
Multi-Epitope DC Vaccines with Melanoma Antigens for Immunotherapy of Melanoma
 , , ,
, , ,  , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. DC-Vaccine Generation
2.3. DEC-205 Binding Assays
2.4. In Vivo Proliferation Assay
2.5. In Vivo DC-Based Vaccination
2.6. Single Cell Suspension
2.7. Flow Cytometry
2.8. Transplantable B16.OVA Melanoma Model
2.9. In Vitro Restimulation
2.10. Statistical Analysis
3. Results
3.1. Cloning of gp100 and trp2 CD8+ T Cell Epitopes into DEC-205 Monoclonal Antibody
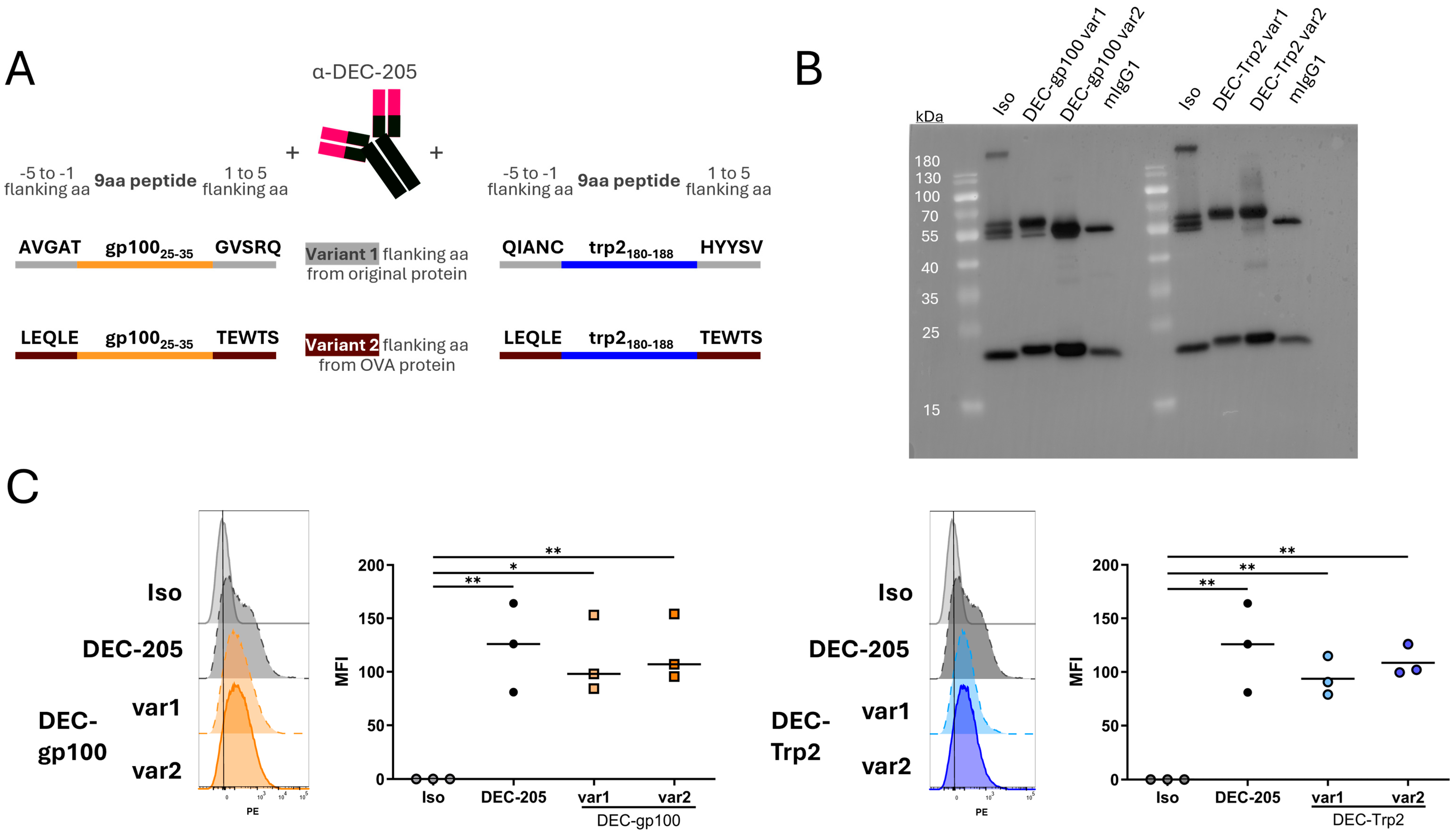
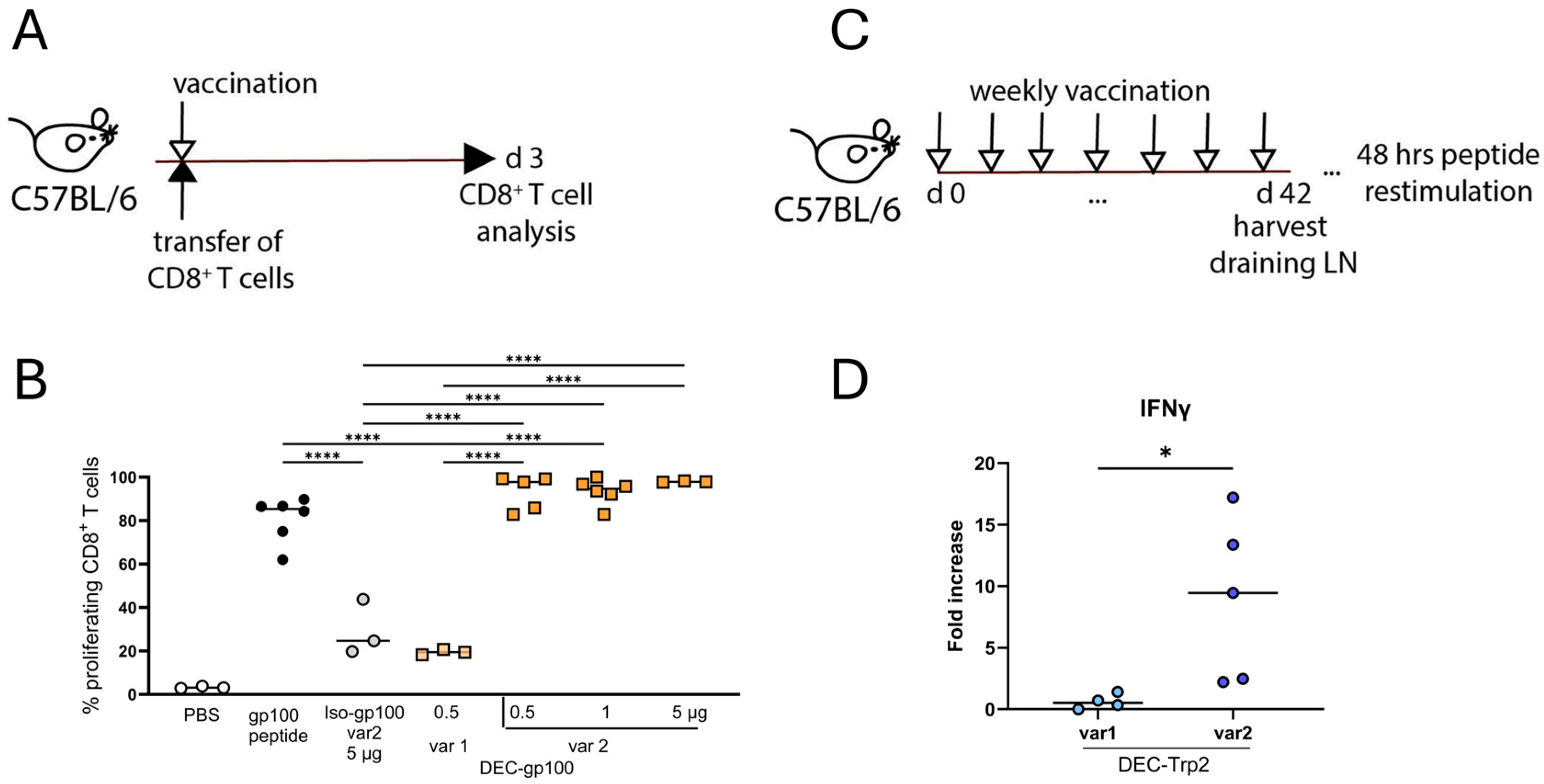
3.2. Vaccine Variants with OVA257–264 SIINFEKL Peptide Flanking Sites Are Cross-Presented Better to CD8+ T Cells
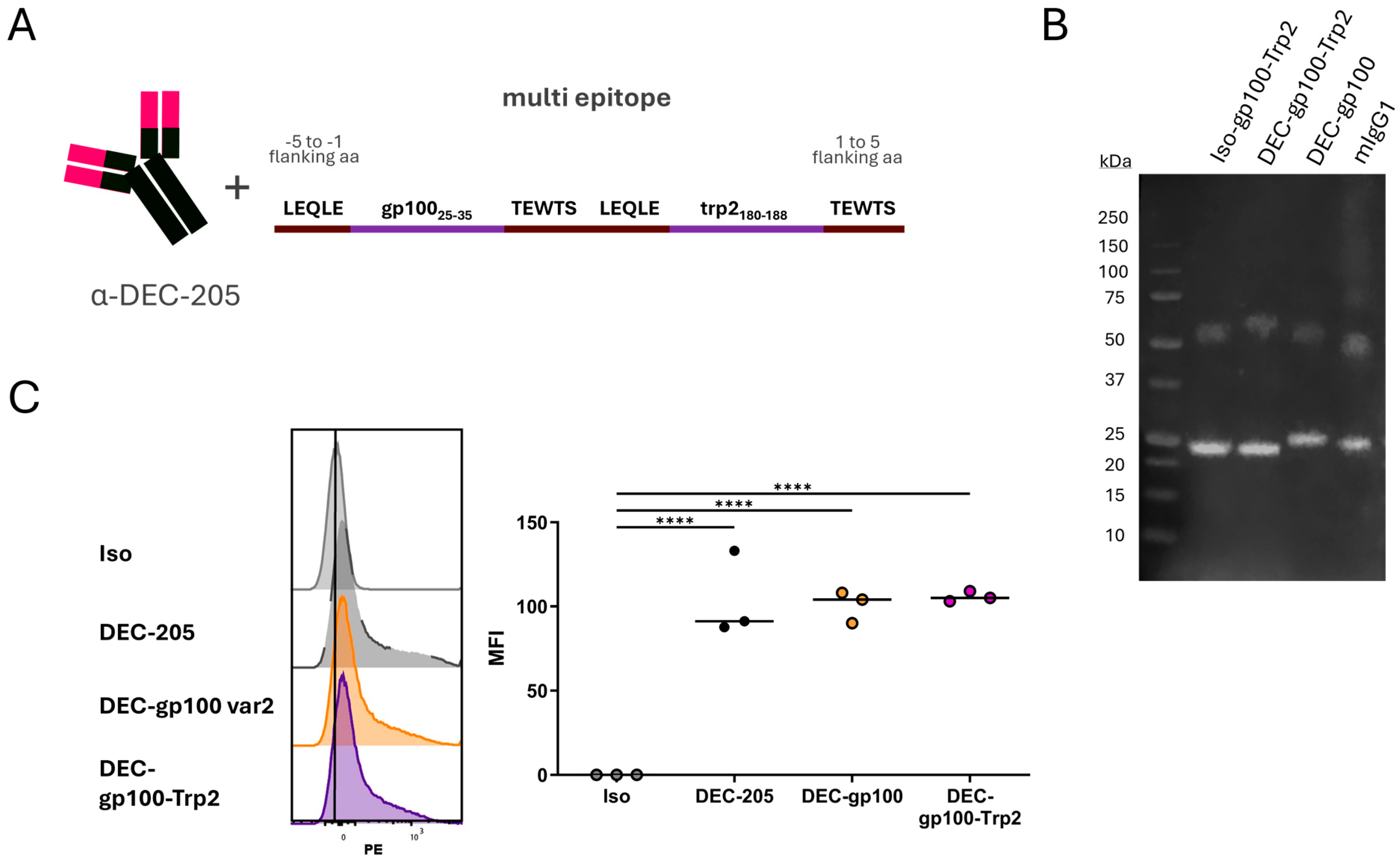
3.3. Cloning of DEC-205 Multi-Epitope Vaccine with gp100 and trp2 Peptides
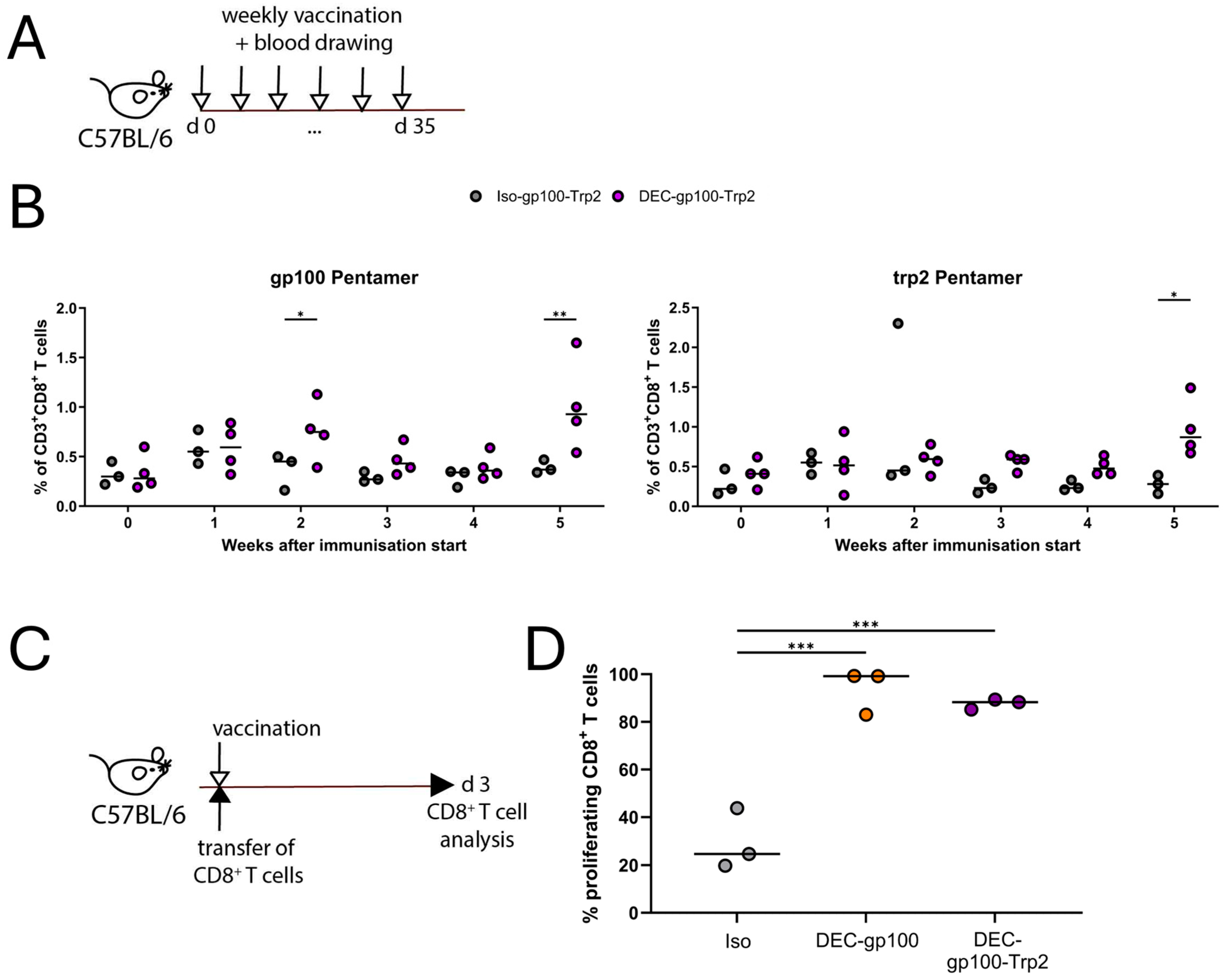
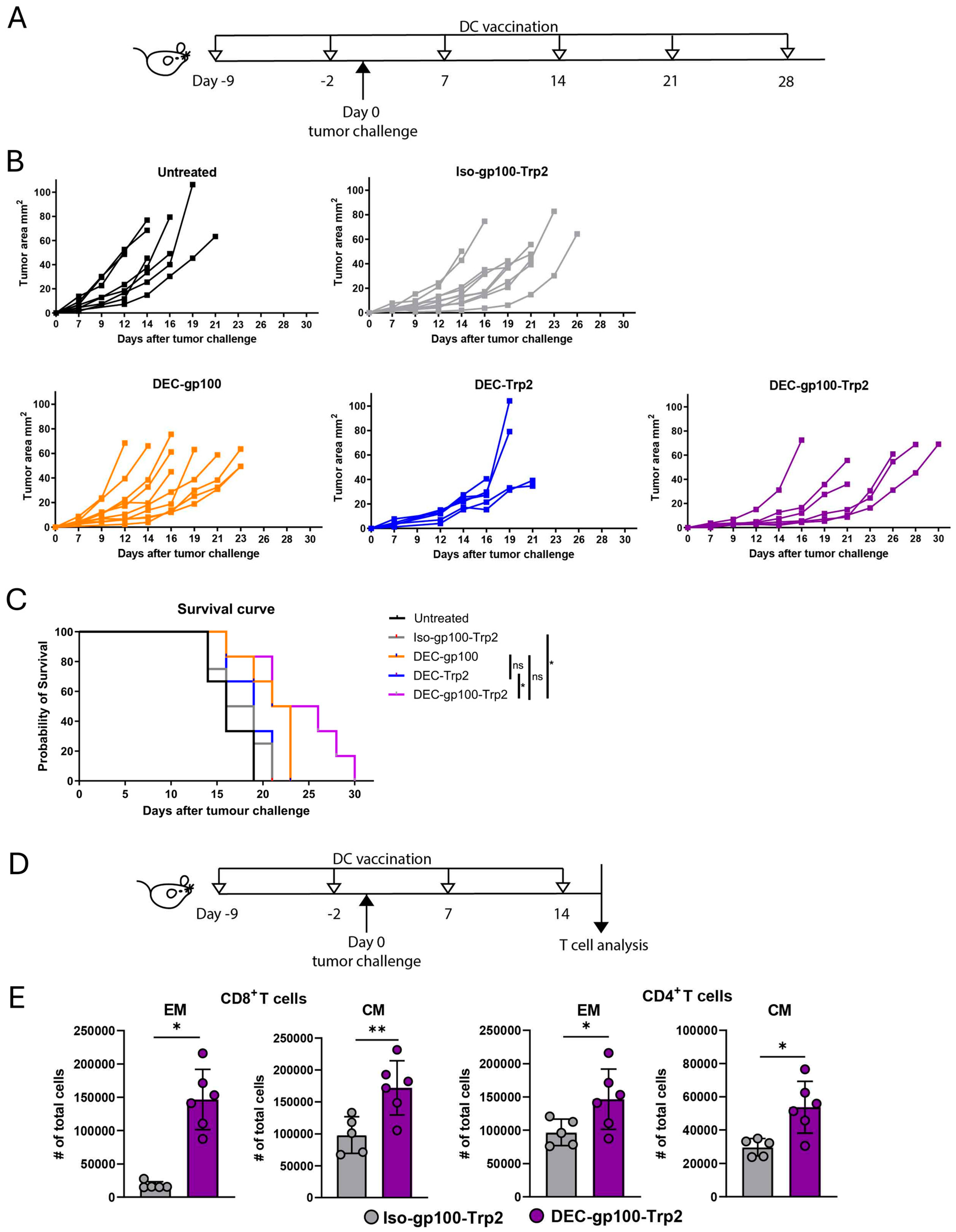
3.4. DEC-gp100-Trp2 Slows Down Against B16-OVA Melanoma Growth by Inducing Memory T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BCA | Bicinchoninic acid |
| BMDC | Bone marrow-derived dendritic cells |
| CFSE | Carboxyfluorescein succinimidyl ester |
| CM | Central memory |
| DC | Dendritic cells |
| EDTA | Ethylenediaminetetraacetic acid |
| ELISA | Enzyme-linked immunosorbent assay |
| EM | Effector memory |
| FCS | Fetal calf serum |
| FPLC | Fast protein liquid chromatography |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| IFNγ | Interferon gamma |
| LAL | Limulus amebocyte lysate |
