
Entry Efficiency, Protease Dependence, and Antibody-Mediated Neutralization of SARS-CoV-2 Sublineages KP.3.1.1 and XEC

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
| Cell Line | Species and Organ | Source (Catalogue No. and RRID) | Culture Medium and Conditions | Special Characteristics/Method |
| BHK-21 | Syrian hamster, kidney | ATCC, Cat# CCL-10, RRID: CVCL_1915 | Dulbecco’s Modified Eagle Medium (DMEM; PAN-Biotech, Aidenbach, Germany) with 10% FCS and 1% penicillin–streptomycin (P/S) at 37 °C, 5% CO2 | Transfected with Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions |
| Vero | African green monkey, kidney, female | ATCC, Cat# CRL-1586, RRID: CVCL_0574 | DMEM (PAN-Biotech, Aidenbach, Germany) with 10% FCS and 1% P/S at 37 °C, 5% CO2 | |
| 293T | Human, kidney, female | DSMZ, Cat# ACC-635, RRID: CVCL_0063 | DMEM (PAN-Biotech, Aidenbach, Germany) with 10% FCS and 1% P/S at 37 °C, 5% CO2 | Transfected via calcium phosphate precipitation method |
| Caco-2 | Human, colon, male | ATCC, Cat# HTB-37, RRID: 0025 | DMEM (PAN-Biotech, Aidenbach, Germany) with 10% FCS and 1% P/S at 37 °C, 5% CO2 | |
| Calu-3 | Human, lung, male | ATCC, Cat# HTB-55, RRID: CVCL_0609 | DMEM (PAN-Biotech, Aidenbach, Germany) with 10% FCS and 1% P/S at 37 °C, 5% CO2 | |
| Calu-3-Omega | Human, lung, male | DMEM/F-12 (Thermo Fisher Scientific, Waltham, MA, USA) with 10% FCS, 1% P/S, NEAA, sodium pyruvate, and 2 µg/mL puromycin at 37 °C, 5% CO2 | Expressing beta-galactosidase omega fragment, generated by retroviral transduction and puromycin selection [25] | |
