
Deep Structural Analysis of Myriads of Omicron Sub-Variants Revealed Hotspot for Vaccine Escape Immunity


 ,
,  and
and 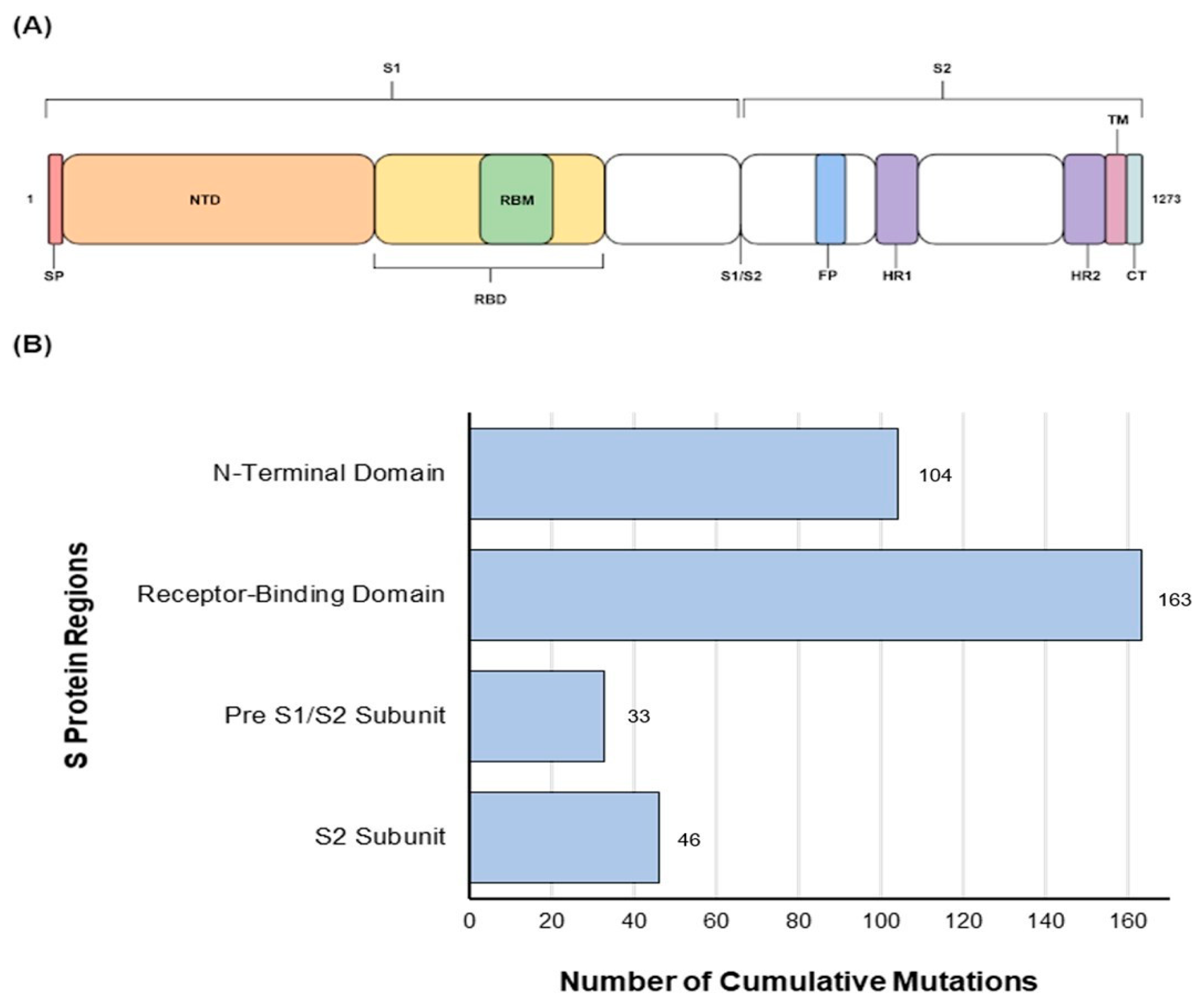{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Complete Genome Sequence Acquisition
2.2. Extraction of the S Gene Sequences