| LPS | Lipopolysaccharide |
| MHC-I | Major histocompatibility complex I |
| MHC-II | Major histocompatibility complex II |
| NY-ESO1 | New York esophageal squamous cell carcinoma-1 |
| OVA | Ovalbumin |
| TCR | T cell receptor |
| TLR | Toll-like receptor |
| TNP | Trinitrophenyl |
References
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of Antigen-Specific Immunity with a Vaccine Targeting NY-ESO-1 to the Dendritic Cell Receptor DEC-205. Sci.Transl. Med. 2014, 6, 232ra251. [Google Scholar]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Erdmann, M.; Haendle, I.; Voland, S.; Berger, T.; Schultz, E.; Strasser, E.; Dankerl, P.; Janka, R.; Schliep, S.; et al. Twelve-year survival and immune correlates in dendritic cell-vaccinated melanoma patients. JCI Insight 2017, 2, e91438. [Google Scholar] [CrossRef]
- Laureano, R.S.; Sprooten, J.; Vanmeerbeerk, I.; Borras, D.M.; Govaerts, J.; Naulaerts, S.; Berneman, Z.N.; Beuselinck, B.; Bol, K.F.; Borst, J.; et al. Trial watch: Dendritic cell (DC)-based immunotherapy for cancer. Oncoimmunology 2022, 11, 2096363. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Tacken, P.J.; de Vries, I.J.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.H.; Heger, L.; Heidkamp, G.F.; Baranska, A.; Luhr, J.J.; Hoffmann, A.; Dudziak, D. Direct Delivery of Antigens to Dendritic Cells via Antibodies Specific for Endocytic Receptors as a Promising Strategy for Future Therapies. Vaccines 2016, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Kastenmuller, W.; Kastenmuller, K.; Kurts, C.; Seder, R.A. Dendritic cell-targeted vaccines--hope or hype? Nat. Rev. Immunol. 2014, 14, 705–711. [Google Scholar] [CrossRef]
