
B and T Cell Bi-Cistronic Multiepitopic Vaccine Induces Broad Immunogenicity and Provides Protection Against SARS-CoV-2

 , , , , , , , , and
, , , , , , , , and 
Abstract
1. Introduction
2. Materials and Methods
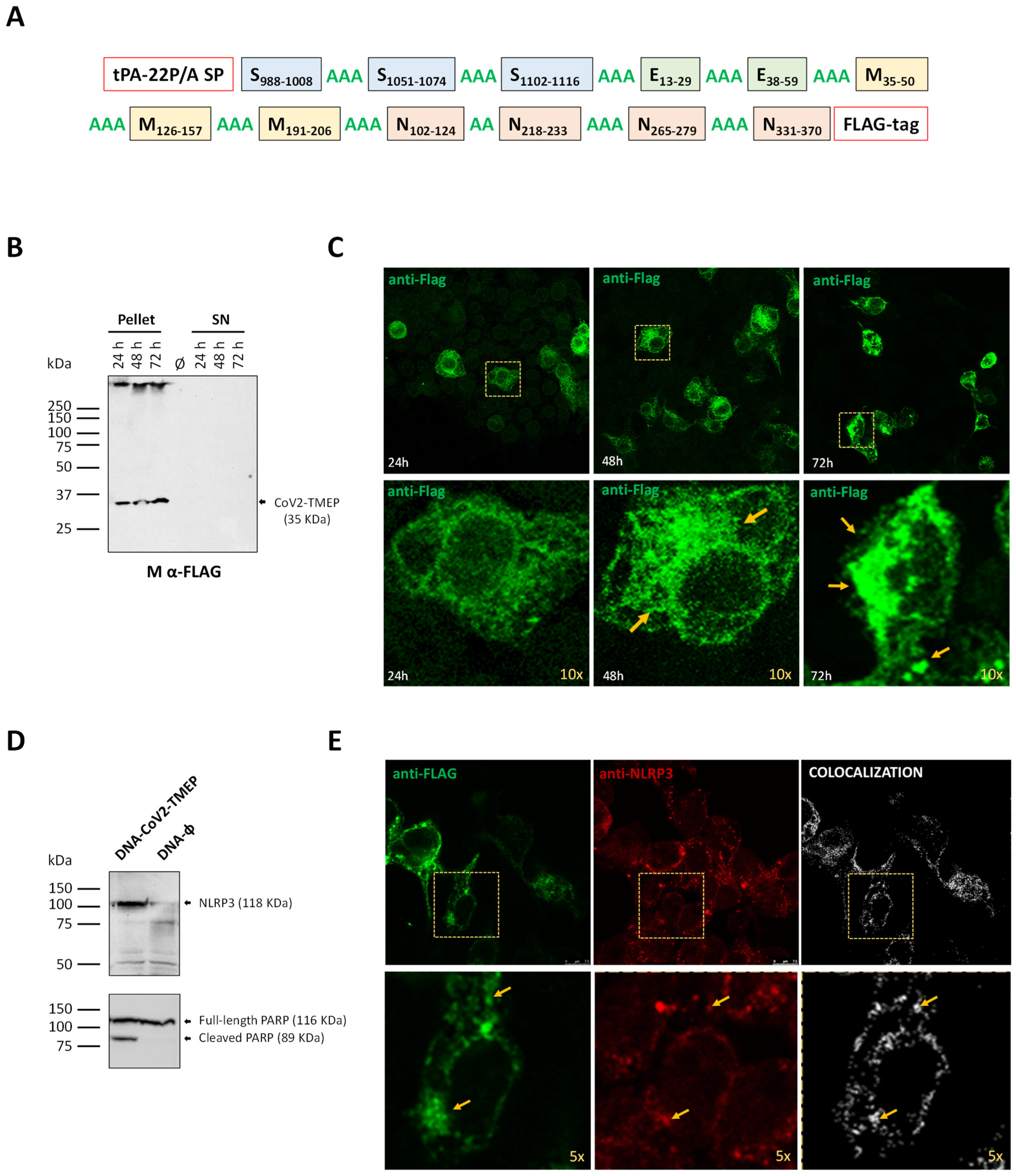
2.1. Design of Chimeric CoV2-TMEP Polyvalent Multiepitopic Immunogen Targeting T Cells
2.2. In Silico Characterization of CoV2-TMEP Synthetic Protein
2.3. Design and In Silico Characterization of the Bi-Cistronic CoV2-BMEP-P2A-TMEP Synthetic Protein
2.4. Cells
2.5. Viruses
2.6. DNA Vectors
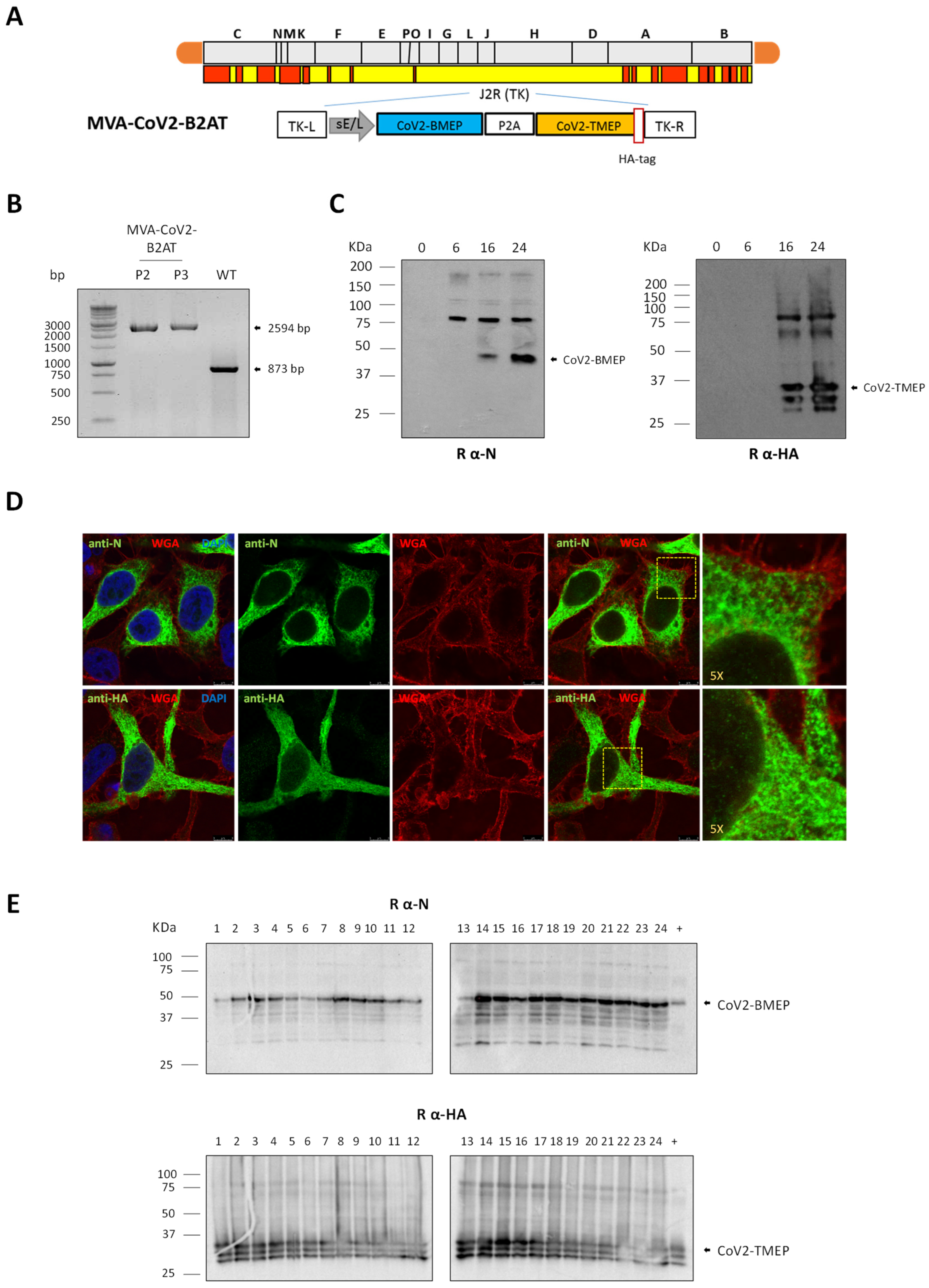
2.7. Construction and In Vitro Characterization of MVA-CoV2-B2AT Recombinant Virus
2.8. Expression Analysis of CoV2-TMEP and CoV2-BMEP-P2A-TMEP Multiepitopic Proteins by Western Blotting
2.9. Subcellular Localization of CoV2-TMEP and CoV2-BMEP-P2A-TMEP Multiepitopic Proteins by Immunofluorescence Microscopy
2.10. Peptides
2.11. Ethics Statement
2.12. Preclinical Evaluation of CoV2-TMEP and CoV2-BMEP-P2A-TMEP Proteins in Mice
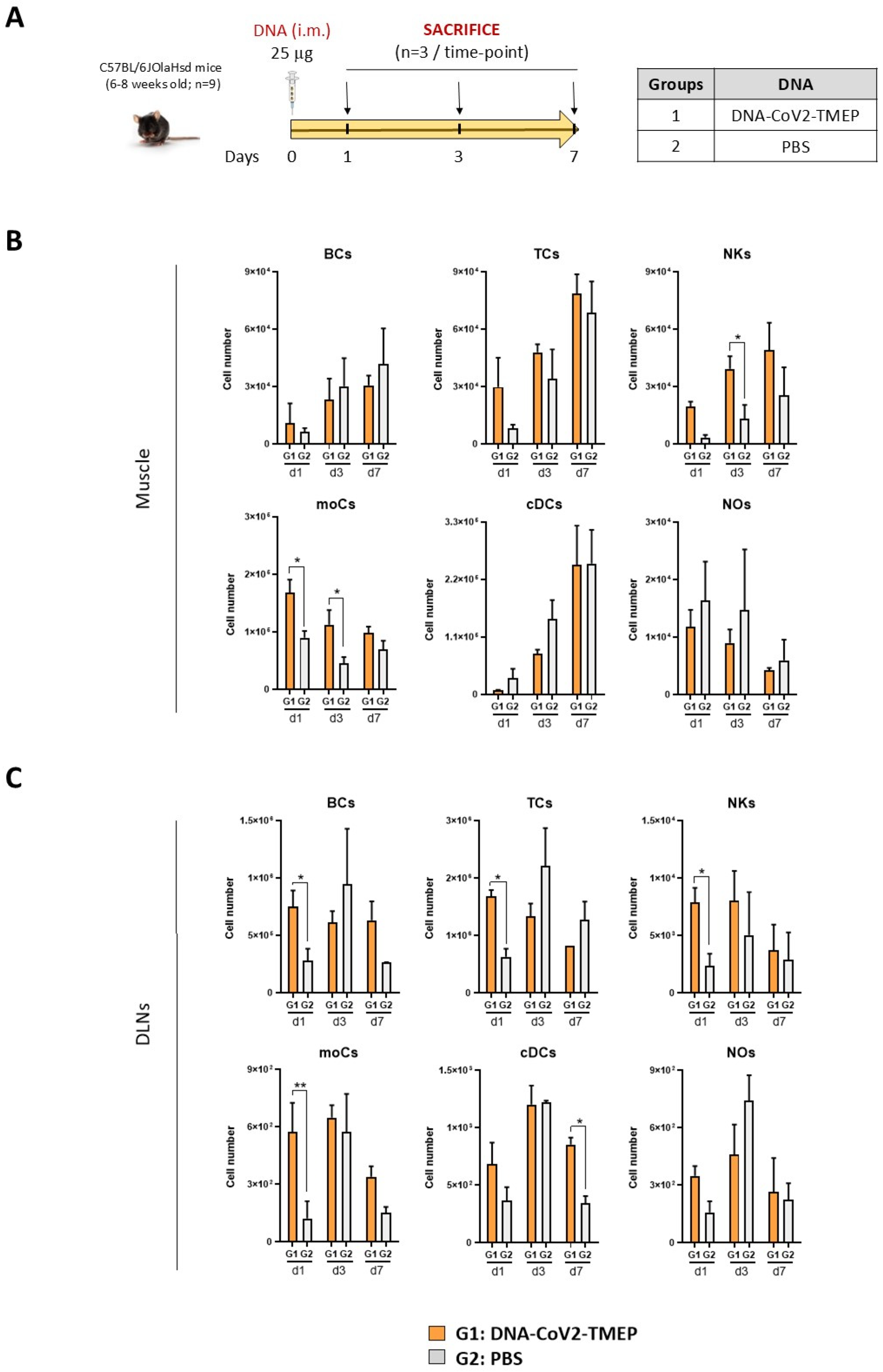
2.12.1. Analysis of the Impact of CoV2-TMEP or CoV2-BMEP-P2A-TMEP Expression on Immune Cell Recruitment by Flow Cytometry
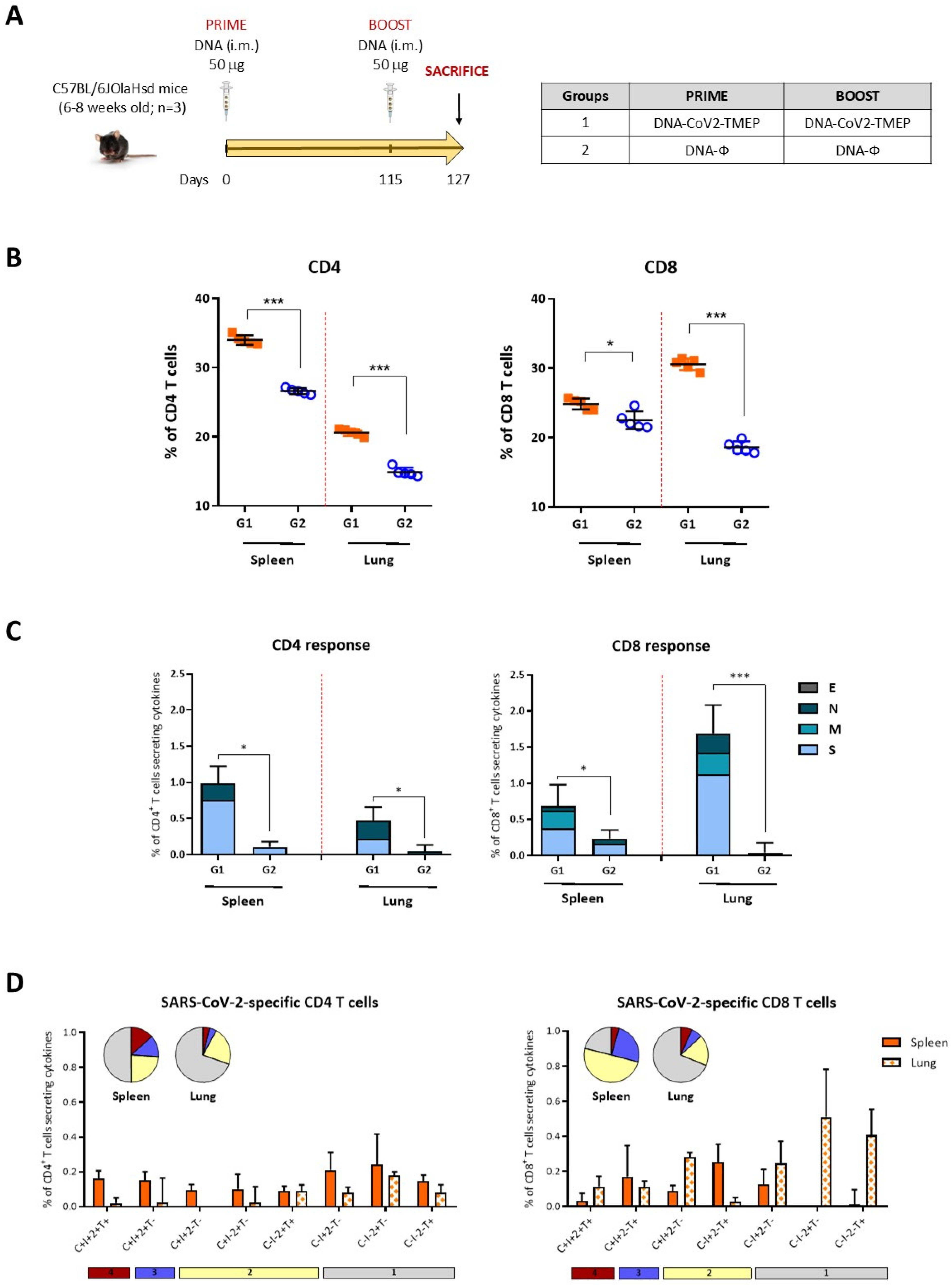
2.12.2. SARS-CoV-2-Specific Immunogenicity Elicited by CoV2-TMEP or CoV2-BMEP-P2A-TMEP Proteins
2.12.3. Efficacy of MVA-CoV2-B2AT against SARS-CoV-2 Infection in Susceptible K18-hACE2 Tg Mice
SARS-CoV-2 Neutralization Assay
Analysis of SARS-CoV-2 RNA by Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
Analysis of SARS-CoV-2 Virus Yields by Plaque Assay
2.13. Data Analysis and Statistics
3. Results
3.1. In Vitro Characterization of the Multiepitopic CoV2-TMEP Protein Expressed from a DNA Vector
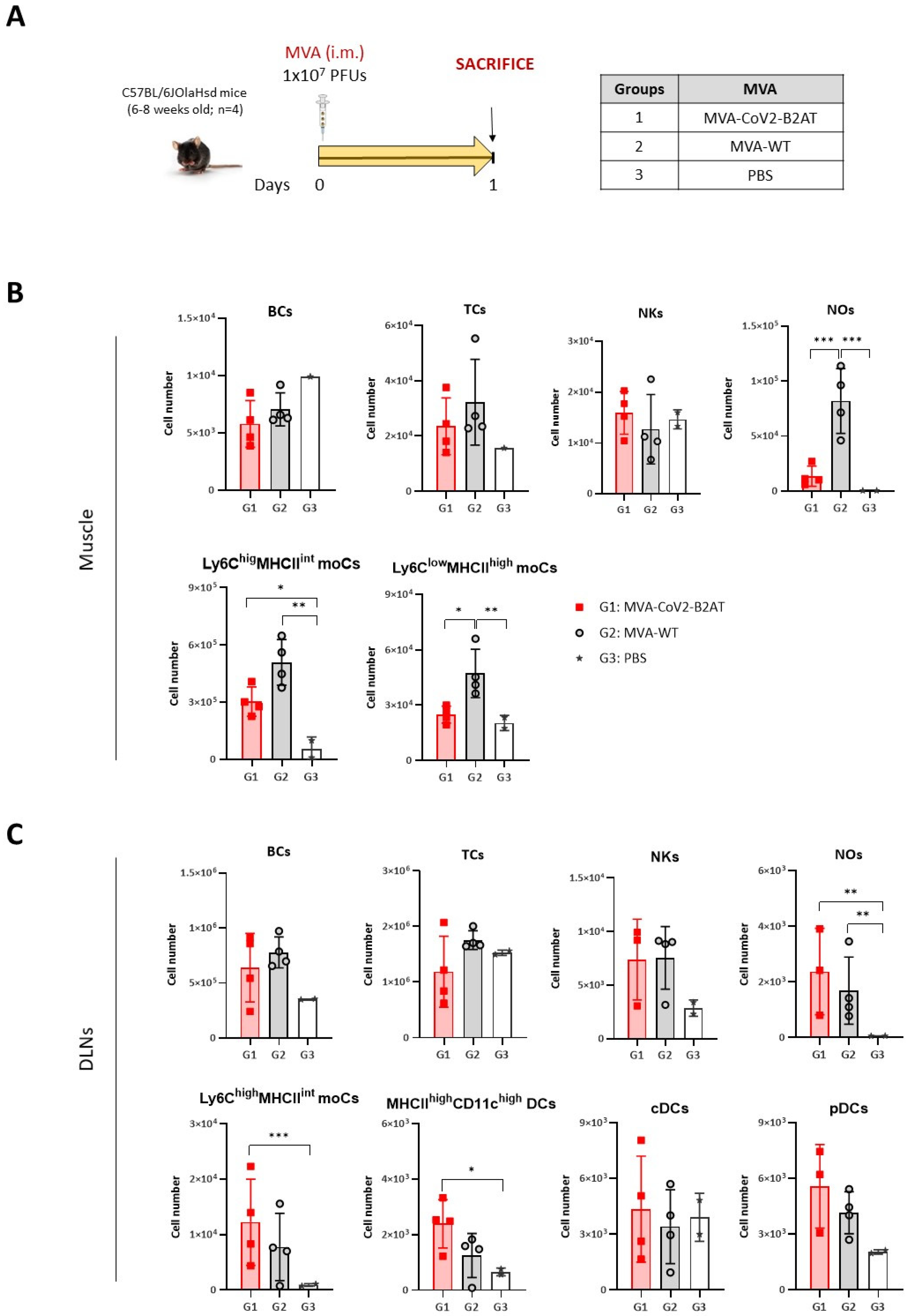
3.2. CoV2-TMEP Expression from a DNA Vector Enhances Immune Cell Recruitment in Muscle and DLNs
3.3. Homologous DNA-CoV2-TMEP Prime/Boost Vaccination Regimen Elicits Robust and Polyfunctional SARS-CoV-2-Specific T Cell Responses in C57BL/6 Mice
3.4. In Vitro Characterization of an MVA-Based Vector Expressing the Bi-Cistronic CoV2-BMEP-P2A-TMEP Multiepitopic Protein
3.5. Effect of CoV2-BMEP-P2A-TMEP Expression from an MVA Vector on Immune Cell Recruitment in Muscle and DLNs
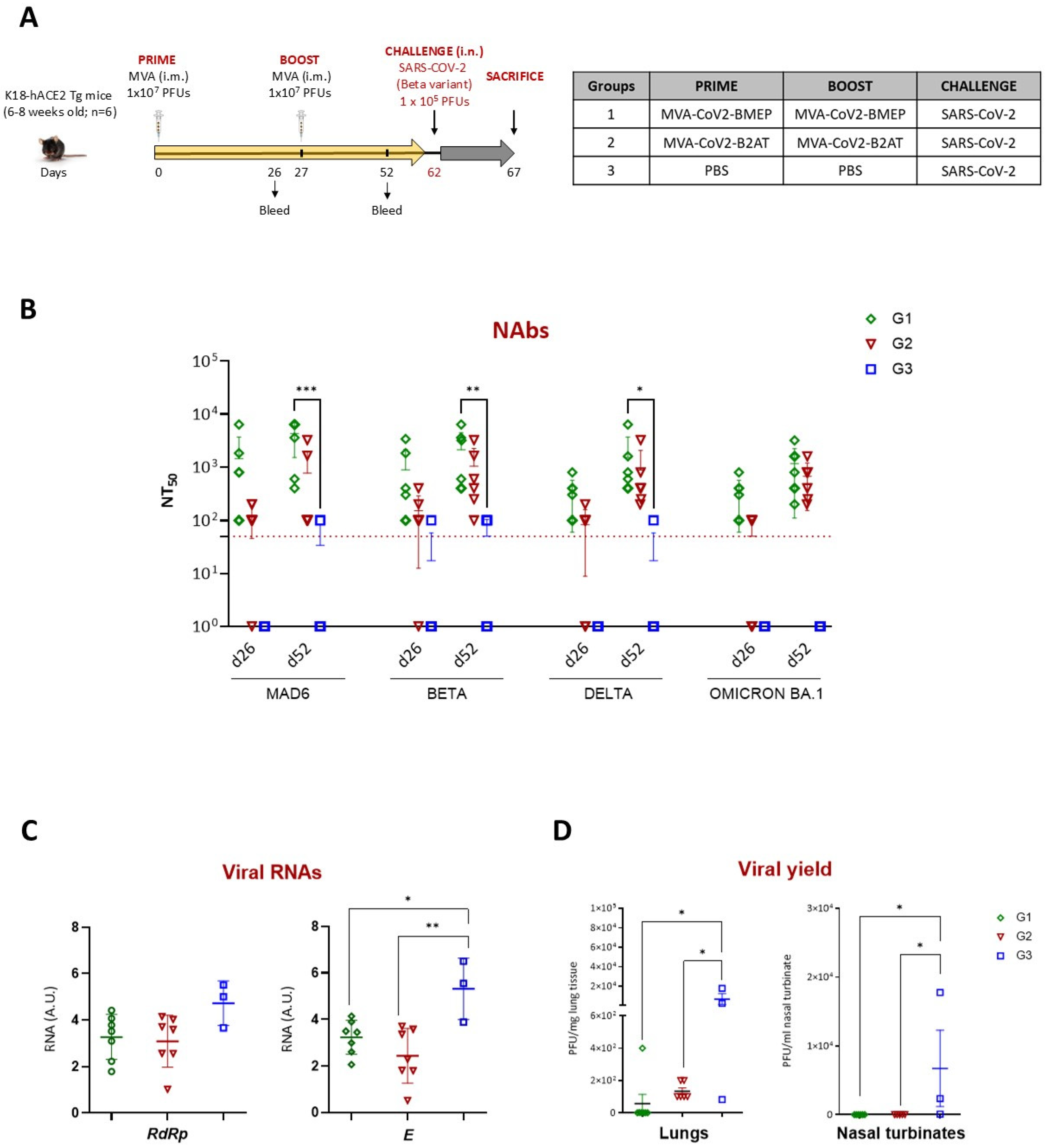
3.6. Homologous MVA-CoV2-B2AT Prime/Boost Vaccination Regimen Effectively Induces Neutralizing Antibodies and Reduces SARS-CoV-2 Beta Variant Replication in K18-hACE2 Tg Mice
3.6.1. Analysis of the SARS-CoV-2-Specific Humoral Response at Pre-Challenge
3.6.2. Restricted SARS-CoV-2 Virus Replication Induced by Vaccination