2.3. Amino Acid Analyses of the S Protein
2.4. RBD 3D Modelling for Amino Acid Substitutions
2.5. Phylogenetic Analysis
3. Results
3.1. Amino Acid Substitution Analyses
3.2. Pre S1/S2 Subunit Substitutions
3.3. S2 Subunit Substitutions
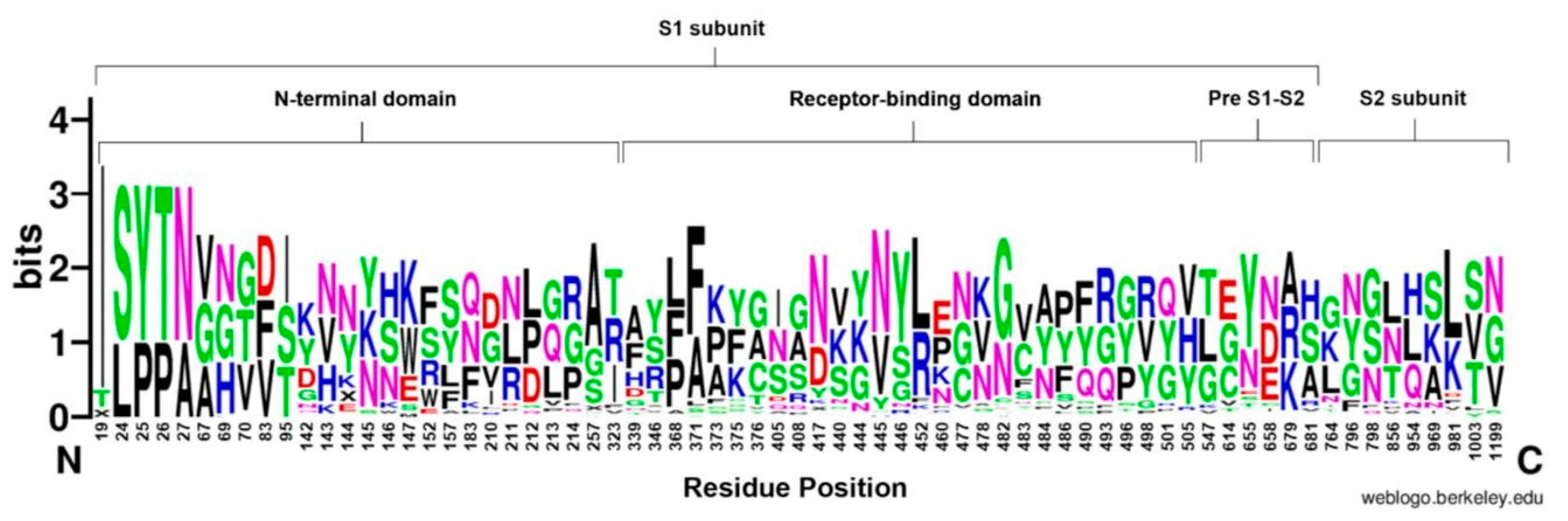
3.4. Amino Acids Conservation and Variation