- Stoitzner, P.; Romani, N.; Rademacher, C.; Probst, H.C.; Mahnke, K. Antigen targeting to dendritic cells: Still a place in future immunotherapy? Eur. J. Immunol. 2022, 52, 1909–1924. [Google Scholar] [CrossRef]
- Mahnke, K.; Guo, M.; Lee, S.; Sepulveda, H.; Swain, S.L.; Nussenzweig, M.; Steinman, R.M. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 2000, 151, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef]
- Dudziak, D.; Kamphorst, A.O.; Heidkamp, G.F.; Buchholz, V.R.; Trumpfheller, C.; Yamazaki, S.; Cheong, C.; Liu, K.; Lee, H.W.; Park, C.G.; et al. Differential antigen processing by dendritic cell subsets in vivo. Science 2007, 315, 107–111. [Google Scholar] [CrossRef]
- Idoyaga, J.; Fiorese, C.; Zbytnuik, L.; Lubkin, A.; Miller, J.; Malissen, B.; Mucida, D.; Merad, M.; Steinman, R.M. Specialized role of migratory dendritic cells in peripheral tolerance induction. J. Clin. Investig. 2013, 123, 844–854. [Google Scholar] [CrossRef]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef]
- Neubert, K.; Lehmann, C.H.; Heger, L.; Baranska, A.; Staedtler, A.M.; Buchholz, V.R.; Yamazaki, S.; Heidkamp, G.F.; Eissing, N.; Zebroski, H.; et al. Antigen delivery to CD11c+CD8− dendritic cells induces protective immune responses against experimental melanoma in mice in vivo. J. Immunol. 2014, 192, 5830–5838. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, K.; Qian, Y.; Fondel, S.; Brueck, J.; Becker, C.; Enk, A.H. Targeting of antigens to activated dendritic cells in vivo cures metastatic melanoma in mice. Cancer Res. 2005, 65, 7007–7012. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, A.; Oks, M.; Nchinda, G.; Yamazaki, S.; Steinman, R.M. Dendritic cell targeting of survivin protein in a xenogeneic form elicits strong CD4+ T cell immunity to mouse survivin. J. Immunol. 2006, 177, 8410–8421. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zaidi, N.; He, L.Z.; Zhang, L.; Kuroiwa, J.M.; Keler, T.; Steinman, R.M. Targeting of the non-mutated tumor antigen HER2/neu to mature dendritic cells induces an integrated immune response that protects against breast cancer in mice. Breast Cancer Res. 2012, 14, R39. [Google Scholar] [CrossRef]
- Schreurs, M.W.; Eggert, A.A.; de Boer, A.J.; Vissers, J.L.; van Hall, T.; Offringa, R.; Figdor, C.G.; Adema, G.J. Dendritic cells break tolerance and induce protective immunity against a melanocyte differentiation antigen in an autologous melanoma model. Cancer Res. 2000, 60, 6995–7001. [Google Scholar]
- Overwijk, W.W.; Theoret, M.R.; Finkelstein, S.E.; Surman, D.R.; de Jong, L.A.; Vyth-Dreese, F.A.; Dellemijn, T.A.; Antony, P.A.; Spiess, P.J.; Palmer, D.C.; et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 2003, 198, 569–580. [Google Scholar] [CrossRef]
