
Isolation and Characterization of E8 Monoclonal Antibodies from Donors Vaccinated with Recombinant Vaccinia Vaccine with Efficient Neutralization of Authentic Monkeypox Virus
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses, Cells, and Mice
2.2. Donors
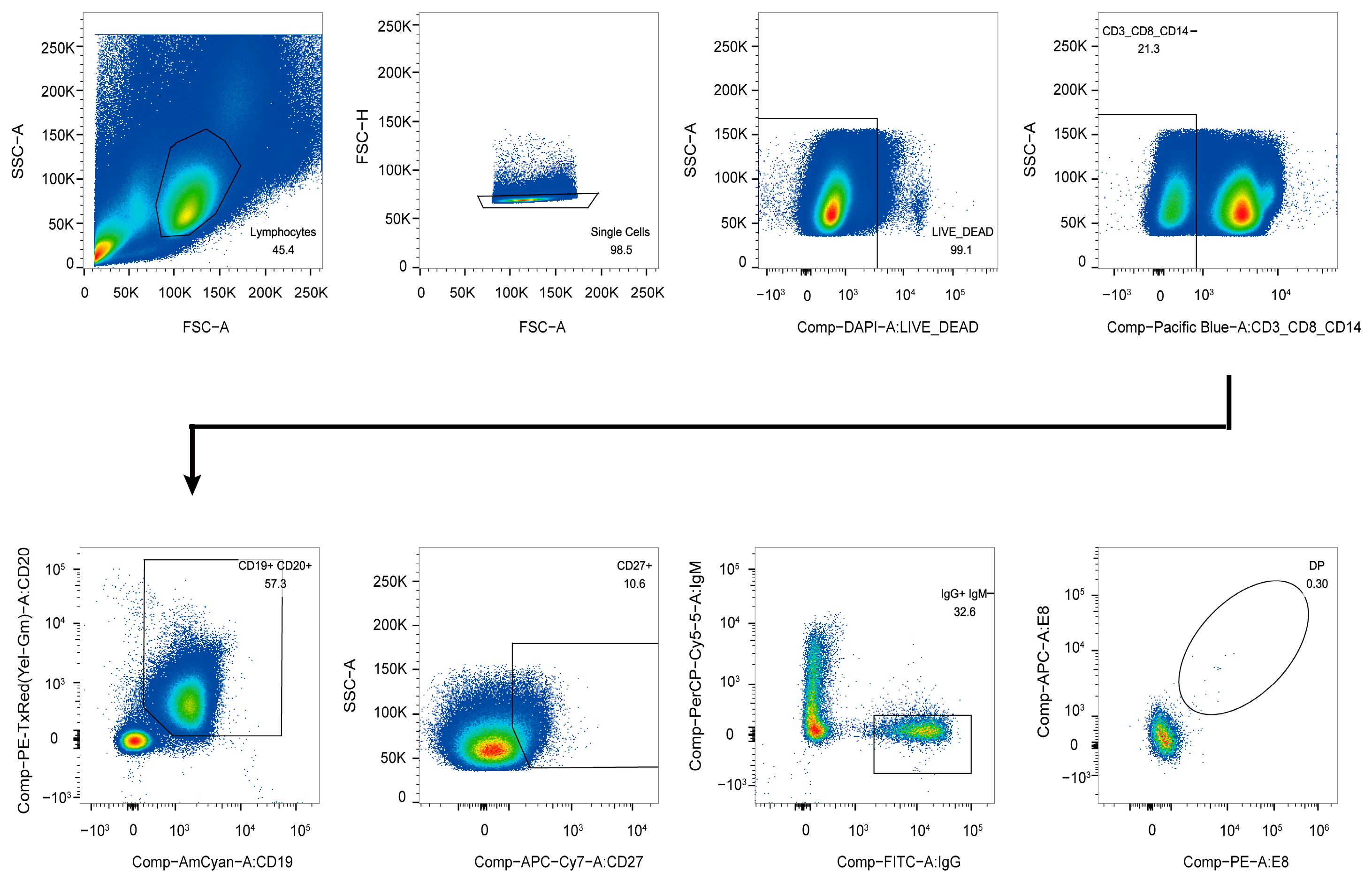
2.3. Antigen-Specific Single B Cell Sorting
2.4. Single B Cell RT-PCR, Family Analysis, and Construction of the Vector
2.5. Co-Transfection of Heavy and Light Chains of Antibodies in 293T Cells and ELISA Screening for Specific Antibodies
2.6. Expression and Purification of Monoclonal Antibodies
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Biolayer Interferometry (BLI)
2.9. Molecular Docking
2.10. VACV IMV and EEV Neutralization Assays
2.11. MPXV Neutralization Assay
2.12. Protection Studies in Mice
2.13. Viral Load
2.14. Statistical Analyses
3. Results
3.1. Isolation of E8-Specific IgG+CD19+ Cells
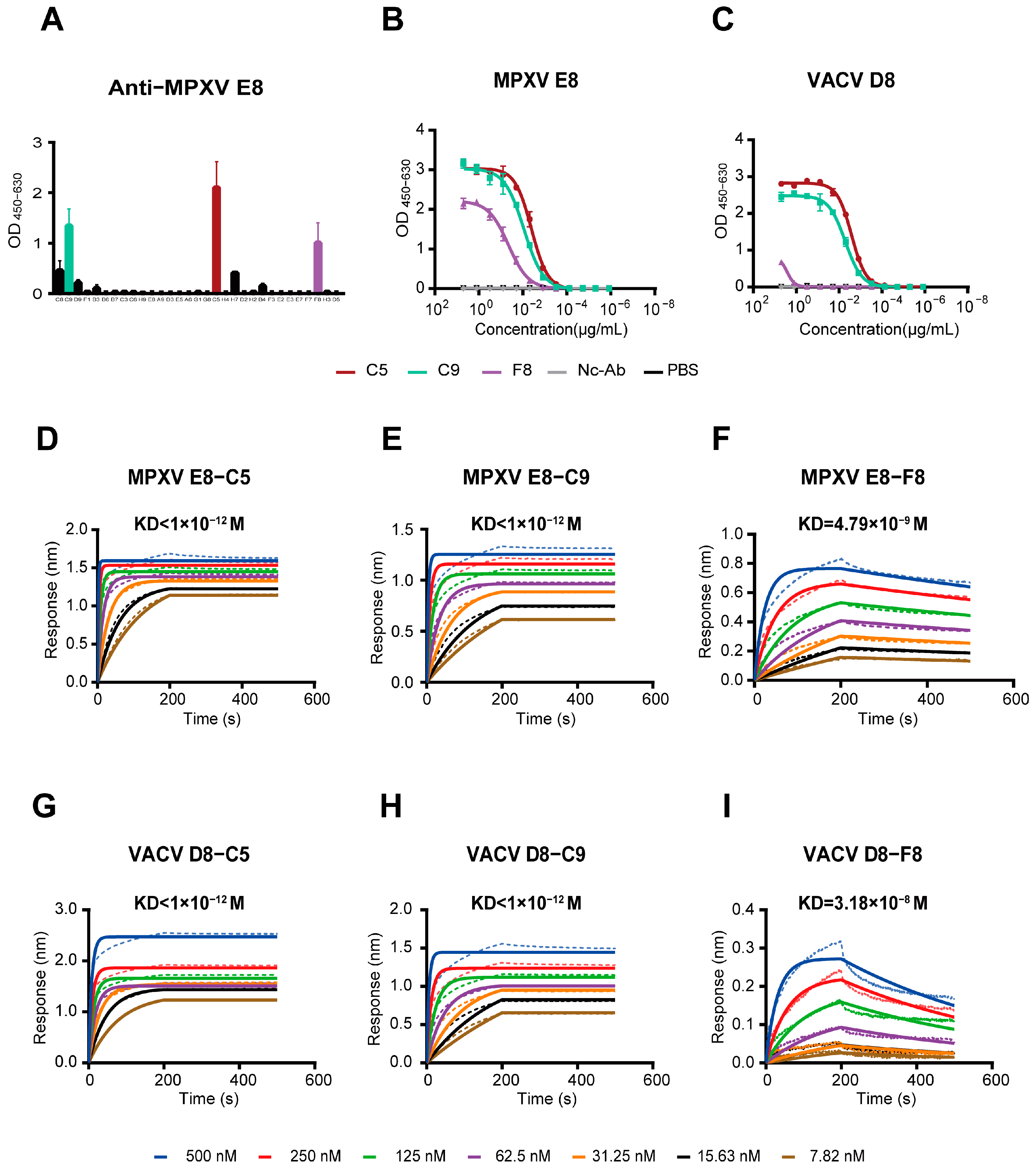
3.2. The mAbs Specific for the MPXV E8 Protein
3.3. Analysis of the Variable-Region Sequences of Monoclonal Antibodies