3.7. MVA-CoV2-B2AT Combined with ISG15 Adjuvant Enhanced Magnitude, Breadth and Polyfunctional Profile of the SARS-CoV-2-Specific T Cell Response in C57BL/6 Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senevirathne, T.H.; Wekking, D.; Swain, J.W.R.; Solinas, C.; De Silva, P. COVID-19: From emerging variants to vaccination. Cytokine Growth Factor Rev. 2024, 76, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.W.; Linderman, S.L.; Moodie, Z.; Czartoski, J.; Lai, L.; Mantus, G.; Norwood, C.; Nyhoff, L.E.; Edara, V.V.; Floyd, K.; et al. Longitudinal analysis shows durable and broad immune memory after SARS-CoV-2 infection with persisting antibody responses and memory B and T cells. Cell Rep. Med. 2021, 2, 100354. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef] [PubMed]
- Heitmann, J.S.; Bilich, T.; Tandler, C.; Nelde, A.; Maringer, Y.; Marconato, M.; Reusch, J.; Jager, S.; Denk, M.; Richter, M.; et al. A COVID-19 peptide vaccine for the induction of SARS-CoV-2 T cell immunity. Nature 2022, 601, 617–622. [Google Scholar] [CrossRef]
- Meyers, L.M.; Gutierrez, A.H.; Boyle, C.M.; Terry, F.; McGonnigal, B.G.; Salazar, A.; Princiotta, M.F.; Martin, W.D.; De Groot, A.S.; Moise, L. Highly conserved, non-human-like, and cross-reactive SARS-CoV-2 T cell epitopes for COVID-19 vaccine design and validation. NPJ Vaccines 2021, 6, 71. [Google Scholar] [CrossRef]
- Parvizpour, S.; Pourseif, M.M.; Razmara, J.; Rafi, M.A.; Omidi, Y. Epitope-based vaccine design: A comprehensive overview of bioinformatics approaches. Drug Discov. Today 2020, 25, 1034–1042. [Google Scholar] [CrossRef]
- Perdiguero, B.; Marcos-Villar, L.; Lopez-Bravo, M.; Sanchez-Cordon, P.J.; Zamora, C.; Valverde, J.R.; Sorzano, C.O.S.; Sin, L.; Alvarez, E.; Ramos, M.; et al. Immunogenicity and efficacy of a novel multi-patch SARS-CoV-2/COVID-19 vaccine candidate. Front. Immunol. 2023, 14, 1160065. [Google Scholar] [CrossRef]
- Falqui, M.; Perdiguero, B.; Coloma, R.; Albert, M.; Marcos-Villar, L.; McGrail, J.P.; Sorzano, C.O.S.; Esteban, M.; Gomez, C.E.; Guerra, S. An MVA-based vector expressing cell-free ISG15 increases IFN-I production and improves HIV-1-specific CD8 T cell immune responses. Front. Cell Infect. Microbiol. 2023, 13, 1187193. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, J.; Lu, R.; Zhang, Y.; Du, M.; Xing, M.; Wu, Z.; Kong, X.; Zhu, Y.; Zhou, X.; et al. Longitudinal immune profiling reveals dominant epitopes mediating long-term humoral immunity in COVID-19-convalescent individuals. J. Allergy Clin. Immunol. 2022, 149, 1225–1241. [Google Scholar] [CrossRef]
- Wang, J.Y.; Song, W.T.; Li, Y.; Chen, W.J.; Yang, D.; Zhong, G.C.; Zhou, H.Z.; Ren, C.Y.; Yu, H.T.; Ling, H. Improved expression of secretory and trimeric proteins in mammalian cells via the introduction of a new trimer motif and a mutant of the tPA signal sequence. Appl. Microbiol. Biotechnol. 2011, 91, 731–740. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Sidney, J.; Sette, A.; Peters, B. TepiTool: A Pipeline for Computational Prediction of T Cell Epitope Candidates. Curr. Protoc. Immunol. 2016, 114, 18.19.11–18.19.24. [Google Scholar] [CrossRef]