- Stoitzner, P.; Tripp, C.H.; Eberhart, A.; Price, K.M.; Jung, J.Y.; Bursch, L.; Ronchese, F.; Romani, N. Langerhans cells cross-present antigen derived from skin. Proc. Natl. Acad. Sci. USA 2006, 103, 7783–7788. [Google Scholar] [CrossRef]
- Tripp, C.H.; Ebner, S.; Ratzinger, G.; Romani, N.; Stoitzner, P. Conditioning of the injection site with CpG enhances the migration of adoptively transferred dendritic cells and endogenous CD8+ T-cell responses. J. Immunother. 2010, 33, 115–125. [Google Scholar] [CrossRef]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef]
- Idoyaga, J.; Cheong, C.; Suda, K.; Suda, N.; Kim, J.Y.; Lee, H.; Park, C.G.; Steinman, R.M. Cutting edge: Langerin/CD207 receptor on dendritic cells mediates efficient antigen presentation on MHC I and II products in vivo. J. Immunol. 2008, 180, 3647–3650. [Google Scholar] [CrossRef]
- Overwijk, W.W.; Tsung, A.; Irvine, K.R.; Parkhurst, M.R.; Goletz, T.J.; Tsung, K.; Carroll, M.W.; Liu, C.; Moss, B.; Rosenberg, S.A.; et al. gp100/pmel 17 is a murine tumor rejection antigen: Induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998, 188, 277–286. [Google Scholar] [PubMed]
- Kreutz, M.; Giquel, B.; Hu, Q.; Abuknesha, R.; Uematsu, S.; Akira, S.; Nestle, F.O.; Diebold, S.S. Antibody-antigen-adjuvant conjugates enable co-delivery of antigen and adjuvant to dendritic cells in cis but only have partial targeting specificity. PLoS ONE 2012, 7, e40208. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; de Vries, I.J.; Figdor, C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef]
