
Adjuvanted RNA Origami—A Tunable Peptide Assembly Platform for Constructing Cancer Nanovaccines

Abstract
1. Introduction
2. Materials and Methods
2.1. Peptide Sequences
2.2. RNA-OG and RNA-OG-DBCO Generation
2.3. Assembly of RNA-OG Peptide Complexes
2.4. Agarose Gel Electrophoresis
2.5. Stability of RNA-OG
2.6. Atomic Force Microscopy (AFM)
2.7. Bone Marrow-Derived Dendritic Cell (BMDC) Generation
2.8. BMDC Ex Vivo Stimulation and Antigen Presentation
2.9. Splenocyte Isolation
2.10. OT-I T Cell Ex Vivo Proliferation Assay
2.11. Mice
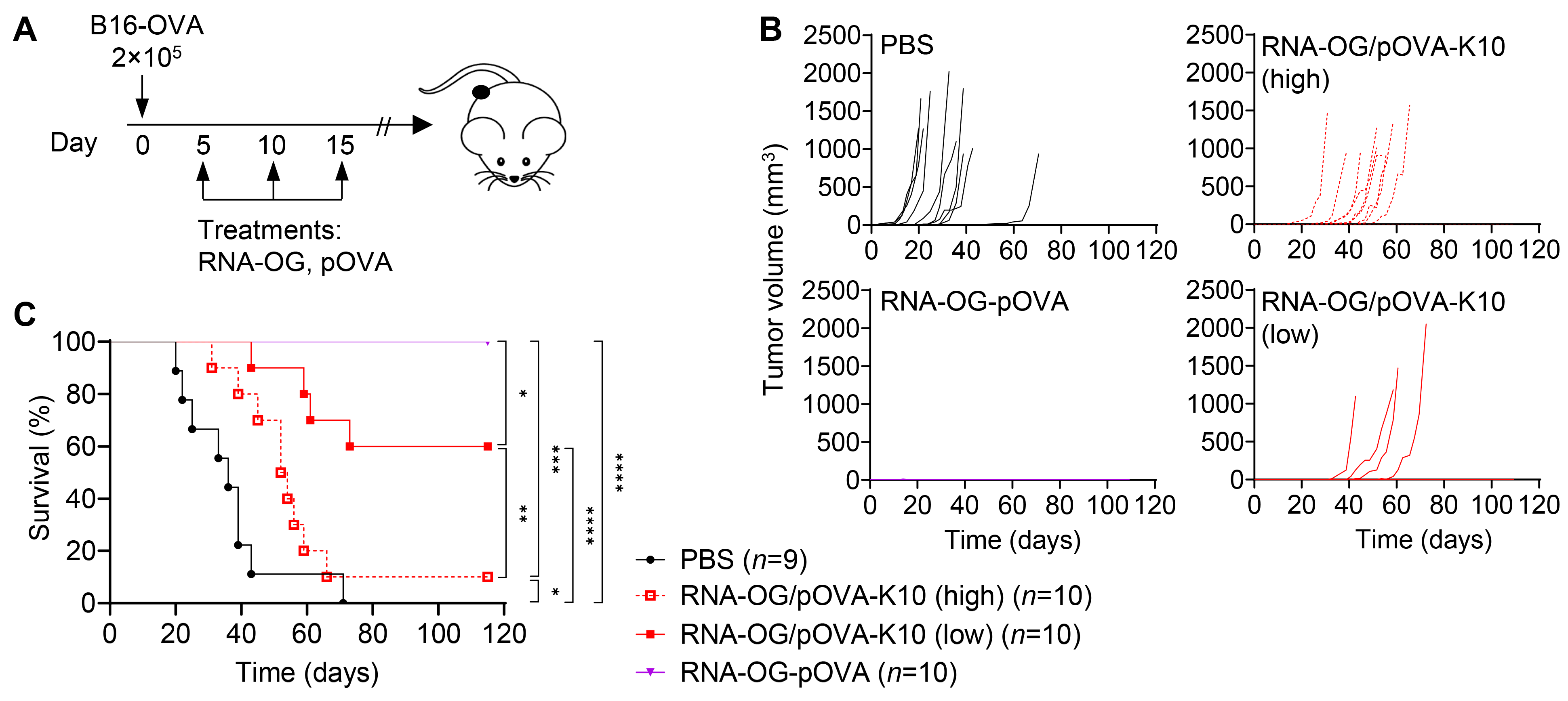
2.12. Tumor Challenge and Treatments
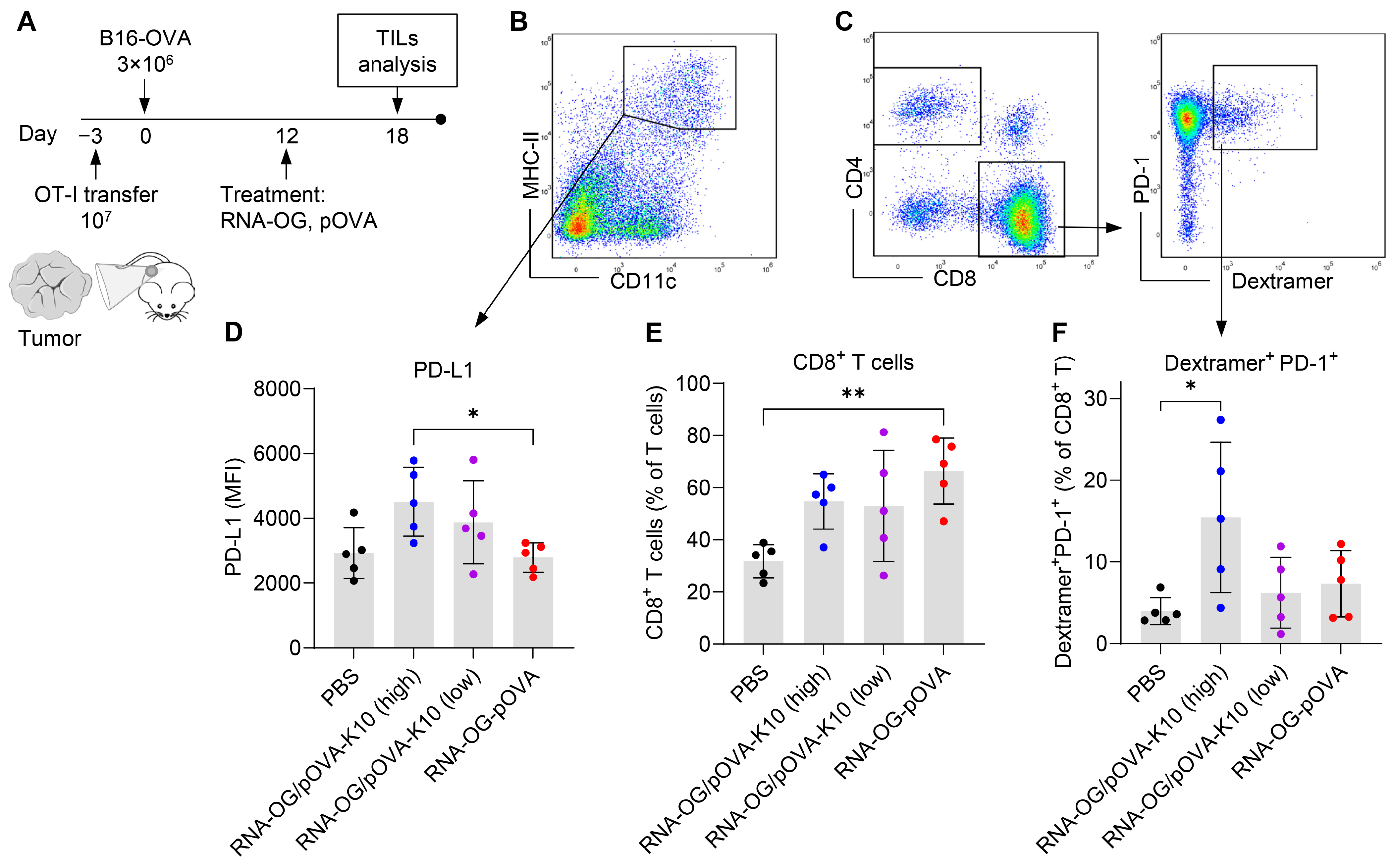
2.13. TILs Analysis
2.14. Tumor Processing
2.15. Flow Cytometry
2.16. Statistical Analysis
3. Results
3.1. RNA-OG Remains Intact Following RNA-OG/Peptide-K10 Assembly
3.2. RNA-OG/pOVA-K10 Promotes Robust DC Maturation and Presentation but Minimal T Cell Proliferation
3.3. Increased Peptide Amount in RNA-OG/pOVA-K10 Reduces Therapeutic Efficacy
3.4. Failed Immunity Initiated at the Tumor Site Attributed to an Early Induction of T Cell Exhaustion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFM | Atomic force microscopy |
| APC | Antigen-presenting cell |
| BMDC | Bone marrow-derived dendritic cell |
| ConA | Concanavalin A |
| DC | Dendritic cell |
| dsRNA | Double-stranded RNA |
| FITC | Fluorescein isothiocyanate |
| HBSS | Hank’s Balanced Salt Solution |
| IACUC | Institutional Animal Care and Use Committee |
| i.p. | Intraperitoneal |
| LN | Lymph node |
| MACS | Magnetic-activated cell sorting |
| PAMP | Pathogen-associated molecular pattern |
| PMB | Polymyxin B |
| pOVA | Ovalbumin peptide, OVA257–264 |
| pOVA-Az | Azide-linked ovalbumin peptide |
| pOVA-K10 | Polylysine-linked ovalbumin peptide |
| PRR | Pattern recognition receptor |
| RBC | Red blood cell |
| RNA-OG | RNA origami |
| RT | Room temperature |
| s.c. | Subcutaneous |
