
The Key Site Variation and Immune Challenges in SARS-CoV-2 Evolution

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structure, Function, and Cell Entry of SARS-CoV-2
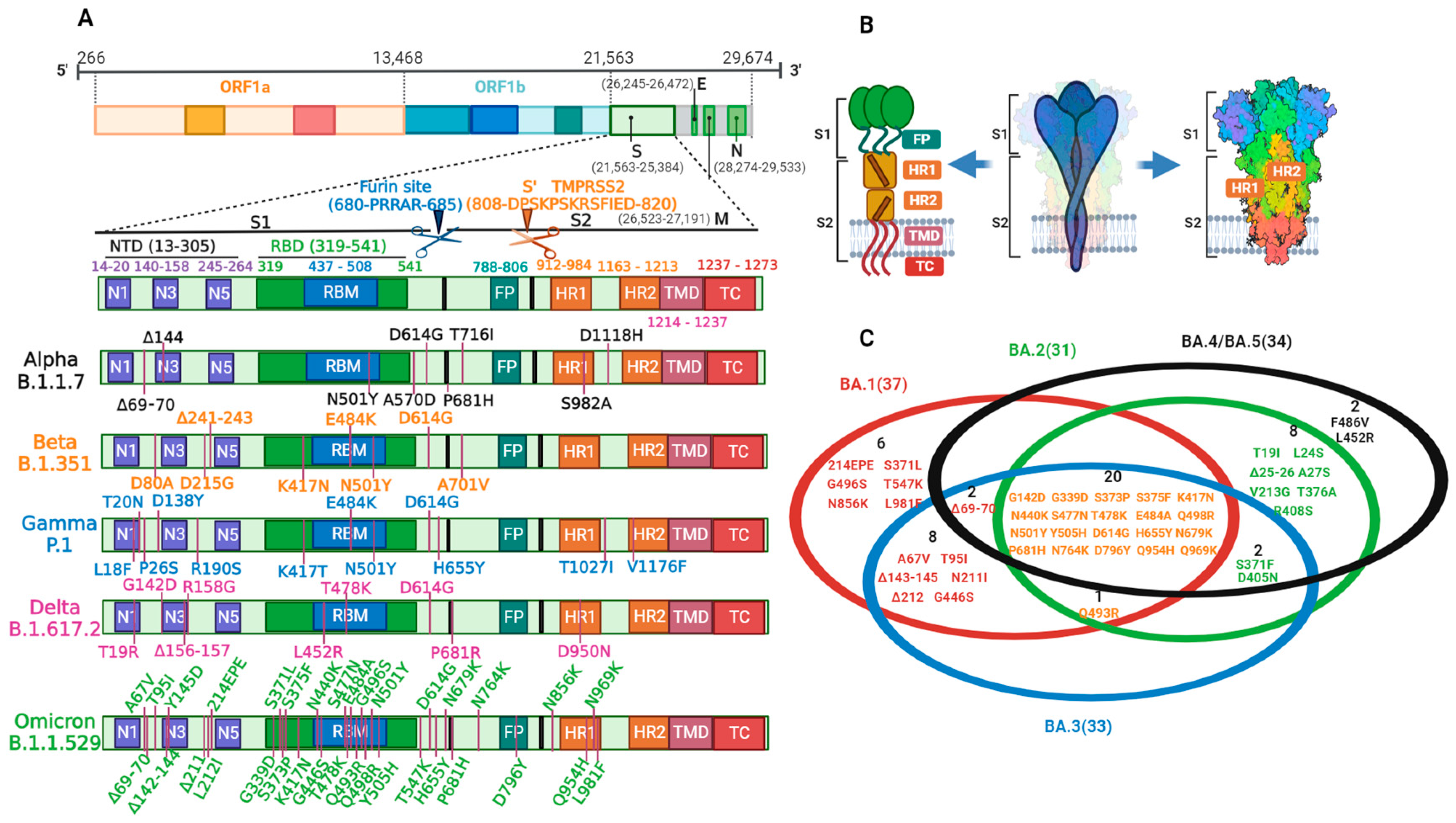
3. SARS-CoV-2 Variation
3.1. Variation Causes
3.2. Variation and Prevalence
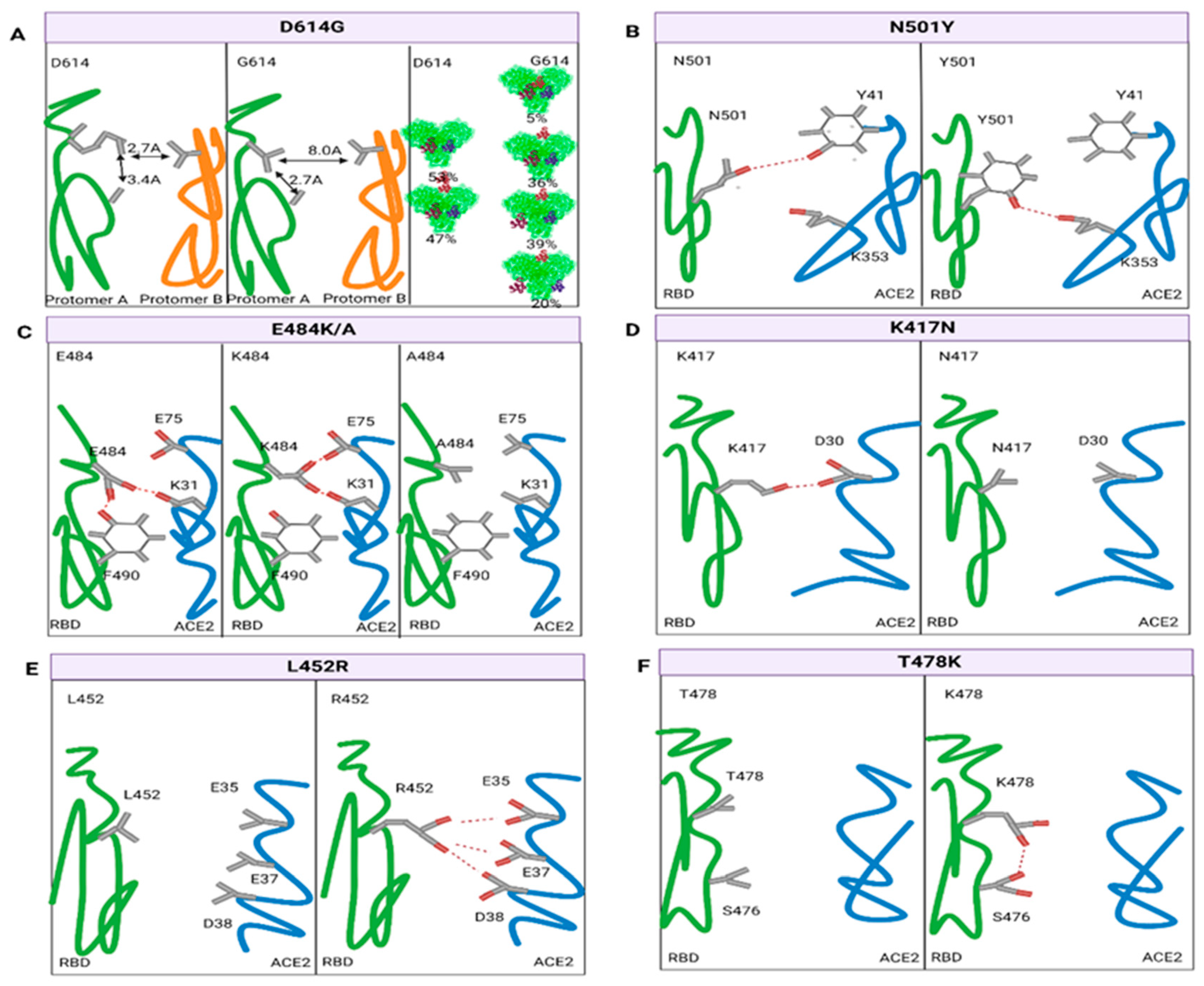
3.3. Variation and Immune Challenge of Key Sites
3.3.1. Variation in the RBD
3.3.2. Variation in Furin Cleavage Sites
3.3.3. Variation in the NTD
4. Prevention and Control Strategies
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Liu, Y.; Ye, Q. Safety and Efficacy of the Common Vaccines against COVID-19. Vaccines 2022, 10, 513. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ye, Q. Nucleic Acid Vaccines against SARS-CoV-2. Vaccines 2022, 10, 1849. [Google Scholar] [CrossRef]
- Chen, P.; Nirula, A.; Heller, B.; Gottlieb, R.L.; Boscia, J.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with COVID-19. N. Engl. J. Med. 2021, 384, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.L.; Nirula, A.; Chen, P.; Boscia, J.; Heller, B.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ye, Q. The variants of SARS-CoV-2 and the challenges of vaccines. J. Med. Virol. 2022, 94, 1366–1372. [Google Scholar] [CrossRef]
- Meng, H.; Mao, J.; Ye, Q. Booster vaccination strategy: Necessity, immunization objectives, immunization strategy, and safety. J. Med. Virol. 2022, 94, 2369–2375. [Google Scholar] [CrossRef]
- Jahirul Islam, M.; Nawal Islam, N.; Siddik Alom, M.; Kabir, M.; Halim, M.A. A review on structural, non-structural, and accessory proteins of SARS-CoV-2: Highlighting drug target sites. Immunobiology 2023, 228, 152302. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K.; Hou, M.H.; Chang, C.F.; Hsiao, C.D.; Huang, T.H. The SARS coronavirus nucleocapsid protein--forms and functions. Antiviral Res. 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, T.; Zhang, J.; Shao, B.; Gong, H.; Wang, Y.; He, X.; Liu, S.; Liu, T.Y. Exploring the Regulatory Function of the N-terminal Domain of SARS-CoV-2 Spike Protein through Molecular Dynamics Simulation. Adv. Theory Simul. 2021, 4, 2100152. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Bosch, B.J.; Martina, B.E.; Van Der Zee, R.; Lepault, J.; Haijema, B.J.; Versluis, C.; Heck, A.J.; De Groot, R.; Osterhaus, A.D.; Rottier, P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 8455–8460. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Blackstone, N.W.; Blackstone, S.R.; Berg, A.T. Variation and multilevel selection of SARS-CoV-2. Evolution 2020, 74, 2429–2434. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Jhun, H.; Park, H.Y.; Hisham, Y.; Song, C.S.; Kim, S. SARS-CoV-2 Delta (B.1.617.2) Variant: A Unique T478K Mutation in Receptor Binding Motif (RBM) of Spike Gene. Immune Netw. 2021, 21, e32. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. Embo J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Tan, J.; Wu, Z.; Hu, P.; Gan, L.; Wang, Y.; Zhang, D. Association between Mutations in Papain-like Protease (PLpro) of SARS-CoV-2 with COVID-19 Clinical Outcomes. Pathogens 2022, 11, 1008. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.U.; Bhattacharjee, A.; Emon, M.T.H.; Chowdhury, Z.M.; Ahammad, I.; Mosaib, M.G.; Moniruzzaman, M.; Rahman, M.H.; Islam, M.N.; Ahmed, I.; et al. Novel mutations in NSP-1 and PLPro of SARS-CoV-2 NIB-1 genome mount for effective therapeutics. J. Genet. Eng. Biotechnol. 2021, 19, 52. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, M.; Dhama, K.; Lee, S.S.; Chakraborty, C. Resistance to nirmatrelvir due to mutations in the Mpro in the subvariants of SARS-CoV-2 Omicron: Another concern? Mol. Ther. Nucleic Acids 2023, 32, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Kang, T.; Ali, H.; Lai, D. Remdesivir Strongly Binds to RNA-Dependent RNA Polymerase, Membrane Protein, and Main Protease of SARS-CoV-2: Indication From Molecular Modeling and Simulations. Front. Pharmacol. 2021, 12, 710778. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H., 3rd; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Chatterjee, S.; Sharma, A.R.; Agoramoorthy, G.; Chakraborty, C. D614G mutation and SARS-CoV-2: Impact on S-protein structure, function, infectivity, and immunity. Appl. Microbiol. Biotechnol. 2021, 105, 9035–9045. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e738. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Omotuyi, I.O.; Nash, O.; Ajiboye, O.B.; Iwegbulam, C.G.; Oyinloye, E.B.; Oyedeji, O.A.; Kashim, Z.A.; Okaiyeto, K. Atomistic simulation reveals structural mechanisms underlying D614G spike glycoprotein-enhanced fitness in SARS-COV-2. J. Comput. Chem. 2020, 41, 2158–2161. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e2316. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F. Neutralising antibody escape of SARS-CoV-2 spike protein: Risk assessment for antibody-based COVID-19 therapeutics and vaccines. Rev. Med. Virol. 2021, 31, e2231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Laffeber, C.; de Koning, K.; Kanaar, R.; Lebbink, J.H.G. Experimental Evidence for Enhanced Receptor Binding by Rapidly Spreading SARS-CoV-2 Variants. J. Mol. Biol. 2021, 433, 167058. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Peng, P.; Wang, K.; Fang, L.; Luo, F.Y.; Jin, A.S.; Liu, B.Z.; Tang, N.; Huang, A.L. Emerging SARS-CoV-2 variants reduce neutralization sensitivity to convalescent sera and monoclonal antibodies. Cell Mol. Immunol. 2021, 18, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chen, Q.; Yang, G.; He, L.; Fan, H.; Deng, Y.Q.; Wang, Y.; Teng, Y.; Zhao, Z.; Cui, Y.; et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 2020, 369, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Zhang, Z.; Gao, X.; Du, P.; Lu, J.; Yan, B.; Wang, C.; Zheng, Y.; Huang, H.; Sun, Q. N501Y mutation imparts cross-species transmission of SARS-CoV-2 to mice by enhancing receptor binding. Signal Transduct. Target. Ther. 2021, 6, 284. [Google Scholar] [CrossRef]