| 293T ACE2 Knockout (293T_ACE2 KO) | Human, kidney, female | DMEM (PAN-Biotech, Aidenbach, Germany) with 10% FCS and 1% P/S at 37 °C, 5% CO2 | Generated via CRISPR-Cas9 system, maintained with 1 µg/mL puromycin [26,27] |
2.2. Plasmids and Expression Constructs
- -
- pCG1-SARS-CoV-2 B.1 S∆18 (GISAID Accession ID: EPI_ISL_425259) [7];
- -
- pCG1-SARS-CoV-2 JN.1 S∆18 (GISAID Accession ID: EPI_ISL_18530042) [20];
- -
- pCG1-SARS-CoV-2 KP.3.1.1 S∆18 (GISAID Accession ID: EPI_ISL_19455032) [28];
- -
- pCG1-SARS-CoV-2 XEC S∆18 (GISAID Accession ID: EPI_ISL_19454087) [28].
2.3. Cell–Cell Fusion Assay
2.4. Production of VSV Pseudoparticles (VSVpp) and Cell Entry Assays
2.5. Immunoblot Analysis of S Protein Processing and Particle Incorporation
2.6. Neutralization Assay
2.7. Statistical Analysis and Quantification of Results
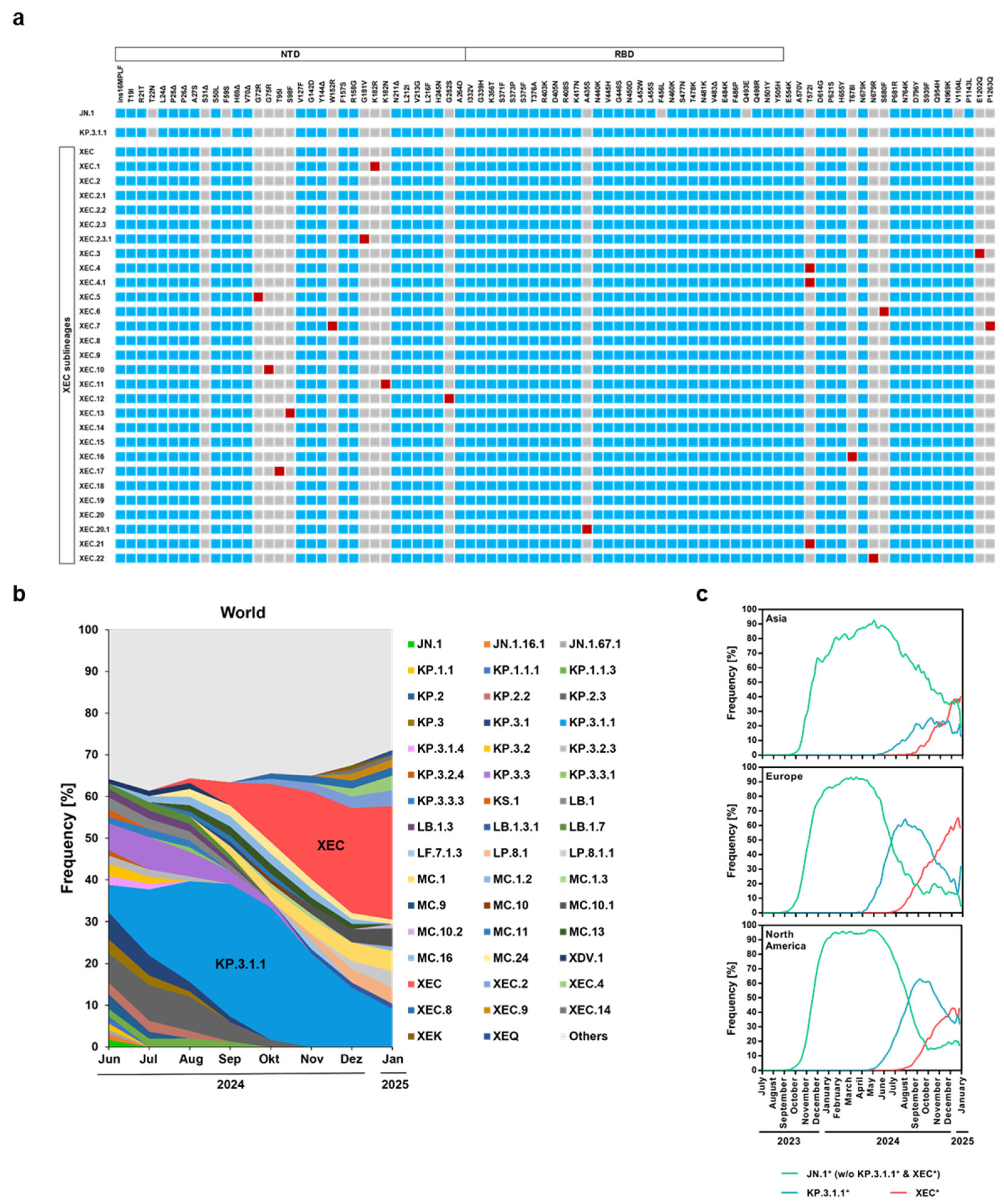
3. Results
3.1. KP.3.1.1 and XEC S Proteins Are Efficiently Cleaved and Facilitate Robust Entry into Several Cell Lines
3.2. Entry into Calu-3 and Caco-2 Cells Is TMPRSS2-Dependent
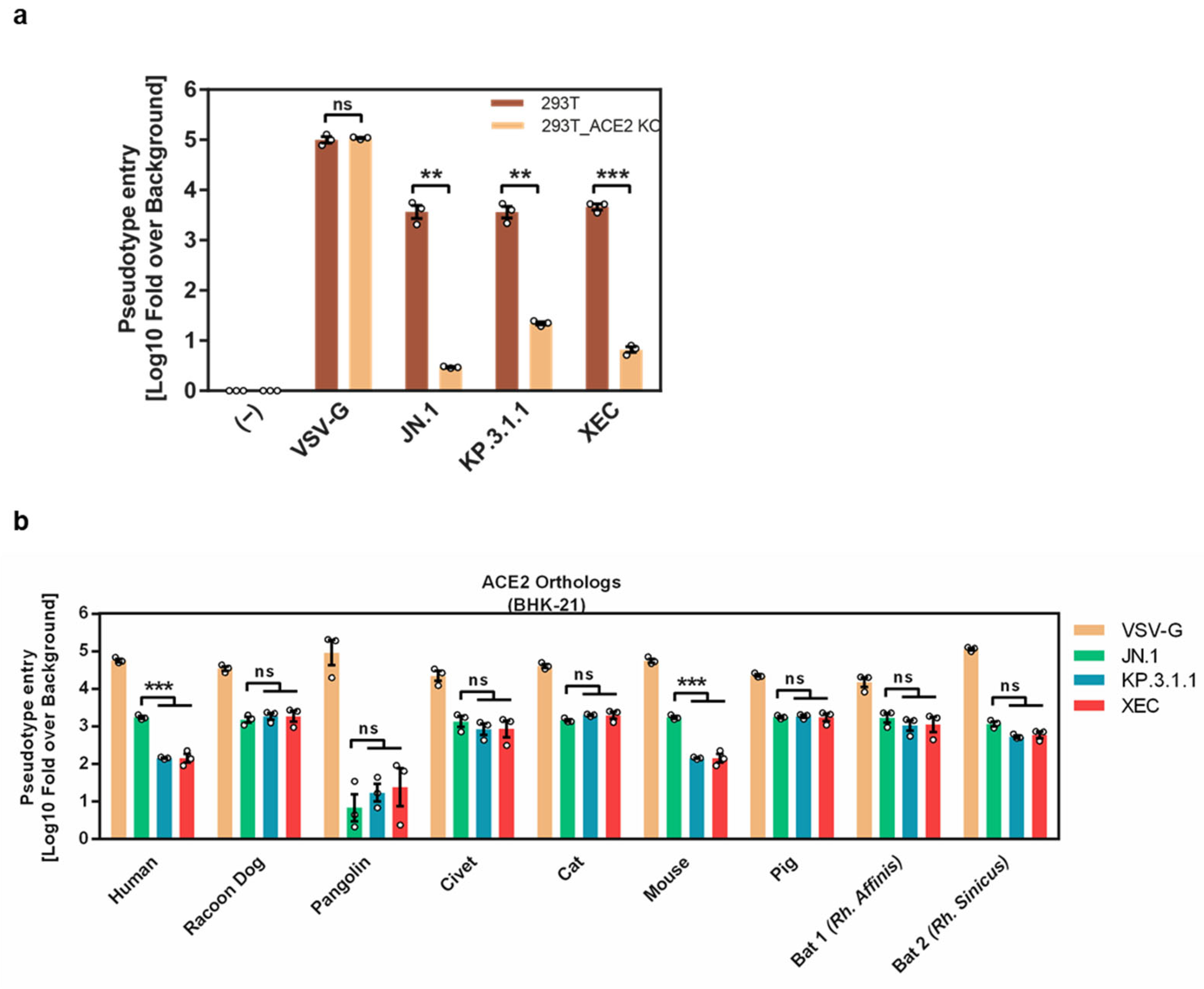
3.3. Differential ACE2 Interactions of KP.3.1.1 and XEC S Proteins
3.4. Reduced Cell–Cell Fusion Mediated by KP.3.1.1 and XEC S Proteins Compared to JN.1 S Protein
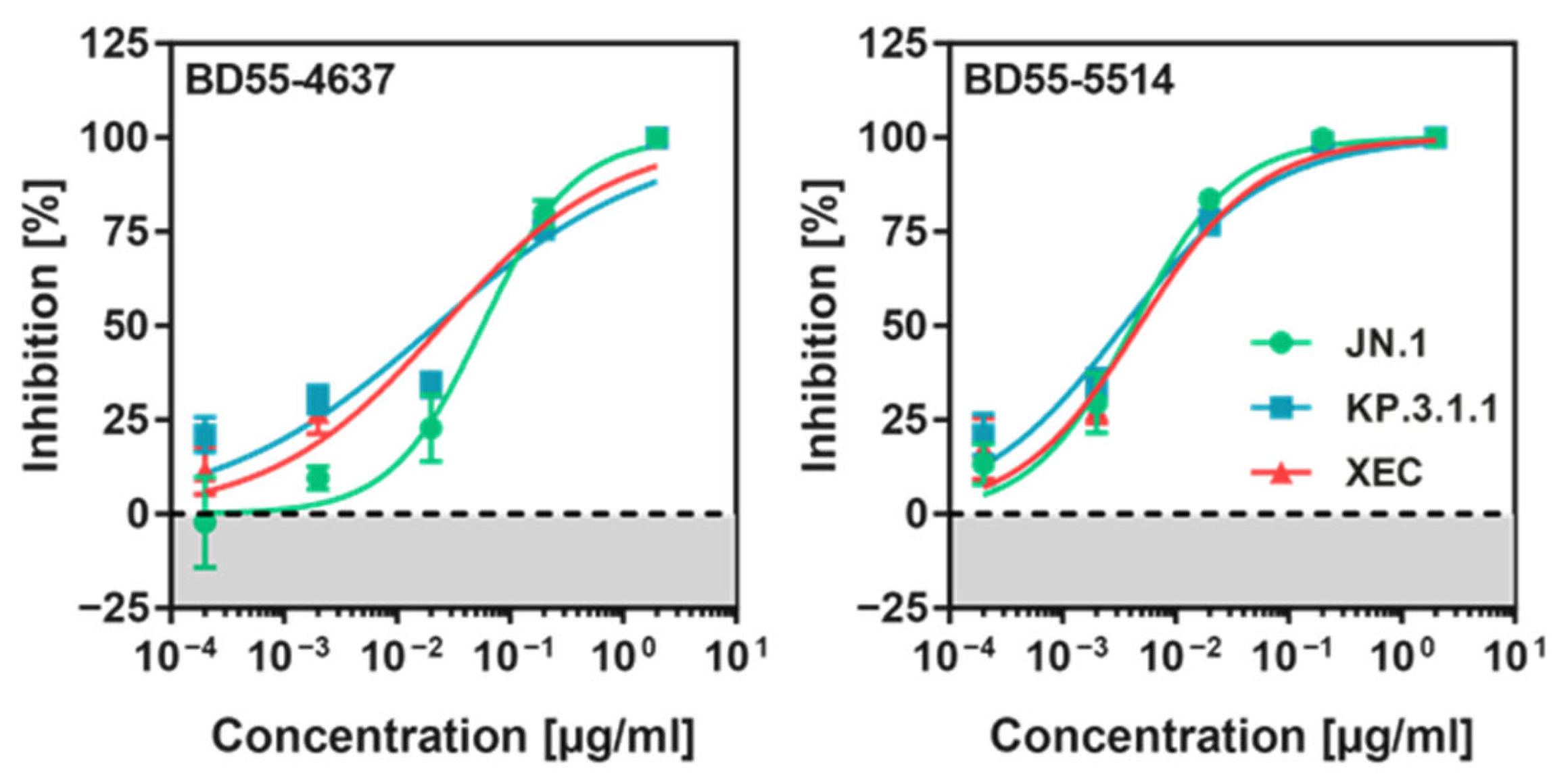
3.5. KP.3.1.1pp and XECpp Are Neutralized by Pre-Clinical Monoclonal Antibodies
4. Discussion