- Donnelly, M.L.L.; Luke, G.; Mehrotra, A.; Li, X.; Hughes, L.E.; Gani, D.; Ryan, M.D. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: A putative ribosomal ‘skip’. J. Gen. Virol. 2001, 82, 1013–1025. [Google Scholar] [CrossRef]
- Guler-Gane, G.; Kidd, S.; Sridharan, S.; Vaughan, T.J.; Wilkinson, T.C.; Tigue, N.J. Overcoming the Refractory Expression of Secreted Recombinant Proteins in Mammalian Cells through Modification of the Signal Peptide and Adjacent Amino Acids. PLoS ONE 2016, 11, e0155340. [Google Scholar] [CrossRef]
- Garcia-Arriaza, J.; Cepeda, V.; Hallengard, D.; Sorzano, C.O.; Kummerer, B.M.; Liljestrom, P.; Esteban, M. A novel poxvirus-based vaccine, MVA-CHIKV, is highly immunogenic and protects mice against chikungunya infection. J. Virol. 2014, 88, 3527–3547. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Garaigorta, U.; Perez, P.; Lazaro-Frias, A.; Zamora, C.; Gastaminza, P.; Del Fresno, C.; Casasnovas, J.M.; Sorzano, C.O.S.; Sancho, D.; et al. COVID-19 vaccine candidates based on modified vaccinia virus Ankara expressing the SARS-CoV-2 spike induce robust T- and B-cell immune responses and full efficacy in mice. J. Virol. 2021, 95, e02260-20. [Google Scholar] [CrossRef]
- Marcos-Villar, L.; Perdiguero, B.; Anthiya, S.; Borrajo, M.L.; Lou, G.; Franceschini, L.; Esteban, I.; Sanchez-Cordon, P.J.; Zamora, C.; Sorzano, C.O.S.; et al. Modulating the immune response to SARS-CoV-2 by different nanocarriers delivering an mRNA expressing trimeric RBD of the spike protein: COVARNA Consortium. NPJ Vaccines 2024, 9, 53. [Google Scholar] [CrossRef]
- Perez, P.; M, Q.M.; Lazaro-Frias, A.; Jimenez de Oya, N.; Blazquez, A.B.; Escribano-Romero, E.; CO, S.S.; Ortego, J.; Saiz, J.C.; Esteban, M.; et al. A Vaccine Based on a Modified Vaccinia Virus Ankara Vector Expressing Zika Virus Structural Proteins Controls Zika Virus Replication in Mice. Sci. Rep. 2018, 8, 17385. [Google Scholar] [CrossRef]
- Perdiguero, B.; Gomez, C.E.; Cepeda, V.; Sanchez-Sampedro, L.; Garcia-Arriaza, J.; Mejias-Perez, E.; Jimenez, V.; Sanchez, C.; Sorzano, C.O.; Oliveros, J.C.; et al. Virological and immunological characterization of novel NYVAC-based HIV/AIDS vaccine candidates expressing clade C trimeric soluble gp140(ZM96) and Gag(ZM96)-Pol-Nef(CN54) as virus-like particles. J. Virol. 2015, 89, 970–988. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.C.; Gherardi, M.M.; Esteban, M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: Virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J. Virol. 