- Rees-Spear, C.; Muir, L.; Griffith, S.A.; Heaney, J.; Aldon, Y.; Snitselaar, J.L.; Thomas, P.; Graham, C.; Seow, J.; Lee, N.; et al. The effect of spike mutations on SARS-CoV-2 neutralization. Cell Rep. 2021, 34, 108890. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Xia, H.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Weaver, S.C.; Swanson, K.A.; Cai, H.; Sarkar, R.; et al. BNT162b2-Elicited Neutralization against New SARS-CoV-2 Spike Variants. N. Engl. J. Med. 2021, 385, 472–474. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e1220. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e610. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Huynh, T. Insights into SARS-CoV-2’s Mutations for Evading Human Antibodies: Sacrifice and Survival. J. Med. Chem. 2022, 65, 2820–2826. [Google Scholar] [CrossRef] [PubMed]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488.e474. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e466. [Google Scholar] [CrossRef] [PubMed]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; García-Sastre, A.; Schotsaert, M. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe 2021, 2, e283–e284. [Google Scholar] [CrossRef] [PubMed]
- Andreano, E.; Piccini, G.; Licastro, D.; Casalino, L.; Johnson, N.V.; Paciello, I.; Dal Monego, S.; Pantano, E.; Manganaro, N.; Manenti, A.; et al. SARS-CoV-2 escape from a highly neutralizing COVID-19 convalescent plasma. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef]
- Wu, K.; Werner, A.P.; Koch, M.; Choi, A.; Narayanan, E.; Stewart-Jones, G.B.E.; Colpitts, T.; Bennett, H.; Boyoglu-Barnum, S.; Shi, W.; et al. Serum Neutralizing Activity Elicited by mRNA-1273 Vaccine. N. Engl. J. Med. 2021, 384, 1468–1470. [Google Scholar] [CrossRef]
- Collier, D.A.; De Marco, A.; Ferreira, I.; Meng, B.; Datir, R.P.; Walls, A.C.; Kemp, S.A.; Bassi, J.; Pinto, D.; Silacci-Fregni, C.; et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 2021, 593, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Abu-Raddad, L.J.; Chemaitelly, H.; Butt, A.A. Effectiveness of the BNT162b2 COVID-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Engl. J. Med. 2021, 385, 187–189. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.T.; Huang, W.H.; Liao, T.L.; Hsiao, T.H.; Chuang, H.N.; Liu, P.Y. SARS-CoV-2 E484K Mutation Narrative Review: Epidemiology, Immune Escape, Clinical Implications, and Future Considerations. Infect. Drug Resist. 2022, 15, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Huang, D.; Lee, C.D.; Wu, N.C.; Jackson, A.M.; Zhu, X.; Liu, H.; Peng, L.; van Gils, M.J.; Sanders, R.W.; et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science 2021, 373, 818–823. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e1111. [Google Scholar] [CrossRef]