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; COVID-19 Genomics UK Consortium; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Zabidi, N.Z.; Liew, H.L.; Farouk, I.A.; Puniyamurti, A.; Yip, A.J.W.; Wijesinghe, V.N.; Low, Z.Y.; Tang, J.W.; Chow, V.T.K.; Lal, S.K. Evolution of SARS-CoV-2 Variants: Implications on Immune Escape, Vaccination, Therapeutic and Diagnostic Strategies. Viruses 2023, 15, 944. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zheng, X.; Zhou, B.; Li, J.; Chen, M.; Deng, R.; Wong, G.; Lavillette, D.; Meng, G. SARS-CoV-2 spike engagement of ACE2 primes S2′ site cleavage and fusion initiation. Proc. Natl. Acad. Sci. USA 2022, 119, e2111199119. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. e278. [Google Scholar] [CrossRef]
- Wang, Q.; Ye, S.; Zhou, Z.; Li, J.; Lv, J.; Hu, B.; Yuan, S.; Qiu, Y.; Ge, X. Key mutations on spike protein altering ACE2 receptor utilization and potentially expanding host range of emerging SARS-CoV-2 variants. J. Med Virol. 2023, 95, e28116. [Google Scholar] [CrossRef]
- Yajima, H.; Nomai, T.; Okumura, K.; Maenaka, K.; Genotype to Phenotype Japan (G2P-Japan) Consortium; Ito, J.; Hashiguchi, T.; Sato, K. Molecular and structural insights into SARS-CoV-2 evolution: From BA.2 to XBB subvariants. mBio 2024, 15, e0322023. [Google Scholar] [CrossRef]
- Ramesh, S.; Govindarajulu, M.; Parise, R.S.; Neel, L.; Shankar, T.; Patel, S.; Lowery, P.; Smith, F.; Dhanasekaran, M.; Moore, T. Emerging SARS-CoV-2 Variants: A Review of Its Mutations, Its Implications and Vaccine Efficacy. Vaccines 2021, 9, 1195. [Google Scholar] [CrossRef]