- Idoyaga, J.; Lubkin, A.; Fiorese, C.; Lahoud, M.H.; Caminschi, I.; Huang, Y.; Rodriguez, A.; Clausen, B.E.; Park, C.G.; Trumpfheller, C.; et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc. Natl. Acad. Sci. USA 2011, 108, 2384–2389. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, M.; Pang, L.; Wang, S.; Kong, Y.; Zhu, X.; Zhou, X.; Wang, X.; Chen, C.; Ning, H.; et al. Identification of a novel DEC-205 binding peptide to develop dendritic cell-targeting nanovaccine for cancer immunotherapy. J. Control. Release 2024, 373, 568–582. [Google Scholar] [CrossRef]
- Terhorst, D.; Fossum, E.; Baranska, A.; Tamoutounour, S.; Malosse, C.; Garbani, M.; Braun, R.; Lechat, E.; Crameri, R.; Bogen, B.; et al. Laser-Assisted Intradermal Delivery of Adjuvant-Free Vaccines Targeting XCR1+ Dendritic Cells Induces Potent Antitumoral Responses. J. Immunol. 2015, 194, 5895–5902. [Google Scholar] [CrossRef]
- Johnson, T.S.; Mahnke, K.; Storn, V.; Schonfeld, K.; Ring, S.; Nettelbeck, D.M.; Haisma, H.J.; Le Gall, F.; Kontermann, R.E.; Enk, A.H. Inhibition of melanoma growth by targeting of antigen to dendritic cells via an anti-DEC-205 single-chain fragment variable molecule. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 8169–8177. [Google Scholar] [CrossRef]
- Sancho, D.; Mourao-Sa, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis e Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Bellmann, L.; Strandt, H.; Zelle-Rieser, C.; Ortner, D.; Tripp, C.H.; Schmid, S.; Ruhl, J.; Cappellano, G.; Schaffenrath, S.; Prokopi, A.; et al. Targeted delivery of a vaccine protein to Langerhans cells in the human skin via the C-type lectin receptor Langerin. Eur. J. Immunol. 2021, 52, 1829–1841. [Google Scholar] [CrossRef]