- Selzer, T.; Albeck, S.; Schreiber, G. Rational design of faster associating and tighter binding protein complexes. Nat. Struct. Biol. 2000, 7, 537–541. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426–3437.e3428. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Toyoshima, Y.; Nemoto, K.; Nakamura, Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. J. Hum. Genet. 2020, 65, 569–575. [Google Scholar] [CrossRef]
- Pondé, R.A.A. Physicochemical effect of the N501Y, E484K/Q, K417N/T, L452R and T478K mutations on the SARS-CoV-2 spike protein RBD and its influence on agent fitness and on attributes developed by emerging variants of concern. Virology 2022, 572, 44–54. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef]
- Kumar, V.; Singh, J.; Hasnain, S.E.; Sundar, D. Possible Link between Higher Transmissibility of Alpha, Kappa and Delta Variants of SARS-CoV-2 and Increased Structural Stability of Its Spike Protein and hACE2 Affinity. Int. J. Mol. Sci. 2021, 22, 9131. [Google Scholar] [CrossRef]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, S.; Sefidbakht, Y.; Uskoković, V. Comparative molecular dynamics study of the receptor-binding domains in SARS-CoV-2 and SARS-CoV and the effects of mutations on the binding affinity. J. Biomol. Struct. Dyn. 2022, 40, 4662–4681. [Google Scholar] [CrossRef]
- Ferreira, I.; Kemp, S.A.; Datir, R.; Saito, A.; Meng, B.; Rakshit, P.; Takaori-Kondo, A.; Kosugi, Y.; Uriu, K.; Kimura, I.; et al. SARS-CoV-2 B.1.617 Mutations L452R and E484Q Are Not Synergistic for Antibody Evasion. J. Infect. Dis. 2021, 224, 989–994. [Google Scholar] [CrossRef]
- Miller, N.L.; Clark, T.; Raman, R.; Sasisekharan, R. Insights on the mutational landscape of the SARS-CoV-2 Omicron variant receptor-binding domain. Cell Rep. Med. 2022, 3, 100527. [Google Scholar] [CrossRef]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Harari, D.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Li, C.; Marton, I.; et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021, 6, 1188–1198. [Google Scholar] [CrossRef]
- Del Rio, C.; Omer, S.B.; Malani, P.N. Winter of Omicron-The Evolving COVID-19 Pandemic. JAMA 2022, 327, 319–320. [Google Scholar] [CrossRef]
- Wilhelm, A.; Widera, M.; Grikscheit, K.; Toptan, T.; Schenk, B.; Pallas, C.; Metzler, M.; Kohmer, N.; Hoehl, S.; Marschalek, R.; et al. Limited neutralisation of the SARS-CoV-2 Omicron subvariants BA.1 and BA.2 by convalescent and vaccine serum and monoclonal antibodies. EBioMedicine 2022, 82, 104158. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef] [PubMed]