| TCRpOVA | pOVA-specific T cell receptor |
| TIL | Tumor-infiltrating leukocyte |
| TLR3 | Toll-like receptor 3 |
| Treg | Regulatory T cell |
References
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards Personalized, Tumour-Specific, Therapeutic Vaccines for Cancer. Nat. Rev. Immunol. 2017, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Toyoshima, Y.; Nakamura, Y. Personalized Immunotherapy in Cancer Precision Medicine. Cancer Biol. Med. 2021, 18, 955–965. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H. A Cell Culture Model for T Lymphocyte Clonal Anergy. Science (1979) 1990, 248, 1349–1356. [Google Scholar] [CrossRef]
- Toes, R.E.M.; van der Voort, E.I.H.; Schoenberger, S.P.; Drijfhout, J.W.; van Bloois, L.; Storm, G.; Kast, W.M.; Offringa, R.; Melief, C.J.M. Enhancement of Tumor Outgrowth Through CTL Tolerization After Peptide Vaccination Is Avoided by Peptide Presentation on Dendritic Cells. J. Immunol. 1998, 160, 4449–4456. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.; Nolte, M.A.; Spörri, R.; Sousa, C.R.E. Inflammatory Signals in Dendritic Cell Activation and the Induction of Adaptive Immunity. Immunol. Rev. 2009, 227, 234–247. [Google Scholar] [CrossRef]
- Mcadam, A.J.; Schweitzer’, N.; Sharpe, A.H.; Mcadum, A.J.; Sthweiizcr, A.N.; Mcadam, A. The Role of B7 Co-Stimulation in Activation and Differentiation OfCD4+and CD8+T Cells. Immunol. Rev. 1998, 165, 231–247. [Google Scholar] [CrossRef]
- Behrens, E.M.; Canna, S.W.; Slade, K.; Rao, S.; Kreiger, P.A.; Paessler, M.; Kambayashi, T.; Koretzky, G.A. Repeated TLR9 Stimulation Results in Macrophage Activation Syndrome–like Disease in Mice. J. Clin. Investig. 2011, 121, 2264–2277. [Google Scholar] [CrossRef]
- Fortier, M.E.; Kent, S.; Ashdown, H.; Poole, S.; Boksa, P.; Luheshi, G.N. The Viral Mimic, Polyinosinic:Polycytidylic Acid, Induces Fever in Rats via an Interleukin-1-Dependent Mechanism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, 759–766. [Google Scholar] [CrossRef]