3.4. Three MPXV Monoclonal Antibodies Exhibit Strong Binding Activity
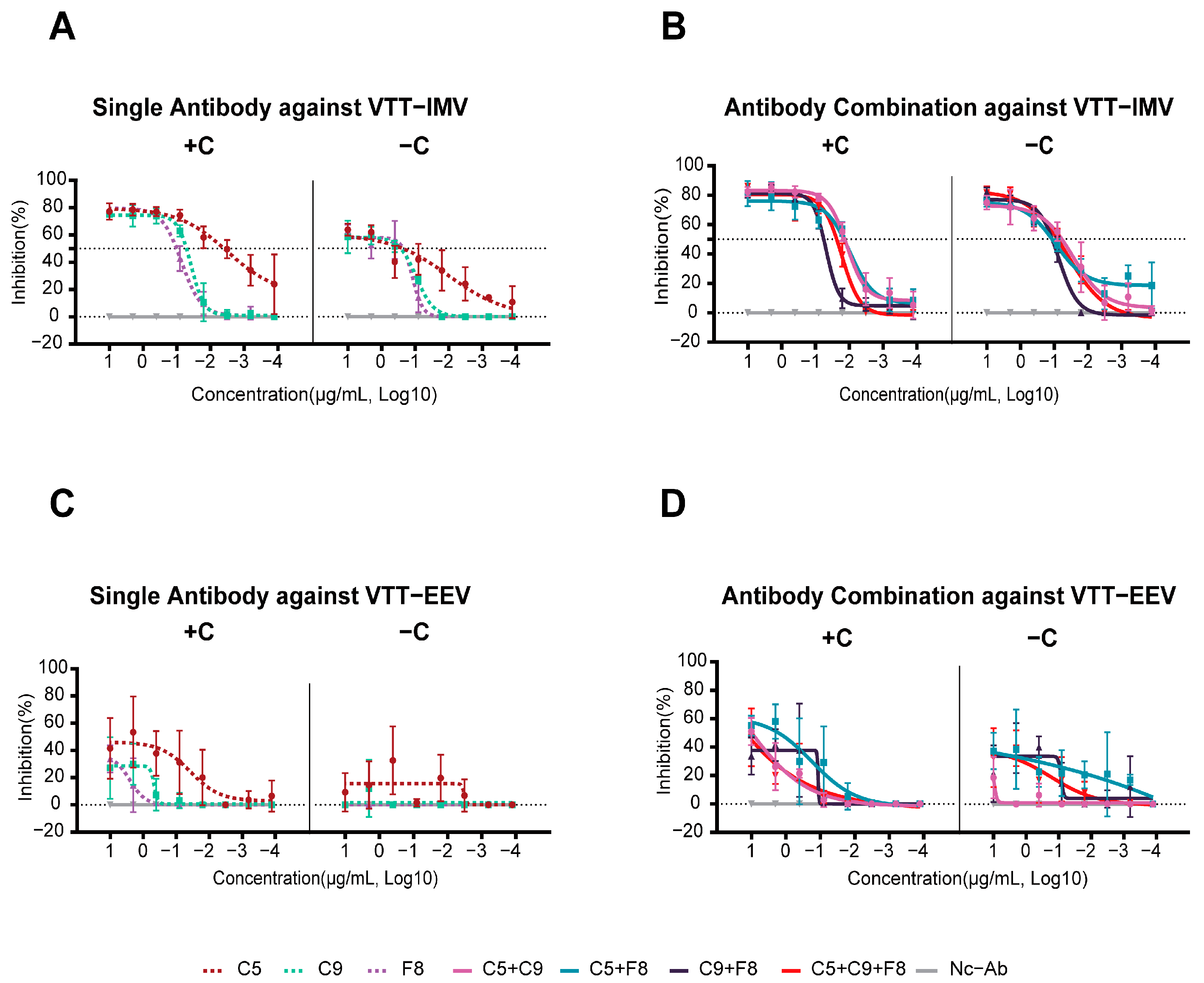
3.5. Potent Neutralizing Activity of the C9 mAb and Its Combination Against the Authentic MPXV
3.6. All Three MPXV Monoclonal Antibodies Were Able to Neutralize the VACV IMV In Vitro
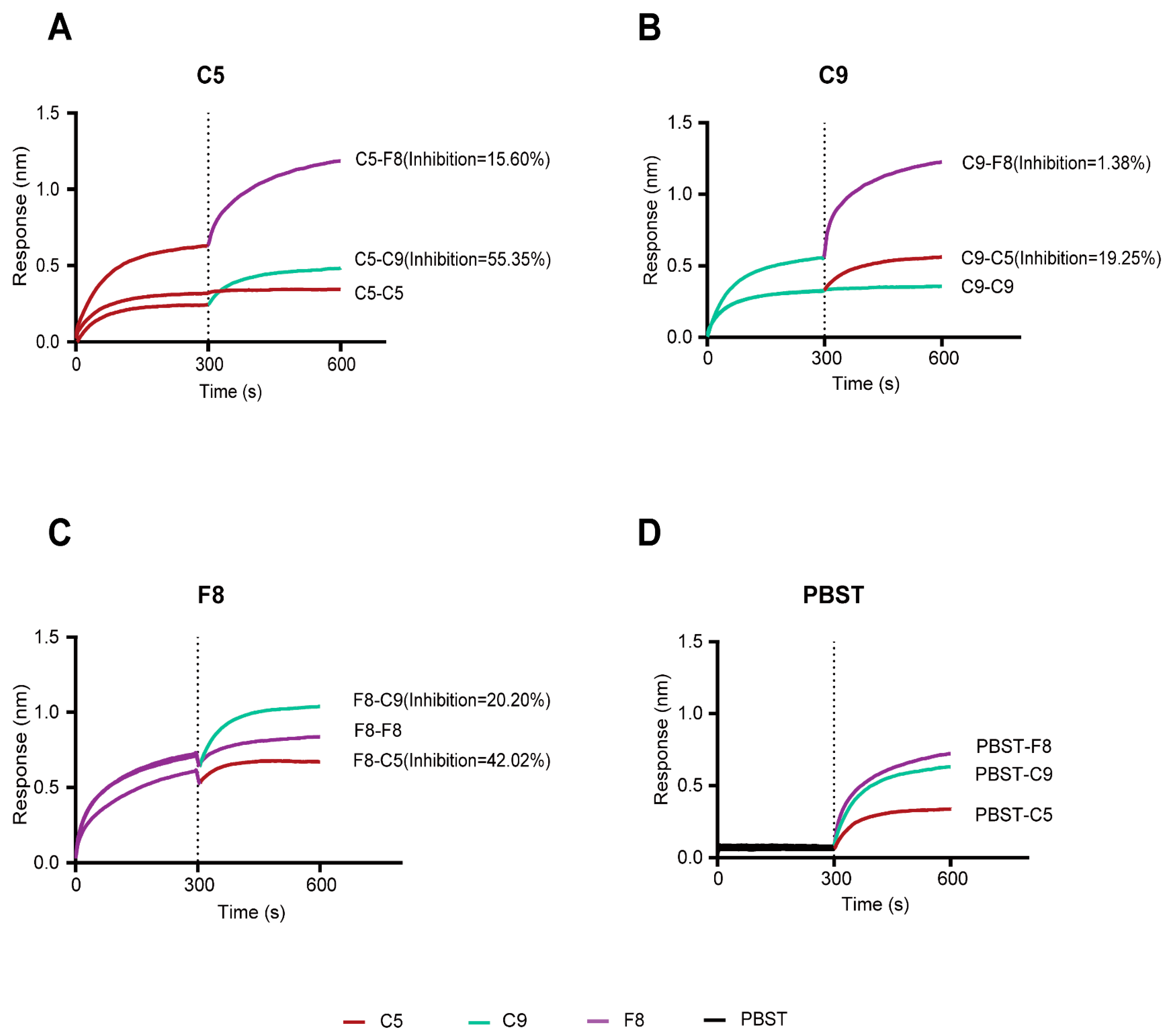
3.7. Integrated Competitive Assays and Structural Analysis of Antibody–MPXV E8 or Antibody–VACV D8 Interactions
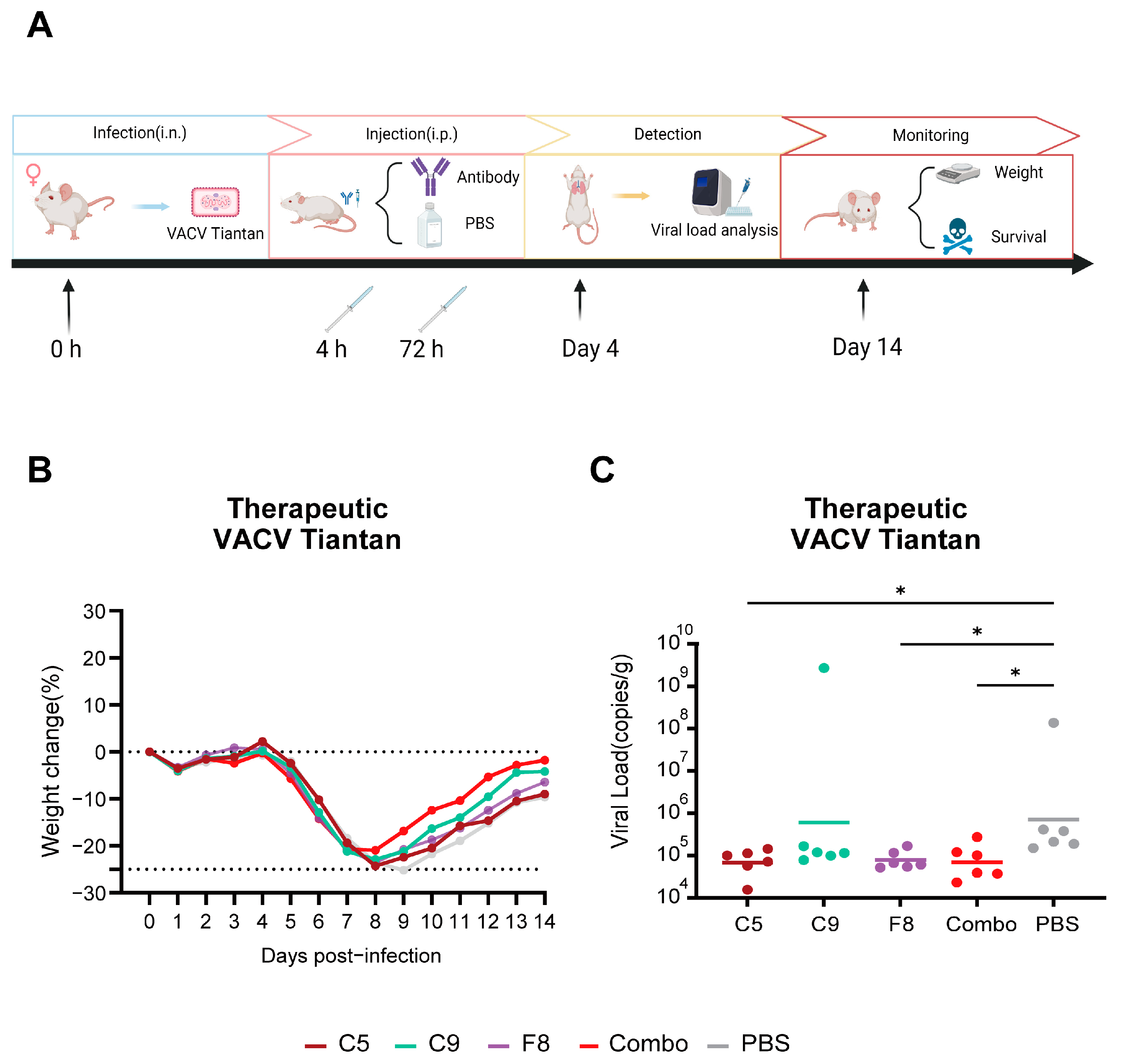
3.8. The Combo Shows Enhanced Protective Efficicency in Therapeutic Mouse Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Likos, A.M.; Sammons, S.A.; Olson, V.A.; Frace, A.M.; Li, Y.; Olsen-Rasmussen, M.; Davidson, W.; Galloway, R.; Khristova, M.L.; Reynolds, M.G.; et al. A tale of two clades: Monkeypox viruses. J. Gen. Virol. 2005, 86, 2661–2672. [Google Scholar] [CrossRef]
- Elsayed, S.; Bondy, L.; Hanage, W.P. Monkeypox Virus Infections in Humans. Clin. Microbiol. Rev. 2022, 35, e0009222. [Google Scholar] [CrossRef] [PubMed]