- Lista, M.J.; Winstone, H.; Wilson, H.D.; Dyer, A.; Pickering, S.; Galao, R.P.; De Lorenzo, G.; Cowton, V.M.; Furnon, W.; Suarez, N.; et al. The P681H Mutation in the Spike Glycoprotein of the Alpha Variant of SARS-CoV-2 Escapes IFITM Restriction and Is Necessary for Type I Interferon Resistance. J. Virol. 2022, 96, e0125022. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Suryadevara, N.; Shrihari, S.; Gilchuk, P.; VanBlargan, L.A.; Binshtein, E.; Zost, S.J.; Nargi, R.S.; Sutton, R.E.; Winkler, E.S.; Chen, E.C.; et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 2021, 184, 2316–2331. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Brejová, B.; Boršová, K.; Hodorová, V.; Čabanová, V.; Reizigová, L.; Paul, E.D.; Čekan, P.; Klempa, B.; Nosek, J.; Vinař, T. A SARS-CoV-2 mutant from B.1.258 lineage with ∆H69/∆V70 deletion in the Spike protein circulating in Central Europe in the fall 2020. Virus Genes 2021, 57, 556–560. [Google Scholar] [CrossRef]
- Yankauckas, M.A.; Morrow, J.E.; Parker, S.E.; Abai, A.; Rhodes, G.H.; Dwarki, V.J.; Gromkowski, S.H. Long-term anti-nucleoprotein cellular and humoral immunity is induced by intramuscular injection of plasmid DNA containing NP gene. DNA Cell Biol. 1993, 12, 771–776. [Google Scholar] [CrossRef]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.M.; et al. COVID-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef]
- Zhang, J.; He, Q.; An, C.; Mao, Q.; Gao, F.; Bian, L.; Wu, X.; Wang, Q.; Liu, P.; Song, L.; et al. Boosting with heterologous vaccines effectively improves protective immune responses of the inactivated SARS-CoV-2 vaccine. Emerg. Microbes Infect. 2021, 10, 1598–1608. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; St Denis, K.J.; Hoelzemer, A.; Lam, E.C.; Nitido, A.D.; Sheehan, M.L.; Berrios, C.; Ofoman, O.; Chang, C.C.; Hauser, B.M.; et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 2022, 185, 457–466.e454. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Ye, Q. The Key Site Variation and Immune Challenges in SARS-CoV-2 Evolution. Vaccines 2023, 11, 1472. https://doi.org/10.3390/vaccines11091472
Liu Y, Ye Q. The Key Site Variation and Immune Challenges in SARS-CoV-2 Evolution. Vaccines. 2023; 11(9):1472. https://doi.org/10.3390/vaccines11091472
Chicago/Turabian StyleLiu, Ying, and Qing Ye. 2023. "The Key Site Variation and Immune Challenges in SARS-CoV-2 Evolution" Vaccines 11, no. 9: 1472. https://doi.org/10.3390/vaccines11091472
APA StyleLiu, Y., & Ye, Q. (2023). The Key Site Variation and Immune Challenges in SARS-CoV-2 Evolution. Vaccines, 11(9), 1472. https://doi.org/10.3390/vaccines11091472