- Hafner, A.M.; Corthésy, B.; Merkle, H.P. Particulate Formulations for the Delivery of Poly(I:C) as Vaccine Adjuvant. Adv. Drug Deliv. Rev. 2013, 65, 1386–1399. [Google Scholar] [CrossRef]
- Howard, G.P.; Verma, G.; Ke, X.; Thayer, W.M.; Hamerly, T.; Baxter, V.K.; Lee, J.E.; Dinglasan, R.R.; Mao, H.Q. Critical Size Limit of Biodegradable Nanoparticles for Enhanced Lymph Node Trafficking and Paracortex Penetration. Nano Res. 2019, 12, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Yousefpour, P.; Ni, K.; Irvine, D.J. Targeted Modulation of Immune Cells and Tissues Using Engineered Biomaterials. Nat. Rev. Bioeng. 2023, 1, 107–124. [Google Scholar] [CrossRef]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-Presentation of Exogenous Antigens on MHC I Molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef]
- Bijker, M.S.; van den Eeden, S.J.F.; Franken, K.L.; Melief, C.J.M.; van der Burg, S.H.; Offringa, R. Superior Induction of Anti-Tumor CTL Immunity by Extended Peptide Vaccines Involves Prolonged, DC-Focused Antigen Presentation. Eur. J. Immunol. 2008, 38, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Van Montfoort, N.; Camps, M.G.; Khan, S.; Filippov, D.V.; Weterings, J.J.; Griffith, J.M.; Geuze, H.J.; Van Hall, T.; Verbeek, J.S.; Melief, C.J.; et al. Antigen Storage Compartments in Mature Dendritic Cells Facilitate Prolonged Cytotoxic T Lymphocyte Cross-Priming Capacity. Proc. Natl. Acad. Sci. USA 2009, 106, 6730–6735. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Poppleton, E.; Urbanek, N.; Chakraborty, T.; Griffo, A.; Monari, L.; Göpfrich, K. RNA Origami: Design, Simulation and Application. RNA Biol. 2023, 20, 510–524. [Google Scholar] [CrossRef]
- McRae, E.K.S.; Rasmussen, H.Ø.; Liu, J.; Bøggild, A.; Nguyen, M.T.A.; Sampedro Vallina, N.; Boesen, T.; Pedersen, J.S.; Ren, G.; Geary, C.; et al. Structure, Folding and Flexibility of Co-Transcriptional RNA Origami. Nat. Nanotechnol. 2023, 18, 808–817. [Google Scholar] [CrossRef]