3.4.1. N-Terminal Domain
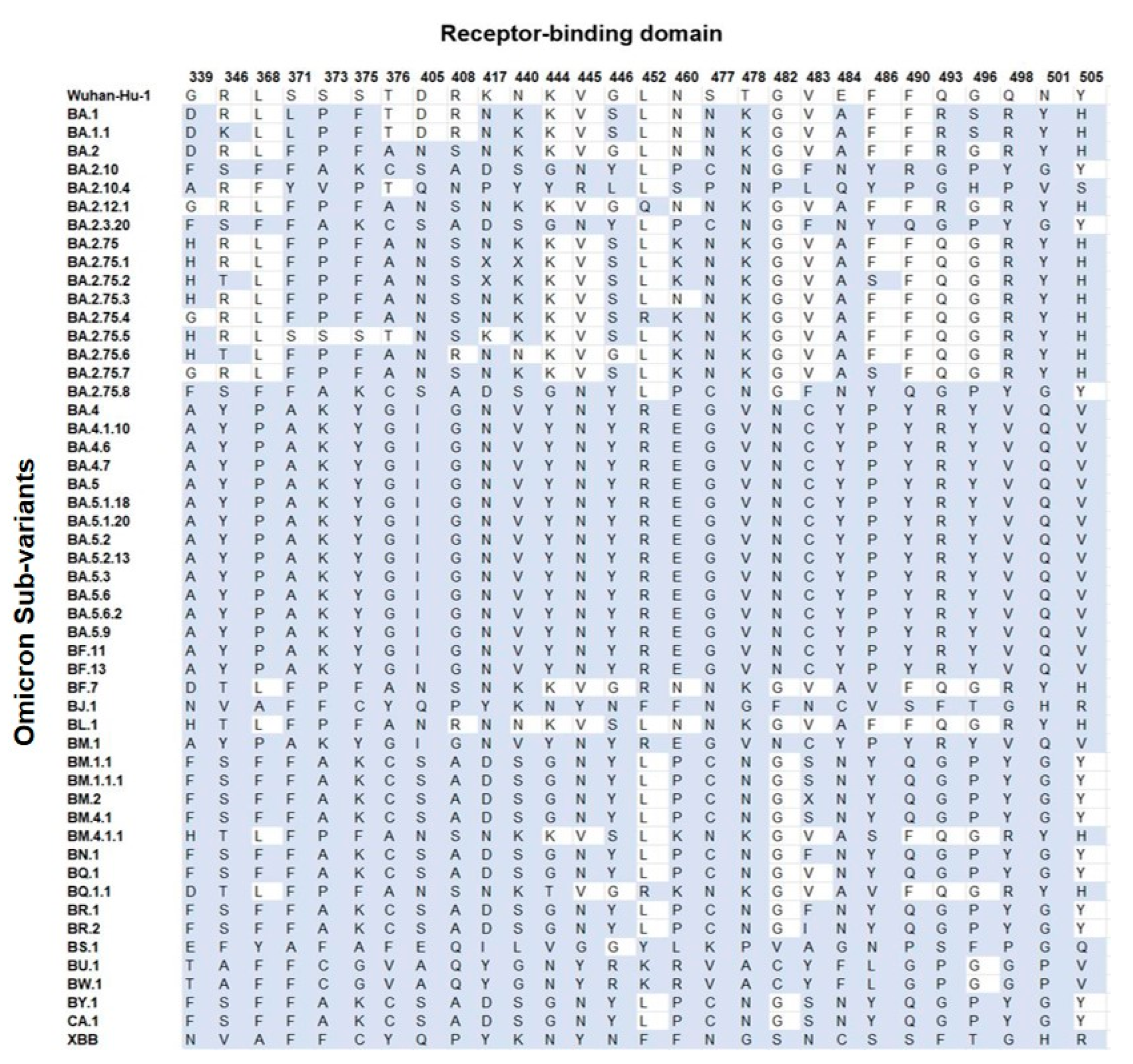
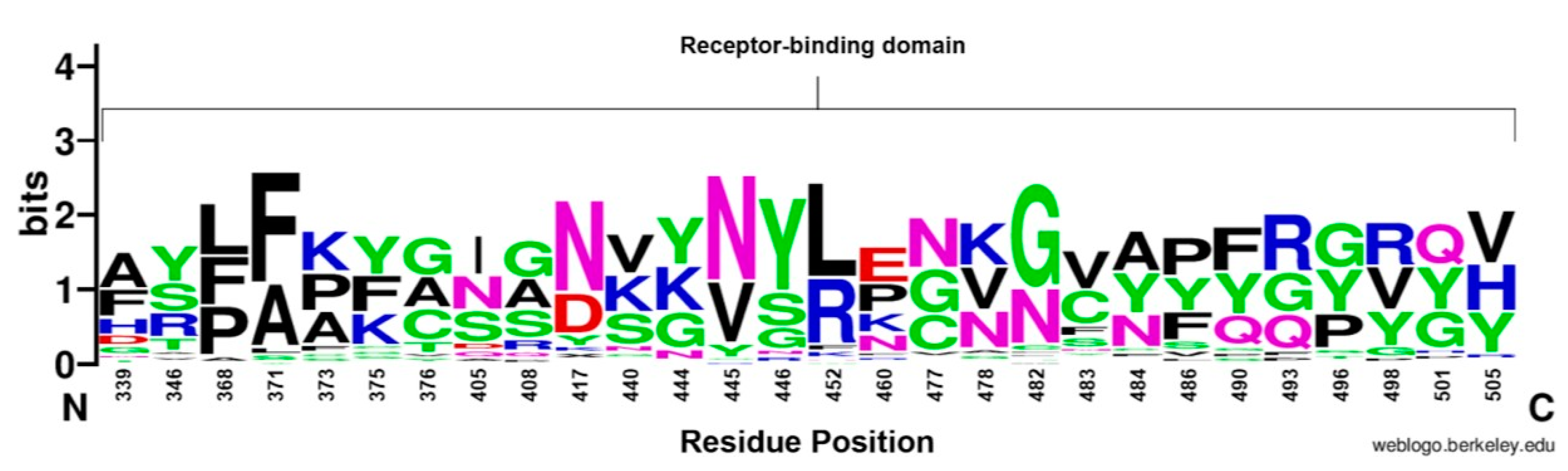
3.4.2. Receptor-Binding Domain
3.4.3. Pre S1/S2 Subunit and S2 Subunit
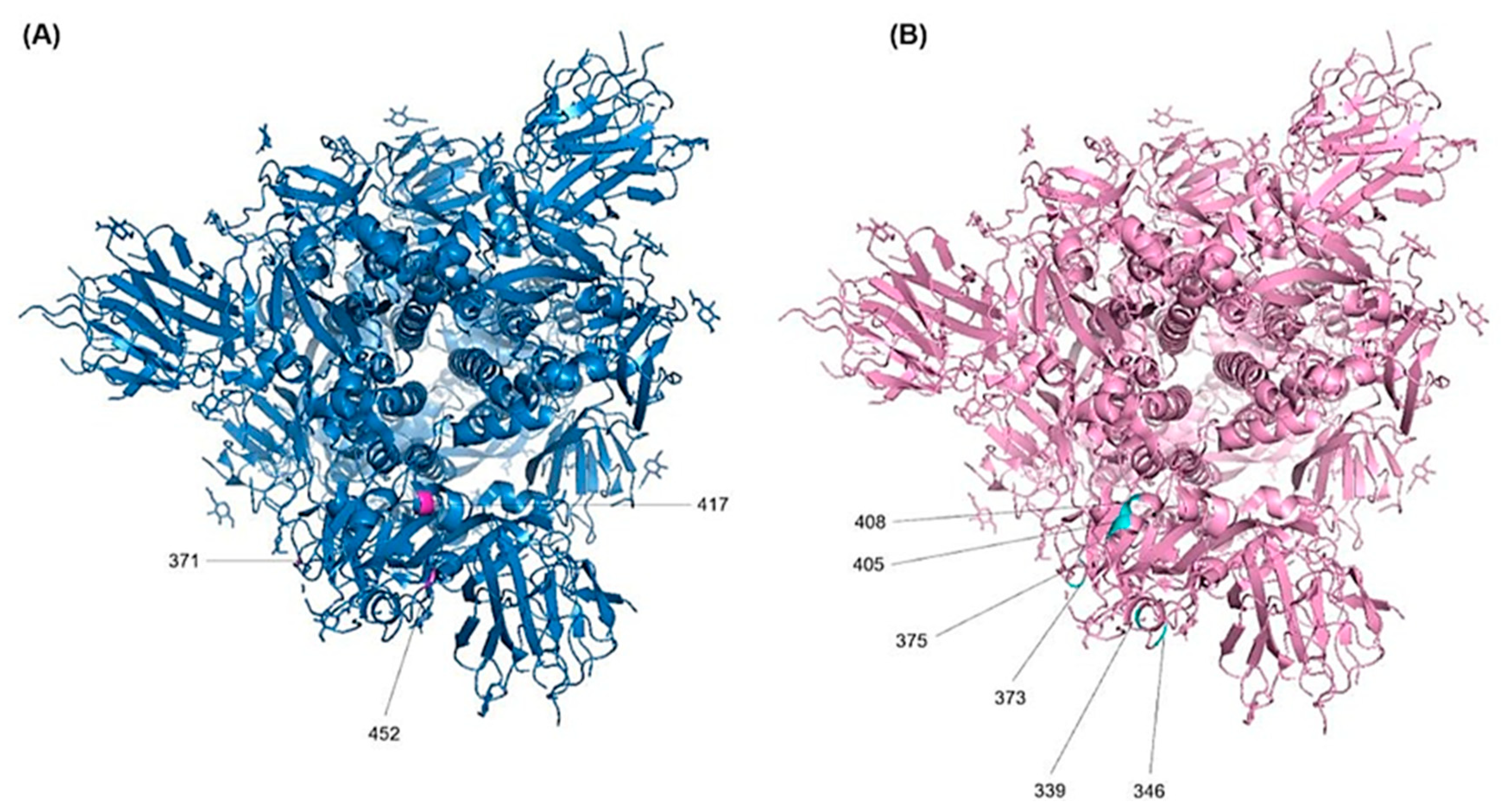
3.4.4. D Visualization for the RBD Mutations
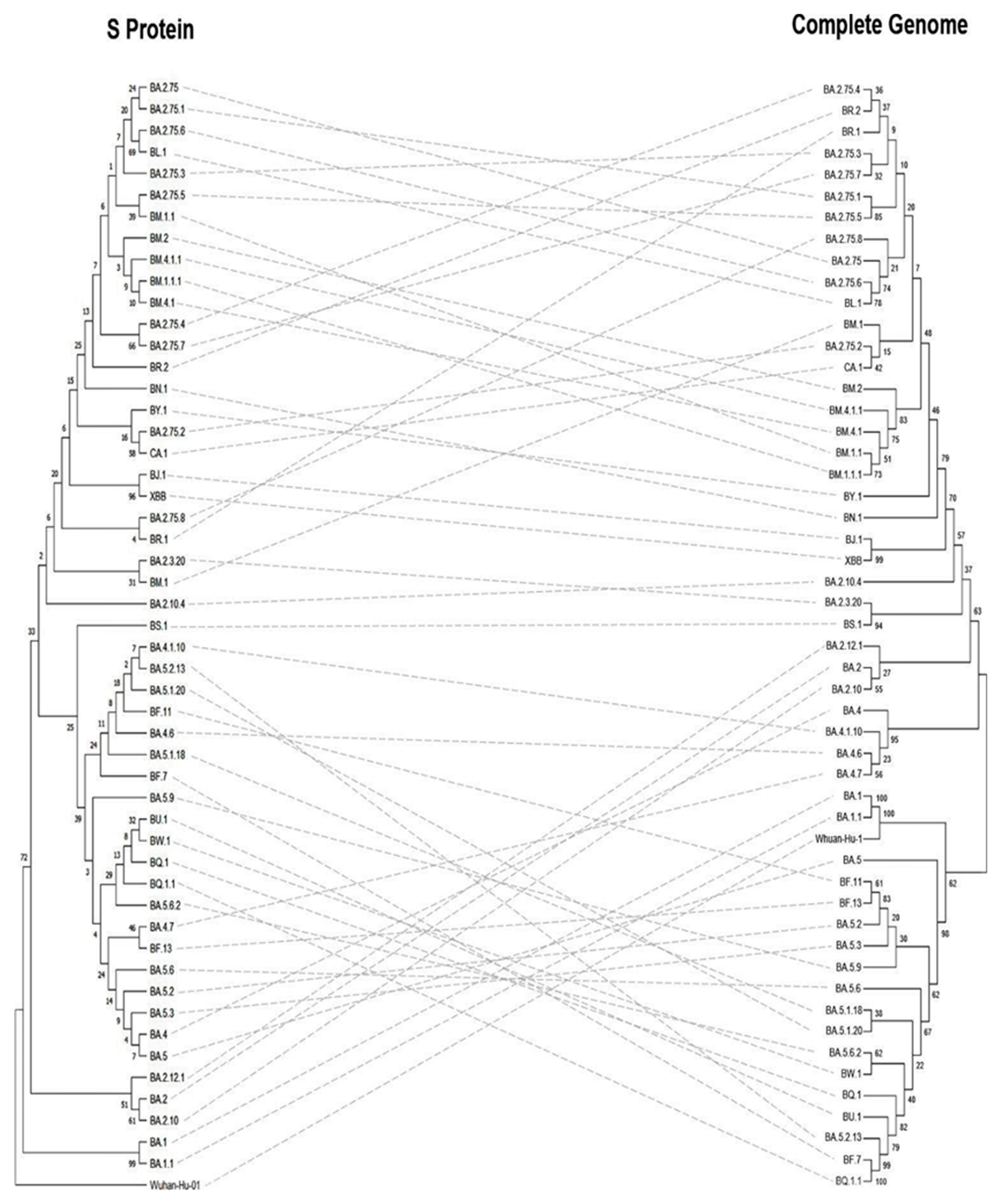
3.5. Phylogeny and Evolution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. WHO Director-General’s Remarks at the Media Briefing on 2019-nCoV on 11 February 2020. 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-2019-ncov-on-11-february-2020 (accessed on 12 February 2023).