- Durski, K.N.; McCollum, A.M.; Nakazawa, Y.; Petersen, B.W.; Reynolds, M.G.; Briand, S.; Khalakdina, A. Emergence of monkeypox in West Africa and Central Africa, 1970–2017. Wkly. Epidemiol. Rec. 2018, 93, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Alakunle, E.; Moens, U.; Nchinda, G.; Okeke, M.I. Monkeypox Virus in Nigeria: Infection Biology, Epidemiology, and Evolution. Viruses 2020, 12, 1257. [Google Scholar] [CrossRef] [PubMed]
- Adegboye, O.A.; Eugenia Castellanos, M.; Alele, F.O.; Pak, A.; Ezechukwu, H.C.; Hou, K.; Emeto, T.I. Travel-Related Monkeypox Outbreaks in the Era of COVID-19 Pandemic: Are We Prepared? Viruses 2022, 14, 1283. [Google Scholar] [CrossRef]
- 2022–24 Mpox (Monkeypox) Outbreak: Global Trends [Internet]. Available online: https://worldhealthorg.shinyapps.io/mpx_global/#1_Overview (accessed on 2 March 2025).
- Masirika, L.M.; Udahemuka, J.C.; Schuele, L.; Ndishimye, P.; Otani, S.; Mbiribindi, J.B.; Marekani, J.M.; Mambo, L.M.; Bubala, N.M.; Boter, M.; et al. Ongoing mpox outbreak in Kamituga, South Kivu province, associated with monkeypox virus of a novel Clade I sub-lineage, Democratic Republic of the Congo, 2024. Euro Surveill. 2024, 29, 2400106. [Google Scholar] [CrossRef]