- Qi, X.; Liu, X.; Matiski, L.; Rodriguez, R.D.V.; Yip, T.; Zhang, F.; Sokalingam, S.; Jiang, S.; Liu, L.; Yan, H.; et al. RNA Origami Nanostructures for Potent and Safe Anticancer Immunotherapy. ACS Nano 2020, 14, 4740. [Google Scholar] [CrossRef]
- Yip, T.; Qi, X.; Yan, H.; Chang, Y. RNA Origami Functions as a Self-Adjuvanted Nanovaccine Platform for Cancer Immunotherapy. ACS Nano 2024, 18, 4056–4067. [Google Scholar] [CrossRef]
- Han, D.; Qi, X.; Myhrvold, C.; Wang, B.; Dai, M.; Jiang, S.; Bates, M.; Liu, Y.; An, B.; Zhang, F.; et al. Single-Stranded DNA and RNA Origami. Science (1979) 2017, 358, eaao2648. [Google Scholar] [CrossRef]
- Jin, H.T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Coop-eration of Tim-3 and PD-1 in CD8 T-Cell Exhaustion during Chronic Viral Infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.X.; et al. PD-L1 on Dendritic Cells Attenuates T Cell Activation and Regulates Response to Immune Checkpoint Blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef] [PubMed]
- Toes, R.E.M.; Offringa, R.; Blom, R.J.J.; Melief, C.J.M.; Kast, W.M. Peptide Vaccination Can Lead to Enhanced Tumor Growth through Specific T-Cell Tolerance Induction. Proc. Natl. Acad. Sci. USA 1996, 93, 7855–7860. [Google Scholar] [CrossRef] [PubMed]
- Bijker, M.S.; van den Eeden, S.J.F.; Franken, K.L.; Melief, C.J.M.; Offringa, R.; van der Burg, S.H. CD8+ CTL Priming by Exact Peptide Epitopes in Incomplete Freund’s Adjuvant Induces a Vanishing CTL Response, Whereas Long Peptides Induce Sustained CTL Reactivity. J. Immunol. 2007, 179, 5033–5040. [Google Scholar] [CrossRef]