- Rastogi, M.; Pandey, N.; Shukla, A.; Singh, S.K. SARS coronavirus 2: From genome to infectome. Respir. Res. 2020, 21, 318. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Rohaim, M.A.; El Naggar, R.F.; Clayton, E.; Munir, M. Structural and functional insights into non-structural proteins of coronaviruses. Microb. Pathog. 2021, 150, 104641. [Google Scholar] [CrossRef]
- Gupta, D.; Sharma, P.; Singh, M.; Kumar, M.; Ethayathulla, A.S.; Kaur, P. Structural and functional insights into the spike protein mutations of emerging SARS-CoV-2 variants. Cell. Mol. Life Sci. 2021, 78, 7967–7989. [Google Scholar] [CrossRef] [PubMed]
- Peisahovics, F.; Rohaim, M.A.; Munir, M. Structural topological analysis of spike proteins of SARS-CoV-2 variants of concern highlight distinctive amino acid substitution patterns. Eur. J. Cell Biol. 2022, 101, 151275. [Google Scholar] [CrossRef]
- Clayton, E.; Ackerley, J.; Aelmans, M.; Ali, N.; Ashcroft, Z.; Ashton, C.; Barker, R.; Budryte, V.; Burrows, C.; Cai, S.; et al. Structural Bases of Zoonotic and Zooanthroponotic Transmission of SARS-CoV-2. Viruses 2022, 14, 418. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Li, Q.; Nie, J.; Wu, J.; Zhang, L.; Ding, R.; Wang, H.; Zhang, Y.; Li, T.; Liu, S.; Zhang, M.; et al. SARS-CoV-2 501Y.V2 variants lack higher infectivity but do have immune escape. Cell 2021, 184, 2362–2371.e9. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A. COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef] [PubMed]
- WHO. Tracking SARS-CoV-2 Variants. 2023. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 12 February 2023).
- Ledford, H. How severe are Omicron infections? Nature 2021, 600, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Burki, T.K. Omicron variant and booster COVID-19 vaccines. Lancet Respir. Med. 2022, 10, e17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA.1) and sub-variants (BA.1.1, BA.2, and BA.3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. J. Med. Virol. 2022, 94, 4780–4791. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations Strengthened SARS- CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Xu, A.; Hong, B.; Lou, F.; Wang, S.; Li, W.; Shafqat, A.; An, X.; Zhao, Y.; Song, L.; Tong, Y.; et al. Sub-lineages of the SARS-CoV-2 Omicron variants: Characteristics and prevention. MedComm 2022, 3, e172. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Where did ‘weird’ Omicron come from? Science 2021, 374, 1179. [Google Scholar] [CrossRef]
- Pulliam, J.R.C.; van Schalkwyk, C.; Govender, N.; von Gottberg, A.; Cohen, C.; Groome, M.J.; Dushoff, J.; Mlisana, K.; Moultrie, H. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. Science 2022, 376, eabn4947. [Google Scholar] [CrossRef]
- Mahase, E. COVID-19: What we know about the BA.4 and BA.5 omicron variants. BMJ 2022, 378, o1969. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Da Silva, S.J.R.; Lima, S.C.; Silva, R.C. Viral load in COVID-19 patients: Implications for prognosis and vaccine efficacy in the context of emerging SARS-CoV-2 variants. Front. Med. 2021, 8, 836826. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Smith, T.; Rohaim, M.A.; Munir, M. Mapping molecular gene signatures mediated by SARS-CoV-2 and large-scale and genome-wide transcriptomics comparative analysis among respiratory viruses of medical importance. Mol. Cell. Probes 2022, 64, 101820. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.; Rohaim, M.A.; Bayoumi, M.; Munir, M. The molecular virology of coronaviruses with special reference to SARS-CoV-2. Adv. Exp. Med. Biol. 2021, 1352, 15–31. [Google Scholar] [PubMed]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kruger, N.; Schulz, S.; Cossmann, A.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Moldenhauer, A.S.; Winkler, M.S.; et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 2022, 185, 447–456.e11. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484.e15. [Google Scholar] [CrossRef]
- Shuai, H.; Chan, J.F.W.; Hu, B.; Chai, Y.; Yuen, T.T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.C.; Liu, H.; et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, L.; Peng, Z.; Chen, L.L.; Meng, X.; Zhang, C.; Ip, J.D.; Chan, W.M.; Chu, A.W.; Chan, K.H.; et al. SARS-CoV-2 Omicron variant shows less efficient replication and fusion activity when compared with Delta variant in TMPRSS2-expressed cells. Emerg. Microb. Infect. 2022, 11, 277–283. [Google Scholar] [CrossRef]
- Suzuki, R.; Yamasoba, D.; Kimura, I.; Wang, L.; Kishimoto, M.; Ito, J.; Morioka, Y.; Nao, N.; Nasser, H.; Uriu, K.; et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 2022, 603, 700–705. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Maslo, C.; Friedland, R.; Toubkin, M.; Laubscher, A.; Akaloo, T.; Kama, B. Characteristics and outcomes of hospitalized patients in South Africa during the COVID-19 omicron wave compared with 10 of 14—SILVA ET AL. previous waves. JAMA 2022, 327, 583–584. [Google Scholar] [CrossRef]