- Rivers, C.; Watson, C.; Phelan, A.L. The Resurgence of Mpox in Africa. JAMA 2024, 332, 1045–1046. [Google Scholar] [CrossRef]
- Elsheikh, R.; Makram, A.M.; Vasanthakumaran, T.; Tomar, S.; Shamim, K.; Tranh, N.D.; Elsheikh, S.S.; Van, N.T.; Huy, N.T. Monkeypox: A comprehensive review of a multifaceted virus. Infect. Med. 2023, 2, 74–88. [Google Scholar] [CrossRef]
- Fink, D.L.; Callaby, H.; Luintel, A.; Beynon, W.; Bond, H.; Lim, E.Y.; Gkrania-Klotsas, E.; Heskin, J.; Bracchi, M.; Rathish, B.; et al. Clinical features and management of individuals admitted to hospital with monkeypox and associated complications across the UK: A retrospective cohort study. Lancet Infect. Dis. 2023, 23, 589–597. [Google Scholar] [CrossRef]
- Miller, M.J.; Cash-Goldwasser, S.; Marx, G.E.; Schrodt, C.A.; Kimball, A.; Padgett, K.; Noe, R.S.; McCormick, D.W.; Wong, J.M.; Labuda, S.M.; et al. Severe Monkeypox in Hospitalized Patients—United States, August 10–October 10, 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1412–1417. [Google Scholar] [CrossRef]