- Li, W.; Xu, Z.; Niu, T.; Xie, Y.; Zhao, Z.; Li, D.; He, Q.; Sun, W.; Shi, K.; Guo, W.; et al. Key mechanistic features of the trade-off between antibody escape and host cell binding in the SARS-CoV-2 Omicron variant spike proteins. EMBO J. 2024, 43, 1484–1498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kempf, A.; Nehlmeier, I.; Cossmann, A.; Richter, A.; Bdeir, N.; Graichen, L.; Moldenhauer, A.-S.; Dopfer-Jablonka, A.; Stankov, M.V.; et al. SARS-CoV-2 BA.2.86 enters lung cells and evades neutralizing antibodies with high efficiency. Cell 2024, 187, 596–608.e17. [Google Scholar] [PubMed]
- Hong, B.; Li, M.; Fan, H. SARS-CoV-2 Omicron subvariants from BA.2 to BA.2.86 and JN.1: Strong lung infection ability and evolving immune escape capacity. Medcomm 2024, 5, e578. [Google Scholar] [CrossRef]
- Tamura, T.; Mizuma, K.; Nasser, H.; Deguchi, S.; Padilla-Blanco, M.; Oda, Y.; Uriu, K.; Tolentino, J.E.; Tsujino, S.; Suzuki, R.; et al. Virological characteristics of the SARS-CoV-2 BA.2.86 variant. Cell Host Microbe 2024, 32, 170–180.e12. [Google Scholar] [CrossRef]
- Qu, P.; Xu, K.; Faraone, J.N.; Goodarzi, N.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Horowitz, J.C.; Mallampalli, R.K.; Saif, L.J.; et al. Immune evasion, infectivity, and fusogenicity of SARS-CoV-2 BA.2.86 and FLip variants. Cell 2024, 187, 585–595.e6. [Google Scholar]