2000, 74, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, B.; Gomez, C.E.; Garcia-Arriaza, J.; Sanchez-Corzo, C.; Sorzano, C.O.S.; Wilmschen, S.; von Laer, D.; Asbach, B.; Schmalzl, C.; Peterhoff, D.; et al. Heterologous Combination of VSV-GP and NYVAC Vectors Expressing HIV-1 Trimeric gp145 Env as Vaccination Strategy to Induce Balanced B and T Cell Immune Responses. Front. Immunol. 2019, 10, 2941. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, B.; Sanchez-Corzo, C.; Sorzano, C.O.S.; Saiz, L.; Mediavilla, P.; Esteban, M.; Gomez, C.E. A Novel MVA-Based HIV Vaccine Candidate (MVA-gp145-GPN) Co-Expressing Clade C Membrane-Bound Trimeric gp145 Env and Gag-Induced Virus-Like Particles (VLPs) Triggered Broad and Multifunctional HIV-1-Specific T Cell and Antibody Responses. Viruses 2019, 11, 160. [Google Scholar] [CrossRef]
- Perdiguero, B.; Sanchez-Corzo, C.; CO, S.S.; Mediavilla, P.; Saiz, L.; Esteban, M.; Gomez, C.E. Induction of Broad and Polyfunctional HIV-1-Specific T Cell Responses by the Multiepitopic Protein TMEP-B Vectored by MVA Virus. Vaccines 2019, 7, 57. [Google Scholar] [CrossRef]
- Manenti, A.; Maggetti, M.; Casa, E.; Martinuzzi, D.; Torelli, A.; Trombetta, C.M.; Marchi, S.; Montomoli, E. Evaluation of SARS-CoV-2 neutralizing antibodies using a CPE-based colorimetric live virus micro-neutralization assay in human serum samples. J. Med. Virol. 2020, 92, 2096–2104. [Google Scholar] [CrossRef]
- Perez, P.; Astorgano, D.; Albericio, G.; Flores, S.; Sanchez-Cordon, P.J.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; Garcia-Arriaza, J. Intranasal administration of a single dose of MVA-based vaccine candidates against COVID-19 induced local and systemic immune responses and protects mice from a lethal SARS-CoV-2 infection. Front. Immunol. 2022, 13, 995235. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 2020, 25, 2000045. [Google Scholar] [CrossRef]
- Najera, J.L.; Gomez, C.E.; Garcia-Arriaza, J.; Sorzano, C.O.; Esteban, M. Insertion of vaccinia virus C7L host range gene into NYVAC-B genome potentiates immune responses against HIV-1 antigens. PLoS ONE 2010, 5, e11406. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Lee, K.H.; Kang, T.B. The Molecular Links between Cell Death and Inflammasome. Cells 2019, 8, 1057. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S. A Detailed Overview of Immune Escape, Antibody Escape, Partial Vaccine Escape of SARS-CoV-2 and Their Emerging Variants With Escape Mutations. Front. Immunol. 2022, 13, 801522. [Google Scholar] [CrossRef]
- Ghildiyal, T.; Rai, N.; Mishra Rawat, J.; Singh, M.; Anand, J.; Pant, G.; Kumar, G.; Shidiki, A. Challenges in Emerging Vaccines and Future Promising Candidates against SARS-CoV-2 Variants. J. Immunol. Res. 2024, 2024, 9125398. [Google Scholar] [CrossRef]