- Nyberg, T.; Ferguson, N.M.; Nash, S.G.; Webster, H.H.; Flaxman, S.; Andrews, N.; Hinsley, W.; Bernal, J.L.; Kall, M.; Bhatt, S.; et al. Comparative analysis of the risks of hospitalisation and death associated with SARS-CoV-2 omicron (B.1.1.529) and delta (B.1.617.2) variants in England: A cohort study. Lancet 2022, 399, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Hirose, R.; Itoh, Y.; Ikegaya, H.; Miyazaki, H.; Watanabe, N.; Yoshida, T.; Bandou, R.; Daidoji, T.; Nakaya, T. Differences in environmental stability among SARS-CoV-2 variants of concern: Both omicron BA.1 and BA.2 have higher stability. Clin. Microbiol. Infect. 2022, 28, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Flemming, A. Omicron, the great escape artist. Nat. Rev. Immunol. 2022, 22, 75. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- VanBlargan, L.A.; Errico, J.M.; Halfmann, P.J.; Zost, S.J.; Crowe, J.E., Jr.; Purcell, L.A.; Kawaoka, Y.; Corti, D.; Fremont, D.H.; Diamond, M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022, 28, 490–495. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Shaw, R.H.; Supasa, P.; Liu, C.; Stuart, A.S.; Pollard, A.J.; Liu, X.; Lambe, T.; Crook, D.; Stuart, D.I.; et al. Reduced neutralisation of SARS-CoV-2 omicron B.1.1.529 variant by post-immunisation serum. Lancet 2022, 399, 234–236. [Google Scholar] [CrossRef]
- Lu, L.; Mok, B.W.Y.; Chen, L.L.; Chan, J.M.C.; Tsang, O.T.Y.; Lam, B.H.S.; Chuang, V.W.M.; Chu, A.W.H.; Chan, W.M.; Ip, J.D.; et al. Neutralization of Severe Acute Respiratory Syndrome Coronavirus 2 Omicron Variant by Sera From BNT162b2 or CoronaVac Vaccine Recipients. Clin. Infect. Dis. 2022, 75, e822–e826. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; St Denis, K.J.; Hoelzemer, A.; Lam, E.C.; Nitido, A.D.; Sheehan, M.L.; Berrios, C.; Ofoman, O.; Chang, C.C.; Hauser, B.M.; et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 2022, 185, 457–466.e4. [Google Scholar] [CrossRef]
- Pérez-Then, E.; Lucas, C.; Monteiro, V.S.; Miric, M.; Brache, V.; Cochon, L.; Vogels, C.B.F.; Malik, A.A.; De la Cruz, E.; Jorge, A.; et al. Neutralizing antibodies against the SARS-CoV-2 Delta and Omicron variants following heterologous CoronaVac plus BNT162b2 booster vaccination. Nat. Med. 2022, 28, 481–485. [Google Scholar] [CrossRef]
- Lusvarghi, S.; Pollett, S.D.; Neerukonda, S.N.; Wang, W.; Wang, R.; Vassell, R.; Epsi, N.J.; Fries, A.C.; Agan, B.K.; Lindholm, D.A.; et al. SARS-CoV-2 BA.1 variant is neutralized by vaccine booster-elicited serum but evades most convalescent serum and therapeutic antibodies. Sci. Transl. Med. 2022, 14, eabn8543. [Google Scholar] [CrossRef]
- Evans, J.P.; Zeng, C.; Carlin, C.; Lozanski, G.; Saif, L.J.; Oltz, E.M.; Gumina, R.J.; Liu, S.L. Neutralizing antibody responses elicited by SARS-CoV-2 mRNA vaccination wane over time and are boosted by breakthrough infection. Sci. Transl. Med. 2022, 14, eabn8057. [Google Scholar] [CrossRef]
- Klinakis, A.; Cournia, Z.; Rampias, T. N-terminal domain mutations of the spike protein are structurally implicated in epitope recognition in emerging SARS-CoV-2 strains. Comput. Struct. Biotechnol. J. 2021, 19, 5556–5567. [Google Scholar] [CrossRef]
- Bayani, F.; Safaei Hashkavaei, N.; Uversky, V.N.; Mozaffari-Jovin, S.; Sefidbakht, Y. Insights into the structural peculiarities of the N-terminal and receptor binding domains of the spike protein from the SARS-CoV-2 Omicron variant. Comput. Biol. Med. 2022, 147, 105735. [Google Scholar] [CrossRef]
- Mukherjee, R.; Satardekar, R. Why are some coronavirus variants more infectious? J. Biosci. 2021, 46, 101. [Google Scholar] [CrossRef]
- Lan, J.; He, X.; Ren, Y.; Wang, Z.; Zhou, H.; Fan, S.; Zhu, C.; Liu, D.; Shao, B.; Liu, T.Y.; et al. Structural insights into the SARS-CoV-2 Omicron RBD-ACE2 interaction. Cell Res. 2022, 32, 593–595. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Binding Interactions between RBD of Spike-Protein and Human ACE2 in Omicron variant. bioRxiv 2022. [Google Scholar] [CrossRef]
- Rath, S.L.; Padhi, A.K.; Mandal, N. Scanning the RBD-ACE2 molecular interactions in Omicron variant. Biochem. Biophys. Res. Commun. 2022, 592, 18–23. [Google Scholar] [CrossRef]
- Dudev, T.; Doudeva, L. How the extra methylene group affects the ligation properties of Glu vs. Asp and Gln vs. Asn amino acids: A DFT/PCM study. J. Mol. Model. 2017, 23, 45. [Google Scholar] [CrossRef]
- Miller, N.L.; Clark, T.; Raman, R.; Sasisekharan, R. Insights on the mutational landscape of the SARS-CoV-2 Omicron variant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Focosi, D.; Quiroga, R.; McConnell, S.; Johnson, M.C.; Casadevall, A. Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge. Int. J. Mol. Sci. 2023, 24, 2264. [Google Scholar] [CrossRef]
- Mittal, A.; Manjunath, K.; Ranjan, R.K.; Kaushik, S.; Kumar, S.; Verma, V. COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020, 16, e1008762. [Google Scholar] [CrossRef]
- Lupala, C.S.; Li, X.; Lei, J.; Chen, H.; Qi, J.; Liu, H.; Su, X. Computational simulations reveal the binding dynamics between human ACE2 and the receptor-binding domain of SARS-CoV-2 spike protein. Quant. Biol. 2021, 9, 61–72. [Google Scholar] [CrossRef]