- Hu, Y.; Zou, J.; Kurhade, C.; Deng, X.; Chang, H.C.; Kim, D.K.; Shi, P.-Y.; Ren, P.; Xie, X. Less neutralization evasion of SARS-CoV-2 BA.2.86 than XBB sublineages and CH.1.1. Emerg. Microbes Infect. 2023, 12, 2271089. [Google Scholar] [CrossRef]
- Yang, S.; Yu, Y.; Xu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Wang, P.; Wang, J.; Liu, J.; Yu, L.; et al. Fast evolution of SARS-CoV-2 BA.2.86 to JN.1 under heavy immune pressure. Lancet Infect. Dis. 2024, 24, e70–e72. [Google Scholar]
- Kaku, Y.; Okumura, K.; Padilla-Blanco, M.; Kosugi, Y.; Uriu, K.; Hinay, A.A.; Chen, L.; Plianchaisuk, A.; Kobiyama, K.; Ishii, K.J.; et al. Virological characteristics of the SARS-CoV-2 JN.1 variant. Lancet Infect. Dis. 2024, 24, e82. [Google Scholar] [CrossRef]
- Li, P.; Liu, Y.; Faraone, J.N.; Hsu, C.C.; Chamblee, M.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Horowitz, J.C.; Mallampalli, R.K.; et al. Distinct patterns of SARS-CoV-2 BA.2.87.1 and JN.1 variants in immune evasion, antigenicity, and cell-cell fusion. mBio 2024, 15, e0075124. [Google Scholar] [CrossRef]
- Zhang, L.; Dopfer-Jablonka, A.; Cossmann, A.; Stankov, M.V.; Graichen, L.; Moldenhauer, A.-S.; Fichter, C.; Aggarwal, A.; Turville, S.G.; Behrens, G.M.; et al. Rapid spread of the SARS-CoV-2 JN.1 lineage is associated with increased neutralization evasion. iScience 2024, 27, 109904. [Google Scholar]
- Murrell, B. SARS-CoV-2 Lineage Competition. Available online: https://github.com/MurrellGroup/lineages (accessed on 30 November 2024).