- Mitjà, O.; Alemany, A.; Marks, M.; Lezama Mora, J.I.; Rodríguez-Aldama, J.C.; Torres Silva, M.S.; Corral Herrera, E.A.; Crabtree-Ramirez, B.; Blanco, J.L.; Girometti, N.; et al. Mpox in people with advanced HIV infection: A global case series. Lancet 2023, 401, 939–949. [Google Scholar] [CrossRef]
- Higgins, E.; Ranganath, N.; Mehkri, O.; Majeed, A.; Walker, J.; Spivack, S.; Bhaimia, E.; Benamu, E.; Hand, J.; Keswani, S.; et al. Clinical features, treatment, and outcomes of mpox in solid organ transplant recipients: A multicenter case series and literature review. Am. J. Transplant. 2023, 23, 1972–1979. [Google Scholar] [CrossRef]
- Siegrist, E.A.; Sassine, J. Antivirals With Activity Against Mpox: A Clinically Oriented Review. Clin. Infect. Dis. 2023, 76, 155–164. [Google Scholar] [CrossRef]
- National Institutes of Health (NIH) [Internet]. The Antiviral Tecovirimat Is Safe but Did Not Improve Clade I Mpox Resolution in Democratic Republic of the Congo. 2024. Available online: https://www.nih.gov/news-events/news-releases/antiviral-tecovirimat-safe-did-not-improve-clade-i-mpox-resolution-democratic-republic-congo (accessed on 9 September 2024).