- Tragni, V.; Preziusi, F.; Laera, L.; Onofrio, A.; Mercurio, I.; Todisco, S.; Volpicella, M.; De Grassi, A.; Pierri, C. Modeling SARS-CoV-2 spike/ACE2 protein–protein interactions for predicting the binding affinity of new spike variants for ACE2, and novel ACE2 structurally related human protein targets, for COVID-19 handling in the 3PM context. EPMA J. 2022, 13, 149–175. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Q.; Ge, J.; Ren, W.; Zhang, R.; Lan, J.; Ju, B.; Su, B.; Yu, F.; Chen, P.; et al. Analysis of SARS-CoV-2 variant mutations reveals neutralization escape mechanisms and the ability to use ACE2 receptors from additional species. Immunity 2021, 54, 1611–1621.e5. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.; Wang, J. Surface charge changes in spike RBD mutations of SARS-CoV-2 and its variant strains alter the virus evasiveness via HSPGs: A review and mechanistic hypothesis. Front. Public Health 2022, 10, 952916. [Google Scholar] [CrossRef]
- Saifi, S.; Ravi, V.; Sharma, S.; Swaminathan, A.; Chauhan, N.S.; Pandey, R. SARS-CoV-2 VOCs, Mutational diversity and clinical outcome: Are they modulating drug efficacy by altered binding strength? Genomics 2022, 114, 110466. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Sanches, P.R.S.; Charlie-Silva, I.; Braz, H.L.B.; Bittar, C.; Freitas Calmon, M.; Rahal, P.; Cilli, E.M. Recent advances in SARS-CoV-2 Spike protein and RBD mutations comparison between new variants Alpha (B.1.1.7, United Kingdom), Beta (B.1.351, South Africa), Gamma (P.1, Brazil) and Delta (B.1.617.2, India). J. Virus Erad. 2021, 7, 100054. [Google Scholar] [CrossRef]
- Laurini, E.; Marson, D.; Aulic, S.; Fermeglia, A.; Pricl, S. Molecular rationale for SARS-CoV-2 spike circulating mutations able to escape bamlanivimab and etesevimab monoclonal antibodies. Sci. Rep. 2021, 11, 20274. [Google Scholar] [CrossRef]
- Guan, Q.; Sadykov, M.; Nugmanova, R.; Carr, M.J.; Arold, S.T.; Pain, A. The genomic variation landscape of globally-circulating clades of SARS-CoV-2 defines a genetic barcoding scheme. bioRxiv 2020. [Google Scholar] [CrossRef]
- Uriu, K.; Ito, J.; Zahradnik, J.; Fujita, S.; Kosugi, Y.; Schreiber, G.; Sato, K. Enhanced transmissibility, infectivity and immune resistance of the SARS-CoV-2 Omicron XBB.1.5 variant. Lancet Infect. Dis. 2023, 23, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wu, L.; Peng, C.; Yang, Y.; Shi, Y.; Gong, L.; Xu, Z.; Zhu, W. Predicting spike protein NTD mutations of SARS-CoV-2 causing immune evasion by molecular dynamics simulations. Phys. Chem. Chem. Phys. 2022, 24, 3410–3419. [Google Scholar] [CrossRef]
- Pastorio, C.; Zech, F.; Noettger, S.; Jung, C.; Jacob, T.; Sanderson, T.; Sparrer, K.M.J.; Kirchhoff, F. Determinants of Spike infectivity, processing, and neutralization in SARS-CoV-2 Omicron subvariants BA.1 and BA.2. Cell Host Microbe 2022, 30, 1255–1268.e5. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Wang, X.; He, X.; Zhao, X.; Zhang, Y.; Jiang, Y.; Li, M.; Cui, Y.; Chen, Y.; Qiao, R.; et al. Antibody evasion of SARS-CoV-2 Omicron BA.1, BA.1.1, BA.2, and BA.3 sub-lineages. Cell Host Microbe 2022, 30, 1077–1083.e4. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Wang, L.; Zhu, Y.; Lu, L.; Jiang, S. Origin, virological features, immune evasion and intervention of SARS-CoV-2 Omicron sublineages. Signal Transduct. Target Ther. 2022, 7, 241. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Gong, S.Y.; Chatterjee, D.; Richard, J.; Prévost, J.; Tauzin, A.; Gasser, R.; Bo, Y.; Vézina, D.; Goyette, G.; Gendron-Lepage, G.; et al. Contribution of single mutations to selected SARS-CoV-2 emerging variants spike antigenicity. Virology 2021, 563, 134–145. [Google Scholar] [CrossRef]
- Asif, A.; Ilyas, I.; Abdullah, M.; Sarfraz, S.; Mustafa, M.; Mahmood, A. The Comparison of Mutational Progression in SARS-CoV-2: A Short Updated Overview. J. Mol. Pathol. 2022, 3, 201–218. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Vankadari, N. Structure of Furin Protease Binding to SARS-CoV-2 Spike Glycoprotein and Implications for Potential Targets and Virulence. J. Phys. Chem. Lett. 2020, 11, 6655–6663. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Le, L.; Andricioaei, I. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2100943118. [Google Scholar] [CrossRef] [PubMed]
- Kuzmina, A.; Ottolenghi, A.; Korovin, D.; Cohen-Lass, I.; Atari, N.; Hu, P.; Mandelboim, M.; Rosental, B.; Rosenberg, E.; Diaz-Griffero, F.; et al. P681 mutations within the polybasic motif of spike dictate fusogenicity and syncytia formation of SARS-CoV-2 variants. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kuzmina, A.; Ottolenghi, A.; Korovin, D.; Cohen-Lass, I.; Atari, N.; Hu, P.; Mandelboim, M.; Rosental, B.; Rosenberg, E.; Diaz-Griffero, F.; et al. Changes within the P681 Spike Residue of the Polybasic Motif Dictate Fusogenicity and Syncytia Formation of Delta and Omicron Variants of SARS- CoV-2 with No Effects on Neutralization or Infectivity. Available online: https://ssrn.com/abstract=4308571 (accessed on 13 February 2023).