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of globally shared SARS-CoV-2 data to identify and characterize new variants. Bioinformatics 2021, 38, 1735–1737. [Google Scholar]
- Li, P.; Faraone, J.N.; Hsu, C.C.; Chamblee, M.; Liu, Y.; Zheng, Y.-M.; Xu, Y.; Carlin, C.; Horowitz, J.C.; Mallampalli, R.K.; et al. Neutralization and Stability of JN.1-derived LB.1, KP.2.3, KP.3 and KP.3.1.1 Subvariants. bioRxiv 2024. bioRxiv:2024.09.04.611219. [Google Scholar] [CrossRef]
- Li, P.; Faraone, J.N.; Hsu, C.C.; Chamblee, M.; Liu, Y.; Zheng, Y.-M.; Xu, Y.; Carlin, C.; Horowitz, J.C.; Mallampalli, R.K. Immune Evasion, Cell-Cell Fusion, and Spike Stability of the SARS-CoV-2 XEC Variant: Role of Glycosylation Mutations at the N-terminal Domain. bioRxiv 2024. bioRxiv:2024.11.12.623078. [Google Scholar] [CrossRef]
- Zhang, L.; Dopfer-Jablonka, A.; Nehlmeier, I.; Kempf, A.; Graichen, L.; Hampel, N.C.; Cossmann, A.; Stankov, M.V.; Ramos, G.M.; Schulz, S.R.; et al. Virological Traits of the SARS-CoV-2 BA.2.87.1 Lineage. Vaccines 2024, 12, 487. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, H.-H.; Krüger, N.; Hörnich, B.; Graichen, L.; Hahn, A.S.; Schulz, S.R.; Jäck, H.-M.; Stankov, M.V.; Behrens, G.M.N.; et al. ACE2-independent sarbecovirus cell entry can be supported by TMPRSS2-related enzymes and can reduce sensitivity to antibody-mediated neutralization. PLOS Pathog. 2024, 20, e1012653. [Google Scholar] [CrossRef]
- Chen, N.; Decker, K.E.; Schulz, S.R.; Kempf, A.; Nehlmeier, I.; Moldenhauer, A.-S.; Dopfer-Jablonka, A.; Behrens, G.M.N.; Stankov, M.V.; Manthey, L.; et al. Comparative Analysis of Host Cell Entry Efficiency and Neutralization Sensitivity of Emerging SARS-CoV-2 Lineages KP.2, KP.2.3, KP.3, and LB.1. Vaccines 2024, 12, 1236. [Google Scholar] [CrossRef]
- Arora, P.; Happle, C.; Kempf, A.; Nehlmeier, I.; Stankov, M.V.; Dopfer-Jablonka, A.; Behrens, G.M.N.; Pöhlmann, S.; Hoffmann, M. Impact of JN.1 booster vaccination on neutralisation of SARS-CoV-2 variants KP.3.1.1 and XEC. Lancet Infect. Dis. 2024, 24, e732–e733. [Google Scholar]
- Rentsch, M.B.; Zimmer, G. A Vesicular Stomatitis Virus Replicon-Based Bioassay for the Rapid and Sensitive Determination of Multi-Species Type I Interferon. PLoS ONE 2011, 6, e25858. [Google Scholar]
- Schmidt, F.; Weisblum, Y.; Muecksch, F.; Hoffmann, H.-H.; Michailidis, E.; Lorenzi, J.C.; Mendoza, P.; Rutkowska, M.; Bednarski, E.; Gaebler, C.; et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 2020, 217, e20201181. [Google Scholar]
- Riepler, L.; Rössler, A.; Falch, A.; Volland, A.; Borena, W.; von Laer, D.; Kimpel, J. Comparison of Four SARS-CoV-2 Neutralization Assays. Vaccines 2020, 9, 13. [Google Scholar] [CrossRef]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeño, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Han, P.; Huang, B.; Xie, Y.; Li, W.; Zhang, D.; Han, P.; Xu, Z.; Bai, B.; Zhou, J.; et al. Broader-species receptor binding and structural bases of Omicron SARS-CoV-2 to both mouse and palm-civet ACE2s. Cell Discov. 2022, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Li, L.; Luo, C.; Xu, Z.; Huang, B.; Ma, S.; Liu, K.; Yu, G.; Gao, G.F. Structural basis of increased binding affinities of spikes from SARS-CoV-2 Omicron variants to rabbit and hare ACE2s reveals the expanding host tendency. mBio 2024, 15, e0298823. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; He, J.; Han, P.; Bai, B.; Li, D.; Cao, J.; Tian, M.; Hu, Y.; Zheng, A.; Niu, S.; et al. Molecular Basis of Mink ACE2 Binding to SARS-CoV-2 and Its Mink-Derived Variants. J. Virol. 2022, 96, e0081422. [Google Scholar] [CrossRef]