- Smith, T.G.; Gigante, C.M.; Wynn, N.T.; Matheny, A.; Davidson, W.; Yang, Y.; Condori, R.E.; O’Connell, K.; Kovar, L.; Williams, T.L.; et al. Tecovirimat Resistance in Mpox Patients, United States, 2022–2023. Emerg. Infect. Dis. 2023, 29, 2426–2432. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, G.; Correia, B.; Fenwick, C.; Joo, V.S.; Perez, L. Antibodies to combat viral infections: Development strategies and progress. Nat. Rev. Drug Discov. 2022, 21, 676–696. [Google Scholar] [CrossRef] [PubMed]
- Gilchuk, I.; Gilchuk, P.; Sapparapu, G.; Lampley, R.; Singh, V.; Kose, N.; Blum, D.L.; Hughes, L.J.; Satheshkumar, P.S.; Townsend, M.B.; et al. Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections. Cell 2016, 167, 684–694.e9. [Google Scholar] [CrossRef]
- Alakunle, E.; Kolawole, D.; Diaz-Cánova, D.; Alele, F.; Adegboye, O.; Moens, U.; Okeke, M.I. A comprehensive review of monkeypox virus and mpox characteristics. Front. Cell. Infect. Microbiol. 2024, 14, 1360586. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Guo, X.; Hong, D.; Gong, Q.; Xie, W.; Li, T.; Wang, J.; Chuai, X.; Chiu, S. Duration of humoral immunity from smallpox vaccination and its cross-reaction with Mpox virus. Signal Transduct. Target. Ther. 2023, 8, 350. [Google Scholar] [CrossRef]
- Rao, A.K. Use of JYNNEOS (Smallpox and Monkeypox Vaccine, Live, Nonreplicating) for Preexposure Vaccination of Persons at Risk for Occupational Exposure to Orthopoxviruses: Recommendations of the Advisory Committee on Immunization Practices—United States, 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 734–742. [Google Scholar] [CrossRef]
- Ramírez, J.C.; Tapia, E.; Esteban, M. Administration to mice of a monoclonal antibody that neutralizes the intracellular mature virus form of vaccinia virus limits virus replication efficiently under prophylactic and therapeutic conditions. J. Gen. Virol. 2002, 83 Pt 5, 1059–1067. [Google Scholar] [CrossRef]
- Wolffe, E.J.; Vijaya, S.; Moss, B. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 1995, 211, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.C.; Chung, C.S.; Chang, W. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 1999, 73, 8750–8761. [Google Scholar] [CrossRef]
- Law, M.; Smith, G.L. Antibody neutralization of the extracellular enveloped form of vaccinia virus. Virology 2001, 280, 132–142. [Google Scholar] [CrossRef]
- Lin, C.L.; Chung, C.S.; Heine, H.G.; Chang, W. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 2000, 74, 3353–3365. [Google Scholar] [CrossRef]
- Qu, Y.; Tai, W.; Ma, E.; Jiang, Q.; Fan, M.; Xiao, W.; Tian, C.; Liu, Y.; Liu, J.; Wang, X.; et al. Generation and characterization of neutralizing antibodies against M1R and B6R proteins of monkeypox virus. Nat. Commun. 2025, 16, 3100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wu, L.; Sun, J.; Liu, D.; Han, P.; Gao, Y.; Zhang, Y.; Xu, Y.; Qu, X.; Wang, H.; et al. Two noncompeting human neutralizing antibodies targeting MPXV B6 show protective effects against orthopoxvirus infections. Nat. Commun. 2024, 15, 4660. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, J.; Wang, F.; Kuang, J.; Peng, Y.; Asghar, S.; Zhao, W.; Yang, Y.; Shen, C. Bispecific antibodies targeting MPXV A29 and B6 demonstrate efficacy against MPXV infection. J. Virol. 2025, e0232024. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ren, Z.; Wang, Y.; Jiang, Y.; Yang, M.; Li, D.; Chen, J.; Liang, Z.; Lin, Y.; Zeng, Z.; et al. Three neutralizing mAbs induced by MPXV A29L protein recognizing different epitopes act synergistically against orthopoxvirus. Emerg. Microbes Infect. 2023, 12, 2223669. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, H.; Cheng, L.; Zhao, C.; Zhou, X.; Liao, X.; Ge, X.; Liu, L.; Lu, X.; Ju, B.; et al. Two long-lasting human monoclonal antibodies cross-react with monkeypox virus A35 antigen. Cell Discov. 2023, 9, 50. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, M.; Zhang, M.; Jing, R.; Zhou, J.; Cao, H.; Liu, C.; Zhu, H.; Ghonaim, A.H.; Rouby, S.R.; et al. The Generation and Characterization of Monoclonal Antibodies against the MPXV A29L Protein. Viruses 2024, 16, 1184. [Google Scholar] [CrossRef]
