2024, 391, 1696–1708. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Trial No. | Virus | Characteristics | Participants | Treatment Protocol | Best Response | Current Status |
|---|---|---|---|---|---|---|
| Phase I/Ib trials | ||||||
| Double-stranded DNA viruses | ||||||
| NCT03767348 [90] | HSV-1 (RP-1) | Expressing human GM-CSF and a fusogenic protein (GALV-GP-R−) | Multiple solid tumors | Monotherapy and in combination with nivolumab | 156 patients ORR: 31.4% CR: 12.2% | Phase I/II study Led to phase III trial (ongoing IGNYTE-3 study) |
| NCT04336241 [92] | HSV-1 (RP-2) | Expresses GM-CSF, fusogenic protein GALV-GP-R-, and anti-CTLA-4 like molecule | Multiple solid tumors (including ICI-refractory uveal melanoma) | Monotherapy (IT) and in combination with nivolumab | Prelim results (uveal melanoma) 17 patients enrolled: Monotherapy: 3 patients Combination: 14 patients Combination arm: ORR: 28.6% (all PR) DCR: 57.1% Median DoR: 5.1 months | Recruiting |
| NCT04735978 [96] | HSV-1 (RP-3) | Expresses anti-CTLA-4 antibody, CD40 ligand, and h4-1BBL | Advanced or metastatic non-neurological solid tumors | Monotherapy (IT) or in combination with IV nivolumab | No results | Recruiting |
| NCT00429312 [98] | Vaccinia virus (JX-594) | Thymidine kinase-deleted vaccinia virus plus GM-CSF (pexastimogene devacirepvec) | Unresectable melanoma or metastatic melanoma | Monotherapy | 10 patients enrolled PR: 2 patients, CR: 1 patient Injected tumor: 5/7 patients Non-injected tumors: 4/7 patients Median survival: 7.1 months | Not in development Tested in pre-ICI era |
| NCT06171178 | Vaccinia virus (ASP1012) | Delivered IV expressing Leptin-IL2 as a payload | Multiple solid tumors including melanoma | Part 1: monotherapy then dose expansion in multiple tumor types | No results posted | Recruiting |
| NCT06444815 | Vaccinia virus (VET-3-TGI) | Expresses CXCR3, IL-12, and a TGFB1-antagonizing mini-monomer | Multiple solid tumors | Groups A and C: monotherapy, IT and IV Groups B and D: IT or IV with pembrolizumab | No results posted | Recruiting |
| NCT05859074 | Vaccinia virus, Ankara strain (MVA) (MQ710) | Induces cGAS/STING pathway and induces Type 1 IFNs | Multiple solid tumors | Monotherapy and in combination with pembrolizumab | No results posted | Recruiting |
| NCT05222932 | Adenovirus (TILT-123) | Tumor Necrosis Factor Alpha- and IL-2-coding oncolytic adenovirus | HNSCC and melanoma | TILT-123 + Avelumab | No results posted | Recruiting |
| NCT05076760 | Adenovirus (MEM-288) | Co-expresses MEM40 (CD40 ligand) and IFNβ | Multiple solid tumors including melanoma | Part 1: MEM-288 monotherapy Part 2: MEM-288 with nivolumab | No results posted | Recruiting |
| RNA viruses | ||||||
| Positive-sense RNA viruses | ||||||
| NCT03408587 [99] | Coxsackievirus A21 (CVA21) | Targets ICAM-1-expressing cancer cells (Gebasaxturev) | Uveal melanoma metastatic to liver | CVA21 + ipilimumab | 11 patients enrolled SD: 3 patients. All patients had PD by week 26 | Not in development (no meaningful responses) |
| NCT02307149 [100] | CVA21 (also known as V937) | Targets ICAM-1-expressing cancer cells | Unresectable melanoma or metastatic melanoma | CVA21 (IT) + ipilimumab | 50 patients enrolled. ORR: 30% (47% in treatment naïve, 21% in anti-PD1 resistant) Median PFS: 6.2 months Median OS: 45.1 months | Phase Ib trial (MITCI study) |
| NCT02565992 [101] | CVA 21 | -- | Unresectable melanoma or metastatic melanoma | CVA21 (IT) + pembrolizumab | 36 patients enrolled ORR: 47%, CR: 22% | Phase Ib study (CAPRA trial) |