- Lin, L.; Li, Q.; Wang, Y.; Shi, Y. Syncytia formation during SARS-CoV-2 lung infection: A disastrous unity to eliminate lymphocytes. Cell Death Differ. 2021, 28, 2019–2021. [Google Scholar] [CrossRef]
- Lo Presti, A.; Di Martino, A.; Faggioni, G.; Giordani, F.; Fillo, S.; Anselmo, A.; Fain, V.V.; Fortunato, A.; Petralito, G.; Molinari, F.; et al. Analysis of Genomic Characteristics of SARS-CoV-2 in Italy, 29 January to 27 March 2020. Viruses 2022, 14, 472. [Google Scholar] [CrossRef]
- Maaroufi, H. The N764K and N856K mutations in SARS-CoV-2 Omicron BA.1 S protein generate potential cleavage sites for SKI-1/S1P protease. bioRxiv 2022, 21, 477298. [Google Scholar] [CrossRef]
- Rehman, S.; Hayat1, F.S.; Norin, S.; Aziz, A.; Rahman, S.U.; Haq, N.U. Epitopes Determination for Omicron: The COVID-19 New Variant. Pak. J. Zool. 2022, 55, 641–648. [Google Scholar] [CrossRef]
- Altaf, M.; Abbasi, A.M.; Amjad, M.S.; Naseer, S.; Umair, M. Wildlife as a Source of SARS-CoV-2 Evolution—A Review. Pak. J. Zool. 2022, 54, 1899–1904. [Google Scholar] [CrossRef]
- Tariq, M.H.; Akram, M.; Sharif, Y. In silico Screening of Bioactive Phytochemicals against Spike Protein of COVID-19. Pak. J. Zool. 2022, 54, 433–438. [Google Scholar]
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 2021, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zai, X.; Zhang, Z.; Xu, J.; Chen, W. Challenges and developments in universal vaccine design against SARS-CoV-2 variants. NPJ Vaccines 2022, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chandrashekar, A.; Sellers, D.; Barrett, J.; Jacob-Dolan, C.; Lifton, M.; McMahan, K.; Sciacca, M.; VanWyk, H.; Wu, C.; et al. Vaccines elicit highly conserved cellular immunity to SARS-CoV-2 Omicron. Nature 2022, 603, 493–496. [Google Scholar] [CrossRef]
- Keeton, R.; Tincho, M.B.; Ngomti, A.; Baguma, R.; Benede, N.; Suzuki, A.; Khan, K.; Cele, S.; Bernstein, M.; Karim, F.; et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 2022, 603, 488–492. [Google Scholar] [CrossRef]
- Coléon, S.; Wiedemann, A.; Surénaud, M.; Lacabaratz, C.; Hue, S.; Prague, M.; Cervantes-Gonzalez, M.; Wang, Z.; Ellis, J.; Sansoni, A.; et al. Design, immunogenicity, and efficacy of a pan-sarbecovirus dendritic-cell targeting vaccine. eBioMedicine 2022, 80, 104062. [Google Scholar] [CrossRef]
- Wang, S.; Wang, C.Y.; Kuo, H.K.; Peng, W.J.; Huang, J.H.; Kuo, B.S.; Lin, F.; Liu, Y.J.; Liu, Z.; Wu, H.T.; et al. A novel RBD-protein/peptide vaccine elicits broadly neutralizing antibodies and protects mice and macaques against SARS-CoV-2. Emerg. Microbes Infect. 2022, 11, 2724–2734. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Tiboni, M.; Casettari, L.; Illum, L. Nasal vaccination against SARS-CoV-2: Synergistic or alternative to intramuscular vaccines? Int. J. Pharm. 2021, 603, 120686. [Google Scholar] [CrossRef]
- Park, J.G.; Oladunni, F.S.; Rohaim, M.A.; Whittingham-Dowd, J.; Tollitt, J.; Hodges, M.D.J.; Fathallah, N.; Assas, M.B.; Alhazmi, W.; Almilaibary, A.; et al. Immunogenicity and protective efficacy of an intranasal live-attenuated vaccine against SARS-CoV-2. iScience 2021, 24, 102941. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerardi, V.; Rohaim, M.A.; Naggar, R.F.E.; Atasoy, M.O.; Munir, M. Deep Structural Analysis of Myriads of Omicron Sub-Variants Revealed Hotspot for Vaccine Escape Immunity. Vaccines 2023, 11, 668. https://doi.org/10.3390/vaccines11030668
Gerardi V, Rohaim MA, Naggar RFE, Atasoy MO, Munir M. Deep Structural Analysis of Myriads of Omicron Sub-Variants Revealed Hotspot for Vaccine Escape Immunity. Vaccines. 2023; 11(3):668. https://doi.org/10.3390/vaccines11030668
Chicago/Turabian StyleGerardi, Valeria, Mohammed A. Rohaim, Rania F. El Naggar, Mustafa O. Atasoy, and Muhammad Munir. 2023. "Deep Structural Analysis of Myriads of Omicron Sub-Variants Revealed Hotspot for Vaccine Escape Immunity" Vaccines 11, no. 3: 668. https://doi.org/10.3390/vaccines11030668
APA StyleGerardi, V., Rohaim, M. A., Naggar, R. F. E., Atasoy, M. O., & Munir, M. (2023). Deep Structural Analysis of Myriads of Omicron Sub-Variants Revealed Hotspot for Vaccine Escape Immunity. Vaccines, 11(3), 668. https://doi.org/10.3390/vaccines11030668