- Sagdat, K.; Batyrkhan, A.; Kanayeva, D. Exploring monkeypox virus proteins and rapid detection techniques. Front. Cell. Infect. Microbiol. 2024, 14, 1414224. [Google Scholar] [CrossRef] [PubMed]
- Tiller, T.; Meffre, E.; Yurasov, S.; Tsuiji, M.; Nussenzweig, M.C.; Wardemann, H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 2008, 329, 112–124. [Google Scholar] [CrossRef]
- Kong, L.; Ju, B.; Chen, Y.; He, L.; Ren, L.; Liu, J.; Hong, K.; Su, B.; Wang, Z.; Ozorowski, G.; et al. Key gp120 Glycans Pose Roadblocks to the Rapid Development of VRC01-Class Antibodies in an HIV-1-Infected Chinese Donor. Immunity 2016, 44, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3 | Nature [Internet]. Available online: https://www.nature.com/articles/s41586-024-07487-w (accessed on 19 January 2025).
- Delano, W.L. PyMOL: An Open-Source Molecular Graphics Tool. 2002. Available online: http://www.researchgate.net/publication/313391141_PyMOL_An_Open-Source_Molecular_Graphics_Tool (accessed on 19 January 2025).
- Lavinder, J.J.; Wine, Y.; Giesecke, C.; Ippolito, G.C.; Horton, A.P.; Lungu, O.I.; Hoi, K.H.; DeKosky, B.J.; Murrin, E.M.; Wirth, M.M.; et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 2259–2264. [Google Scholar] [CrossRef]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Klein, F.; Diskin, R.; Scheid, J.F.; Gaebler, C.; Mouquet, H.; Georgiev, I.S.; Pancera, M.; Zhou, T.; Incesu, R.-B.; Fu, B.Z.; et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 2013, 153, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Gaebler, C.; Muecksch, F.; Lorenzi, J.C.C.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.O.; Gazumyan, A.; Finkin, S.; et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 2020, 584, 437–442. [Google Scholar] [CrossRef]
- Matho, M.H.; de Val, N.; Miller, G.M.; Brown, J.; Schlossman, A.; Meng, X.; Crotty, S.; Peters, B.; Xiang, Y.; Hsieh-Wilson, L.C.; et al. Murine anti-vaccinia virus D8 antibodies target different epitopes and differ in their ability to block D8 binding to CS-E. PLoS Pathog. 2014, 10, e1004495. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, Y.; Deng, Y.; Gao, W.; Huang, B.; Yao, W.; Zhao, Y.; Zhang, Q.; Huang, M.; Liu, M.; et al. Long-lasting humoral and cellular memory immunity to vaccinia virus Tiantan provides pre-existing immunity against mpox virus in Chinese population. Cell Rep. 2024, 43, 113609. [Google Scholar] [CrossRef]
- Huang, Q.; Wang, Y.; Zhao, T.; Wang, Y.; Wang, X.; Li, S.; Su, W.; Ren, X.; Zhang, X.; Liu, J.; et al. Examination of the cross-reactivity between vaccinia virus Tiantan strain and monkeypox virus. J. Virol. Methods 2023, 320, 114772. [Google Scholar] [CrossRef]
- Marchi, S.; Piccini, G.; Cantaloni, P.; Guerrini, N.; Zannella, R.; Coluccio, R.; Benincasa, L.; Solfanelli, N.; Remarque, E.J.; Viviani, S.; et al. Evaluation of monkeypox- and vaccinia-virus neutralizing antibodies before and after smallpox vaccination: A sero-epidemiological study. J. Med. Virol. 2024, 96, e29728. [Google Scholar] [CrossRef]
- Das, R.; Bhattarai, A.; Karn, R.; Tamang, B. Computational investigations of potential inhibitors of monkeypox virus envelope protein E8 through molecular docking and molecular dynamics simulations. Sci. Rep. 2024, 14, 19585. [Google Scholar] [CrossRef]
- Maroli, N. Riding the Wave: Unveiling the Conformational Waves from RBD of SARS-CoV-2 Spike Protein to ACE2. J. Phys. Chem. B 2023, 127, 8525–8536. [Google Scholar] [CrossRef] [PubMed]