- Tobias, J.; Steinberger, P.; Wilkinson, J.; Klais, G.; Kundi, M.; Wiedermann, U. SARS-CoV-2 Vaccines: The Advantage of Mucosal Vaccine Delivery and Local Immunity. Vaccines 2024, 12, 795. [Google Scholar] [CrossRef]
- Cankat, S.; Demael, M.U.; Swadling, L. In search of a pan-coronavirus vaccine: Next-generation vaccine design and immune mechanisms. Cell Mol. Immunol. 2024, 21, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Nathan, A.; Rossin, E.J.; Kaseke, C.; Park, R.J.; Khatri, A.; Koundakjian, D.; Urbach, J.M.; Singh, N.K.; Bashirova, A.; Tano-Menka, R.; et al. Structure-guided T cell vaccine design for SARS-CoV-2 variants and sarbecoviruses. Cell 2021, 184, 4401–4413.e10. [Google Scholar] [CrossRef] [PubMed]
- Vishwanath, S.; Carnell, G.W.; Ferrari, M.; Asbach, B.; Billmeier, M.; George, C.; Sans, M.S.; Nadesalingam, A.; Huang, C.Q.; Paloniemi, M.; et al. A computationally designed antigen eliciting broad humoral responses against SARS-CoV-2 and related sarbecoviruses. Nat. Biomed. Eng. 2023; Epub ahead of print. [Google Scholar] [CrossRef]
- Mortezaee, K.; Majidpoor, J. Cellular immune states in SARS-CoV-2-induced disease. Front. Immunol. 2022, 13, 1016304. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef]
- Wang, L.; Nicols, A.; Turtle, L.; Richter, A.; Duncan, C.J.; Dunachie, S.J.; Klenerman, P.; Payne, R.P. T cell immune memory after COVID-19 and vaccination. BMJ Med. 2023, 2, e000468. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Miller, H.; Byazrova, M.G.; Cndotti, F.; Benlagha, K.; Camara, N.O.S.; Shi, J.; Forsman, H.; Lee, P.; Yang, L.; et al. The characterization of CD8(+) T-cell responses in COVID-19. Emerg. Microbes Infect. 2024, 13, 2287118. [Google Scholar] [CrossRef] [PubMed]
- Geers, D.; Shamier, M.C.; Bogers, S.; den Hartog, G.; Gommers, L.; Nieuwkoop, N.N.; Schmitz, K.S.; Rijsbergen, L.C.; van Osch, J.A.T.; Dijkhuizen, E.; et al. SARS-CoV-2 variants of concern partially escape humoral but not T-cell responses in COVID-19 convalescent donors and vaccinees. Sci. Immunol. 2021, 6, eabj1750. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Methot, N.; Yu, E.D.; Zhang, Y.; Dan, J.M.; Goodwin, B.; Rubiro, P.; Sutherland, A.; Wang, E.; et al. Impact of SARS-CoV-2 variants on the total CD4(+) and CD8(+) T cell reactivity in infected or vaccinated individuals. Cell Rep. Med. 2021, 2, 100355. [Google Scholar] [CrossRef]
- Bonifacius, A.; Tischer-Zimmermann, S.; Dragon, A.C.; Gussarow, D.; Vogel, A.; Krettek, U.; Godecke, N.; Yilmaz, M.; Kraft, A.R.M.; Hoeper, M.M.; et al. COVID-19 immune signatures reveal stable antiviral T cell function despite declining humoral responses. Immunity 2021, 54, 340–354.e6. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680.e2. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Pujadas, E.; Chaudhry, F.; McBride, R.; Richter, F.; Zhao, S.; Wajnberg, A.; Nadkarni, G.; Glicksberg, B.S.; Houldsworth, J.; Cordon-Cardo, C. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir. Med. 2020, 8, e70. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.A.; Diamond, M.S. Host cell-intrinsic innate immune recognition of SARS-CoV-2. Curr. Opin. Virol. 2022, 52, 30–38. [Google Scholar] [CrossRef]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef] [PubMed]