- Ma, X.; Serna, A.; Xu, R.H.; Sigal, L.J. The amino acid sequences flanking an antigenic determinant can strongly affect MHC class I cross-presentation without altering direct presentation. J. Immunol. 2009, 182, 4601–4607. [Google Scholar] [CrossRef]
- Small, M.; Kraal, G. In vitro evidence for participation of DEC-205 expressed by thymic cortical epithelial cells in clearance of apoptotic thymocytes. Int. Immunol. 2003, 15, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 2018, 49, 1148–1161.E7. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Di Pilato, M.; Garris, C.; Mempel, T.R. Dendritic cells as shepherds of T cell immunity in cancer. Immunity 2023, 56, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Chen, D.S.; Powles, T.; Turley, S.J. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity 2023, 56, 2188–2205. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; De Vleeschouwer, S.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Garg, A.D. Trial watch: Dendritic cell vaccination for cancer immunotherapy. Oncoimmunology 2019, 8, e1638212. [Google Scholar] [CrossRef]
- Nava, S.; Lisini, D.; Frigerio, S.; Bersano, A. Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect. Int. J. Mol. Sci. 2021, 22, 12339. [Google Scholar] [CrossRef]
- Sarkizova, S.; Klaeger, S.; Le, P.M.; Li, L.W.; Oliveira, G.; Keshishian, H.; Hartigan, C.R.; Zhang, W.; Braun, D.A.; Ligon, K.L.; et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020, 38, 199–209. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seretis, A.; Amon, L.; Tripp, C.H.; Cappellano, G.; Hornsteiner, F.; Dieckmann, S.; Vierthaler, J.; Ortner-Tobider, D.; Kanduth, M.; Steindl, R.; et al. Multi-Epitope DC Vaccines with Melanoma Antigens for Immunotherapy of Melanoma. Vaccines 2025, 13, 346. https://doi.org/10.3390/vaccines13040346
Seretis A, Amon L, Tripp CH, Cappellano G, Hornsteiner F, Dieckmann S, Vierthaler J, Ortner-Tobider D, Kanduth M, Steindl R, et al. Multi-Epitope DC Vaccines with Melanoma Antigens for Immunotherapy of Melanoma. Vaccines. 2025; 13(4):346. https://doi.org/10.3390/vaccines13040346
Chicago/Turabian StyleSeretis, Athanasios, Lukas Amon, Christoph H. Tripp, Giuseppe Cappellano, Florian Hornsteiner, Sophie Dieckmann, Janine Vierthaler, Daniela Ortner-Tobider, Markus Kanduth, Rita Steindl, and et al. 2025. "Multi-Epitope DC Vaccines with Melanoma Antigens for Immunotherapy of Melanoma" Vaccines 13, no. 4: 346. https://doi.org/10.3390/vaccines13040346
APA StyleSeretis, A., Amon, L., Tripp, C. H., Cappellano, G., Hornsteiner, F., Dieckmann, S., Vierthaler, J., Ortner-Tobider, D., Kanduth, M., Steindl, R., Boon, L., den Haan, J. M. M., Lehmann, C. H. K., Dudziak, D., & Stoitzner, P. (2025). Multi-Epitope DC Vaccines with Melanoma Antigens for Immunotherapy of Melanoma. Vaccines, 13(4), 346. https://doi.org/10.3390/vaccines13040346