- da Silva, P.C.C.; Martinez, L. Extended Conformational Selection in the Antigen-Antibody Interaction of the PfAMA1 Protein. J. Phys. Chem. B 2024, 128, 8400–8408. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.; Baravalle, R.; Holyfield, R.; Kalms, J.; Wright, H.; Seewooruthun, C.; Muskett, F.W.; Scott-Tucker, A.; Merritt, A.; Henry, A.; et al. Identification and characterisation of anti-IL-13 inhibitory single domain antibodies provides new insights into receptor selectivity and attractive opportunities for drug discovery. Front. Immunol. 2023, 14, 1216967. [Google Scholar] [CrossRef]
- Carroll, M.C. The complement system in regulation of adaptive immunity. Nat. Immunol. 2004, 5, 981–986. [Google Scholar] [CrossRef]
- Hudson, P.N.; Self, J.; Weiss, S.; Braden, Z.; Xiao, Y.; Girgis, N.M.; Emerson, G.; Hughes, C.; Sammons, S.A.; Isaacs, S.N.; et al. Elucidating the role of the complement control protein in monkeypox pathogenicity. PLoS ONE 2012, 7, e35086. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Leung, M.K.; Hauhart, R.; Buller, R.M.L.; Bertram, P.; Wang, X.; Rosengard, A.M.; Kotwal, G.J.; Atkinson, J.P. Structure and regulatory profile of the monkeypox inhibitor of complement: Comparison to homologs in vaccinia and variola and evidence for dimer formation. J. Immunol. 2006, 176, 3725–3734. [Google Scholar] [CrossRef]
- Li, Y.; Hou, J.; Sun, Z.; Hu, J.; Thilakavathy, K.; Wang, Y.; Shao, Z.; Lu, Y.; Wang, W.; Xiong, C. Monkeypox virus 2022, gene heterogeneity and protein polymorphism. Signal Transduct. Target. Ther. 2023, 8, 278. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V Gene | D Gene | J Gene | CDR3 | SHM | ||
|---|---|---|---|---|---|---|
| Heavy Chain | C5H | Homsap IGHV1-2*02 F | Homsap IGHD3-22*01 F | Homsap IGHJ4*02 F | ARTPPDLSGFH | 4.86% |
| C9H | Homsap IGHV4-34*01 F | Homsap IGHD6-6*01 F | Homsap IGHJ4*02 F | ARTPATITARRRFDY | 5.26% | |
| F8H | Homsap IGHV1-24*01 F | Homsap IGHD2-21*02 F | Homsap IGHJ5*02 F | ATAVYCGGDCYSPWFDP | 5.56% | |
| Light Chain | C5L | Homsap IGKV2-28*01 F | - | Homsap IGKJ3*01 F | MQTLQTPLT | 5.92% |
| C9L | Homsap IGKV1-39*01 F | - | Homsap IGKJ4*01 F | QQSYTTPLT | 2.94% | |
| F8L | Homsap IGKV3-15*01 F | - | Homsap IGKJ2*01 F | QQYNNWPPDT | 2.94% |
| Antibody | IC50 (μg/mL) | |
|---|---|---|
| −C | +C | |
| C5 | >100 | >100 |
| C9 | 3.2 | 3.0 |
| F8 | >100 | >100 |
| Combo | 0.4 | 2.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Wang, S.; Hao, Y.; Shen, X.; Zhang, J.; Wang, S.; Zhang, J.; Fu, Y.; Chen, R.; Wang, D.; et al. Isolation and Characterization of E8 Monoclonal Antibodies from Donors Vaccinated with Recombinant Vaccinia Vaccine with Efficient Neutralization of Authentic Monkeypox Virus. Vaccines 2025, 13, 471. https://doi.org/10.3390/vaccines13050471
Shi Y, Wang S, Hao Y, Shen X, Zhang J, Wang S, Zhang J, Fu Y, Chen R, Wang D, et al. Isolation and Characterization of E8 Monoclonal Antibodies from Donors Vaccinated with Recombinant Vaccinia Vaccine with Efficient Neutralization of Authentic Monkeypox Virus. Vaccines. 2025; 13(5):471. https://doi.org/10.3390/vaccines13050471
Chicago/Turabian StyleShi, Yutao, Shuhui Wang, Yanling Hao, Xiuli Shen, Jun Zhang, Shuo Wang, Junjie Zhang, Yuyu Fu, Ran Chen, Dong Wang, and et al. 2025. "Isolation and Characterization of E8 Monoclonal Antibodies from Donors Vaccinated with Recombinant Vaccinia Vaccine with Efficient Neutralization of Authentic Monkeypox Virus" Vaccines 13, no. 5: 471. https://doi.org/10.3390/vaccines13050471
APA StyleShi, Y., Wang, S., Hao, Y., Shen, X., Zhang, J., Wang, S., Zhang, J., Fu, Y., Chen, R., Wang, D., Shao, Y., Li, D., & Liu, Y. (2025). Isolation and Characterization of E8 Monoclonal Antibodies from Donors Vaccinated with Recombinant Vaccinia Vaccine with Efficient Neutralization of Authentic Monkeypox Virus. Vaccines, 13(5), 471. https://doi.org/10.3390/vaccines13050471