- Persson, G.; Restori, K.H.; Emdrup, J.H.; Schussek, S.; Klausen, M.S.; Nicol, M.J.; Katkere, B.; Rono, B.; Kirimanjeswara, G.; Sorensen, A.B. DNA immunization with in silico predicted T-cell epitopes protects against lethal SARS-CoV-2 infection in K18-hACE2 mice. Front. Immunol. 2023, 14, 1166546. [Google Scholar] [CrossRef]
- van Bergen, J.; Camps, M.G.; Pardieck, I.N.; Veerkamp, D.; Leung, W.Y.; Leijs, A.A.; Myeni, S.K.; Kikkert, M.; Arens, R.; Zondag, G.C.; et al. Multiantigen pan-sarbecovirus DNA vaccines generate protective T cell immune responses. JCI Insight 2023, 8, e172488. [Google Scholar] [CrossRef]
- Notarbartolo, S.; Ranzani, V.; Bandera, A.; Gruarin, P.; Bevilacqua, V.; Putignano, A.R.; Gobbini, A.; Galeota, E.; Manara, C.; Bombaci, M.; et al. Integrated longitudinal immunophenotypic, transcriptional and repertoire analyses delineate immune responses in COVID-19 patients. Sci. Immunol. 2021, 6, eabg5021. [Google Scholar] [CrossRef]
- Bergamaschi, L.; Mescia, F.; Turner, L.; Hanson, A.L.; Kotagiri, P.; Dunmore, B.J.; Ruffieux, H.; De Sa, A.; Huhn, O.; Morgan, M.D.; et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity 2021, 54, 1257–1275.e8. [Google Scholar] [CrossRef]
- Grau-Exposito, J.; Sanchez-Gaona, N.; Massana, N.; Suppi, M.; Astorga-Gamaza, A.; Perea, D.; Rosado, J.; Falco, A.; Kirkegaard, C.; Torrella, A.; et al. Peripheral and lung resident memory T cell responses against SARS-CoV-2. Nat. Commun. 2021, 12, 3010. [Google Scholar] [CrossRef]
- Wagner, K.I.; Mateyka, L.M.; Jarosch, S.; Grass, V.; Weber, S.; Schober, K.; Hammel, M.; Burrell, T.; Kalali, B.; Poppert, H.; et al. Recruitment of highly cytotoxic CD8(+) T cell receptors in mild SARS-CoV-2 infection. Cell Rep. 2022, 38, 110214. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, J.; Yang, S.; Wang, L.; Chen, X.; Yang, Y.; Li, R.; Pan, Z.; Zhao, J.; Li, Z.; et al. The dichotomous and incomplete adaptive immunity in COVID-19 patients with different disease severity. Signal Transduct. Target. Ther. 2021, 6, 113. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Szabo, P.A.; Dogra, P.; Gray, J.I.; Wells, S.B.; Connors, T.J.; Weisberg, S.P.; Krupska, I.; Matsumoto, R.; Poon, M.M.L.; Idzikowski, E.; et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 2021, 54, 797–814.e6. [Google Scholar] [CrossRef]