- Yao, W.; Li, Y.; Ma, D.; Hou, X.; Wang, H.; Tang, X.; Cheng, D.; Zhang, H.; Du, C.; Pan, H.; et al. Evolution of SARS-CoV-2 Spikes shapes their binding affinities to animal ACE2 orthologs. Microbiol. Spectr. 2023, 11, e0267623. [Google Scholar] [CrossRef]
- Bussani, R.; Schneider, E.; Zentilin, L.; Collesi, C.; Ali, H.; Braga, L.; Volpe, M.C.; Colliva, A.; Zanconati, F.; Berlot, G.; et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBioMedicine 2020, 61, 103104. [Google Scholar] [CrossRef]
- Rajah, M.M.; Bernier, A.; Buchrieser, J.; Schwartz, O. The Mechanism and Consequences of SARS-CoV-2 Spike-Mediated Fusion and Syncytia Formation. J. Mol. Biol. 2021, 434, 167280. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Zhang, Z.; Yisimayi, A.; Hao, X.; Bao, L.; Yuan, F.; Yu, Y.; Du, S.; Wang, J.; et al. Rational identification of potent and broad sarbecovirus-neutralizing antibody cocktails from SARS convalescents. Cell Rep. 2022, 41, 111845. [Google Scholar] [CrossRef]
- Liu, J.; Yu, Y.; Jian, F.; Yang, S.; Song, W.; Wang, P.; Yu, L.; Shao, F.; Cao, Y. Enhanced immune evasion of SARS-CoV-2 variants KP.3.1.1 and XEC through N-terminal domain mutations. Lancet Infect. Dis. 2024, 25, e6–e7. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, X.; Shi, J.; Wang, Y.; Liu, H.; Hu, Y.-F.; Hu, B.; Shuai, H.; Yuen, T.T.-T.; Chai, Y.; et al. Lineage-specific pathogenicity, immune evasion, and virological features of SARS-CoV-2 BA.2.86/JN.1 and EG.5.1/HK.3. Nat. Commun. 2024, 15, 8728. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arora, P.; Kempf, A.; Nehlmeier, I.; Schulz, S.R.; Jäck, H.-M.; Hoffmann, M.; Pöhlmann, S. Entry Efficiency, Protease Dependence, and Antibody-Mediated Neutralization of SARS-CoV-2 Sublineages KP.3.1.1 and XEC. Vaccines 2025, 13, 385. https://doi.org/10.3390/vaccines13040385
Arora P, Kempf A, Nehlmeier I, Schulz SR, Jäck H-M, Hoffmann M, Pöhlmann S. Entry Efficiency, Protease Dependence, and Antibody-Mediated Neutralization of SARS-CoV-2 Sublineages KP.3.1.1 and XEC. Vaccines. 2025; 13(4):385. https://doi.org/10.3390/vaccines13040385
Chicago/Turabian StyleArora, Prerna, Amy Kempf, Inga Nehlmeier, Sebastian R. Schulz, Hans-Martin Jäck, Markus Hoffmann, and Stefan Pöhlmann. 2025. "Entry Efficiency, Protease Dependence, and Antibody-Mediated Neutralization of SARS-CoV-2 Sublineages KP.3.1.1 and XEC" Vaccines 13, no. 4: 385. https://doi.org/10.3390/vaccines13040385
APA StyleArora, P., Kempf, A., Nehlmeier, I., Schulz, S. R., Jäck, H.-M., Hoffmann, M., & Pöhlmann, S. (2025). Entry Efficiency, Protease Dependence, and Antibody-Mediated Neutralization of SARS-CoV-2 Sublineages KP.3.1.1 and XEC. Vaccines, 13(4), 385. https://doi.org/10.3390/vaccines13040385