| Negative-sense RNA viruses | ||||||
| NCT03865212 [102] | Vesicular stomatitis virus | Includes genes for human IFN-β and TYPR1 (expressed in melanocytes and melanoma cells) | Metastatic uveal melanoma | Monotherapy—multiple dose levels | 12 patients SD: 4 patients. PD: 8 patients Two patients treated with ICIs later on had durable responses | Not in development (no meaningful responses) |
| Phase II/III trials | ||||||
| Double-stranded DNA viruses | ||||||
| NCT03190824 (Phase IIA) | Adenovirus (OBP-301) | Replicates selectively in cancer cells by introducing hTERT promotor | Unresectable or metastatic melanoma | Monotherapy | -- | Status unknown. Primarily developed in liver and GI cancers |
| NCT02272855 [103] | HSV-1 (HF-10, now called TBI-1401) | Mutated virus without any foreign genes | Ipilimumab-naïve unresectable melanoma | HF-10 + ipilimumab | 46 patients enrolled ORR at 24 weeks: 41% SD: 68% Median PFS: 19 months Median OS: 21.8 months | Current development unknown in melanoma |
| NCT06581406 | HSV-1 (RP-2) | Expresses GM-CSF, fusogenic protein GALV-GP-R-, and anti-CTLA-4-like molecule | ICI-naïve metastatic uveal melanoma | Arm 1: IT RP-2 + Nivolumab Arm 2: ipilimumab 3 mg/kg and nivolumab 1 mg/kg | Recruiting | Ongoing phase II/III open-label trial |
| NCT06264180 | HSV-1 (RP-1) | Expressing human GM-CSF and a fusogenic protein (GALV-GP-R−) | Ipilimumab- and nivolumab-refractory and BRAF-MEK inhibitor-refractory cutaneous melanoma | Arm 1: nivolumab + RP-1 Arm 2: investigators’ choice (except TIL) | Recruiting | Randomized phase III trial |
| RNA viruses | ||||||
| Positive-sense RNA viruses | ||||||
| NCT01227551 [104] | CVA 21 (also known as V937) | -- | Unresectable or metastatic melanoma | IT monotherapy | 57 patients enrolled: all endpoints at 6 months PFS rate: 38.6% Durable ORR: 21.1% Best ORR: 28.1% 12: month PFS: 32.9% 12: month OS: 75.4% | Led to combination phase II study in combination with pembrolizumab |
| NCT04152863 (Phase II) | CVA 21 (also known as V937) | -- | Unresectable or metastatic melanoma | CVA 21 either IV or IT with pembrolizumab IV | 28 patients in each arm IV V937 + IV Pembro: 46.4% IT V937 + IV Pembro: 39.3% IV Pembro alone: 34.5% | Phase II trial terminated early due to business reasons (no publications) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, A.; Rao, R.; Tejawinata, F.; Shamita, G.A.N.; Herpel, M.S.; Yoshida, A.; Goolamier, G.; Sidiropoulos, J.; Sheng, I.Y.; Abboud, S.-T.; et al. Advances in Oncolytic Viral Therapy in Melanoma: A Comprehensive Review. Vaccines 2025, 13, 727. https://doi.org/10.3390/vaccines13070727
Garg A, Rao R, Tejawinata F, Shamita GAN, Herpel MS, Yoshida A, Goolamier G, Sidiropoulos J, Sheng IY, Abboud S-T, et al. Advances in Oncolytic Viral Therapy in Melanoma: A Comprehensive Review. Vaccines. 2025; 13(7):727. https://doi.org/10.3390/vaccines13070727
Chicago/Turabian StyleGarg, Ayushi, Rohit Rao, Felicia Tejawinata, Gazi Amena Noor Shamita, McKay S. Herpel, Akihiro Yoshida, Gordon Goolamier, Jessica Sidiropoulos, Iris Y. Sheng, Salim-Tamuz Abboud, and et al. 2025. "Advances in Oncolytic Viral Therapy in Melanoma: A Comprehensive Review" Vaccines 13, no. 7: 727. https://doi.org/10.3390/vaccines13070727
APA StyleGarg, A., Rao, R., Tejawinata, F., Shamita, G. A. N., Herpel, M. S., Yoshida, A., Goolamier, G., Sidiropoulos, J., Sheng, I. Y., Abboud, S.-T., Rothermel, L. D., Azar, N., & Mangla, A. (2025). Advances in Oncolytic Viral Therapy in Melanoma: A Comprehensive Review. Vaccines, 13(7), 727. https://doi.org/10.3390/vaccines13070727