- Su, Y.; Chen, D.; Yuan, D.; Lausted, C.; Choi, J.; Dai, C.L.; Voillet, V.; Duvvuri, V.R.; Scherler, K.; Troisch, P.; et al. Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell 2020, 183, 1479–1495.e20. [Google Scholar] [CrossRef]
- Ferrantelli, F.; Chiozzini, C.; Manfredi, F.; Leone, P.; Spada, M.; Di Virgilio, A.; Giovannelli, A.; Sanchez, M.; Cara, A.; Michelini, Z.; et al. Strong SARS-CoV-2 N-Specific CD8(+) T Immunity Induced by Engineered Extracellular Vesicles Associates with Protection from Lethal Infection in Mice. Viruses 2022, 14, 329. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Peng, Y.; Felce, S.L.; Dong, D.; Penkava, F.; Mentzer, A.J.; Yao, X.; Liu, G.; Yin, Z.; Chen, J.L.; Lu, Y.; et al. An immunodominant NP(105-113)-B*07:02 cytotoxic T cell response controls viral replication and is associated with less severe COVID-19 disease. Nat. Immunol. 2022, 23, 50–61. [Google Scholar] [CrossRef]
- Mendoza-Ramirez, N.J.; Garcia-Cordero, J.; Shrivastava, G.; Cedillo-Barron, L. The Key to Increase Immunogenicity of Next-Generation COVID-19 Vaccines Lies in the Inclusion of the SARS-CoV-2 Nucleocapsid Protein. J. Immunol. Res. 2024, 2024, 9313267. [Google Scholar] [CrossRef]
- Brasu, N.; Elia, I.; Russo, V.; Montacchiesi, G.; Stabile, S.A.; De Intinis, C.; Fesi, F.; Gizzi, K.; Macagno, M.; Montone, M.; et al. Memory CD8(+) T cell diversity and B cell responses correlate with protection against SARS-CoV-2 following mRNA vaccination. Nat. Immunol. 2022, 23, 1445–1456. [Google Scholar] [CrossRef]
- Neale, I.; Ali, M.; Kronsteiner, B.; Longet, S.; Abraham, P.; Deeks, A.S.; Brown, A.; Moore, S.C.; Stafford, L.; Dobson, S.L.; et al. CD4+ and CD8+ T cells and antibodies are associated with protection against Delta vaccine breakthrough infection: A nested case-control study within the PITCH study. mBio 2023, 14, e0121223. [Google Scholar] [CrossRef]
- Tarke, A.; Potesta, M.; Varchetta, S.; Fenoglio, D.; Iannetta, M.; Sarmati, L.; Mele, D.; Dentone, C.; Bassetti, M.; Montesano, C.; et al. Early and Polyantigenic CD4 T Cell Responses Correlate with Mild Disease in Acute COVID-19 Donors. Int. J. Mol. Sci. 2022, 23, 7155. [Google Scholar] [CrossRef] [PubMed]
- Koonpaew, S.; Kaewborisuth, C.; Srisutthisamphan, K.; Wanitchang, A.; Thaweerattanasinp, T.; Saenboonrueng, J.; Poonsuk, S.; Jengarn, J.; Viriyakitkosol, R.; Kramyu, J.; et al. A Single-Cycle Influenza A Virus-Based SARS-CoV-2 Vaccine Elicits Potent Immune Responses in a Mouse Model. Vaccines 2021, 9, 850. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Kudchodkar, S.B.; Gil, A.; Jeon, B.; Park, G.H.; Cho, Y.; Lee, H.; Cheong, M.S.; Kim, W.; Hwang, Y.-H.; et al. Immune Responses of a Novel Bi-Cistronic SARS-CoV-2 DNA Vaccine Following Intradermal Immunization With Suction Delivery. Front. Virol. 2022, 2, 891540. [Google Scholar] [CrossRef]