- Varypataki, E.M.; Benne, N.; Bouwstra, J.; Jiskoot, W.; Ossendorp, F. Efficient Eradication of Established Tumors in Mice with Cationic Liposome-Based Synthetic Long-Peptide Vaccines. Cancer Immunol. Res. 2017, 5, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Zwaveling, S.; Mota, S.C.F.; Nouta, J.; Johnson, M.; Lipford, G.B.; Offringa, R.; van der Burg, S.H.; Melief, C.J.M. Es-tablished Human Papillomavirus Type 16-Expressing Tumors Are Effectively Eradicated Following Vaccination with Long Peptides. J. Immunol. 2002, 169, 350–358. [Google Scholar] [CrossRef]
- Chen, X.; Yang, J.; Wang, L.; Liu, B. Personalized Neoantigen Vaccination with Synthetic Long Peptides: Recent Ad-vances and Future Perspectives. Theranostics 2020, 10, 6011–6023. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Assembly Strategy | Molar Ratio (RNA-OG:Peptide) | Vaccine Dosage (RNA-OG:pOVA Peptide) |
|---|---|---|---|
| RNA-OG-peptide | Covalent linkage | 1:13 | 40 μg:0.9 μg |
| RNA-OG/peptide-K10low | Physical assembly | 1:13 | 40 μg:1.8 μg |
| RNA-OG/peptide-K10high | Physical assembly | 1:≥100 | 40 μg:≥14 μg |
| RNA-OG + peptidelow | Non-assembled | 1:13 | 40 μg:0.8 μg |
| RNA-OG + peptidehigh | Non-assembled | 1:100 | 40 μg:5.8 μg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yip, T.; Tu, X.; Qi, X.; Yan, H.; Chang, Y. Adjuvanted RNA Origami—A Tunable Peptide Assembly Platform for Constructing Cancer Nanovaccines. Vaccines 2025, 13, 560. https://doi.org/10.3390/vaccines13060560
Yip T, Tu X, Qi X, Yan H, Chang Y. Adjuvanted RNA Origami—A Tunable Peptide Assembly Platform for Constructing Cancer Nanovaccines. Vaccines. 2025; 13(6):560. https://doi.org/10.3390/vaccines13060560
Chicago/Turabian StyleYip, Theresa, Xinyi Tu, Xiaodong Qi, Hao Yan, and Yung Chang. 2025. "Adjuvanted RNA Origami—A Tunable Peptide Assembly Platform for Constructing Cancer Nanovaccines" Vaccines 13, no. 6: 560. https://doi.org/10.3390/vaccines13060560
APA StyleYip, T., Tu, X., Qi, X., Yan, H., & Chang, Y. (2025). Adjuvanted RNA Origami—A Tunable Peptide Assembly Platform for Constructing Cancer Nanovaccines. Vaccines, 13(6), 560. https://doi.org/10.3390/vaccines13060560