- Kim, W.J.; Roberts, C.C.; Song, J.Y.; Yoon, J.G.; Seong, H.; Hyun, H.J.; Lee, H.; Gil, A.; Oh, Y.; Park, J.E.; et al. Safety and immunogenicity of the bi-cistronic GLS-5310 COVID-19 DNA vaccine delivered with the GeneDerm suction device. Int. J. Infect. Dis. 2023, 128, 112–120. [Google Scholar] [CrossRef]
- Xing, M.; Wang, Y.; Wang, X.; Liu, J.; Dai, W.; Hu, G.; He, F.; Zhao, Q.; Li, Y.; Sun, L.; et al. Broad-spectrum vaccine via combined immunization routes triggers potent immunity to SARS-CoV-2 and its variants. J. Virol. 2023, 97, e0072423. [Google Scholar] [CrossRef]
- Moradian, H.; Gossen, M.; Lendlein, A. Co-delivery of genes can be confounded by bicistronic vector design. MRS Commun. 2022, 12, 145–153. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Viral Protein | Start | End |
|---|---|---|---|
| EAEVQIDRLITGRLQSLQTYV | S | 988 | 1008 |
| SFPQSAPHGVVFLHVTYVPAQEKN | S | 1051 | 1074 |
| WFVTQRNFYEPQIIT | S | 1102 | 1116 |
| IVNSVLLFLAFVVFLLV | E | 13 | 29 |
| RLCAYCCNIVNVSLVKPSFYVY | E | 38 | 59 |
| LQFAYANRNRFLYIIK | M | 35 | 50 |
| GTILTRPLLESELVIGAVILRGHLRIAGHHLG | M | 126 | 157 |
| SGFAAYSRYRIGNYKL | M | 191 | 206 |
| KDLSPRWYFYYLGTGPEAGLPYG | N | 102 | 124 |
| ALALLLLDRLNQLESK | N | 218 | 233 |
| TKAYNVTQAFGRRGP | N | 265 | 279 |
| LTYTGAIKLDDKDPNFKDQVILLNKHIDAYKTFPPTEPKK | N | 331 | 370 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perdiguero, B.; Álvarez, E.; Marcos-Villar, L.; Sin, L.; López-Bravo, M.; Valverde, J.R.; Sorzano, C.Ó.S.; Falqui, M.; Coloma, R.; Esteban, M.; et al. B and T Cell Bi-Cistronic Multiepitopic Vaccine Induces Broad Immunogenicity and Provides Protection Against SARS-CoV-2. Vaccines 2024, 12, 1213. https://doi.org/10.3390/vaccines12111213
Perdiguero B, Álvarez E, Marcos-Villar L, Sin L, López-Bravo M, Valverde JR, Sorzano CÓS, Falqui M, Coloma R, Esteban M, et al. B and T Cell Bi-Cistronic Multiepitopic Vaccine Induces Broad Immunogenicity and Provides Protection Against SARS-CoV-2. Vaccines. 2024; 12(11):1213. https://doi.org/10.3390/vaccines12111213
Chicago/Turabian StylePerdiguero, Beatriz, Enrique Álvarez, Laura Marcos-Villar, Laura Sin, María López-Bravo, José Ramón Valverde, Carlos Óscar S. Sorzano, Michela Falqui, Rocío Coloma, Mariano Esteban, and et al. 2024. "B and T Cell Bi-Cistronic Multiepitopic Vaccine Induces Broad Immunogenicity and Provides Protection Against SARS-CoV-2" Vaccines 12, no. 11: 1213. https://doi.org/10.3390/vaccines12111213
APA StylePerdiguero, B., Álvarez, E., Marcos-Villar, L., Sin, L., López-Bravo, M., Valverde, J. R., Sorzano, C. Ó. S., Falqui, M., Coloma, R., Esteban, M., Guerra, S., & Gómez, C. E. (2024). B and T Cell Bi-Cistronic Multiepitopic Vaccine Induces Broad Immunogenicity and Provides Protection Against SARS-CoV-2. Vaccines, 12(11), 1213. https://doi.org/10.3390/vaccines12111213