

Prophylactic Vaccination and Intratumoral Boost with HER2-Expressing Oncolytic Herpes Simplex Virus Induces Robust and Persistent Immune Response against HER2-Positive Tumor Cells

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Lines and Reagents
2.2. Recombinant Virus Construction
2.3. Cell Viability Assay
2.4. ELISA Assay to Detect Anti-HER2 and Anti-gD IgG in Mouse Serum
2.5. Quantification of the HER2 Payload
2.6. Splenocyte Isolation
2.7. ELISpot Assay
2.8. Quantification of Viral Copies by qPCR
2.9. Staining of Serum Anti-HER2 Antibody Binding to HER2-Expressing Tumor Cells
2.10. Antibody-Dependent Cell-Mediated Cytotoxicity Assay
2.11. Antibody-Dependent Cell-Mediated Phagocytosis Assay
2.12. Complement-Dependent Cytotoxicity Assay
2.13. In Vivo Studies
2.14. Statistical Analysis
3. Results
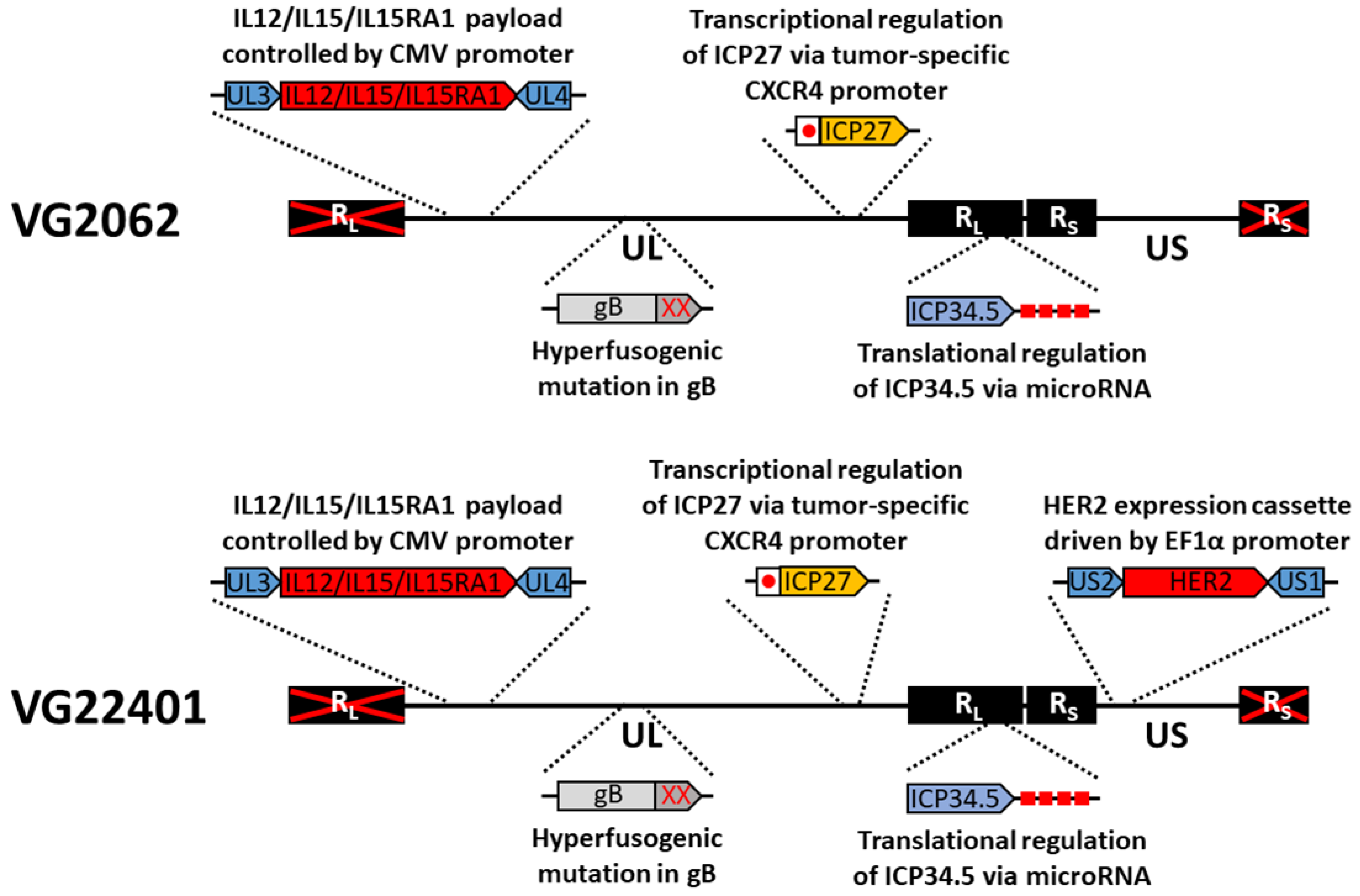
3.1. Construction and Characterization of HER2-Expressing Virus
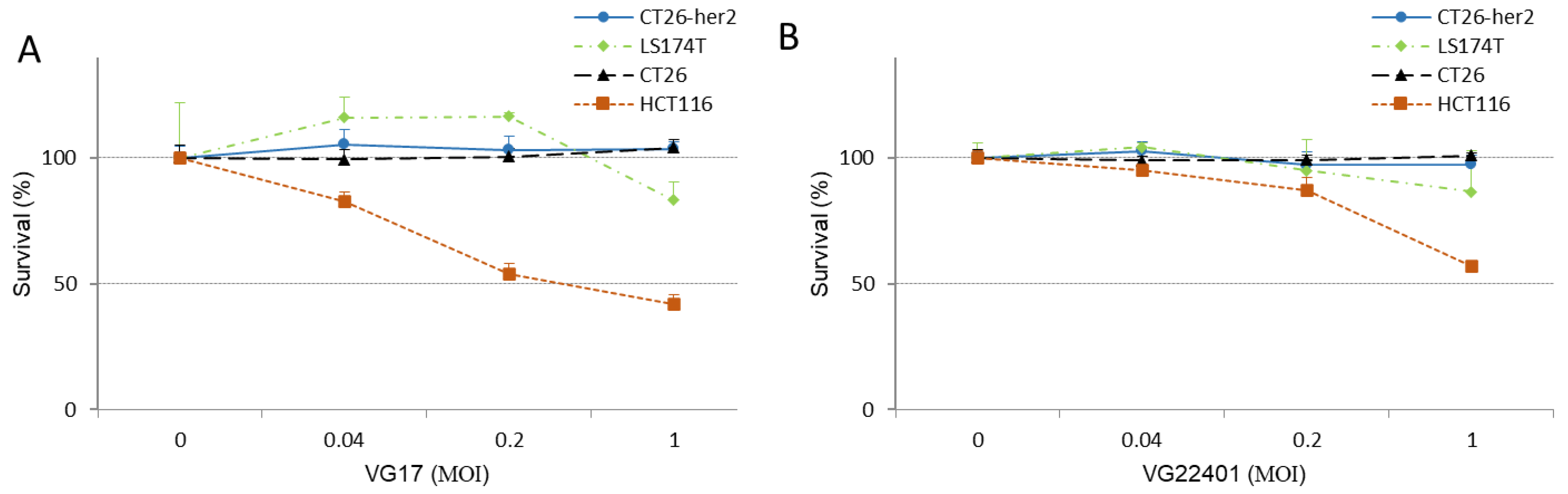
3.2. HER2-Expressing HSV-1 Displays an Antitumor Effect in Human and Mouse Colorectal Cancer Cells
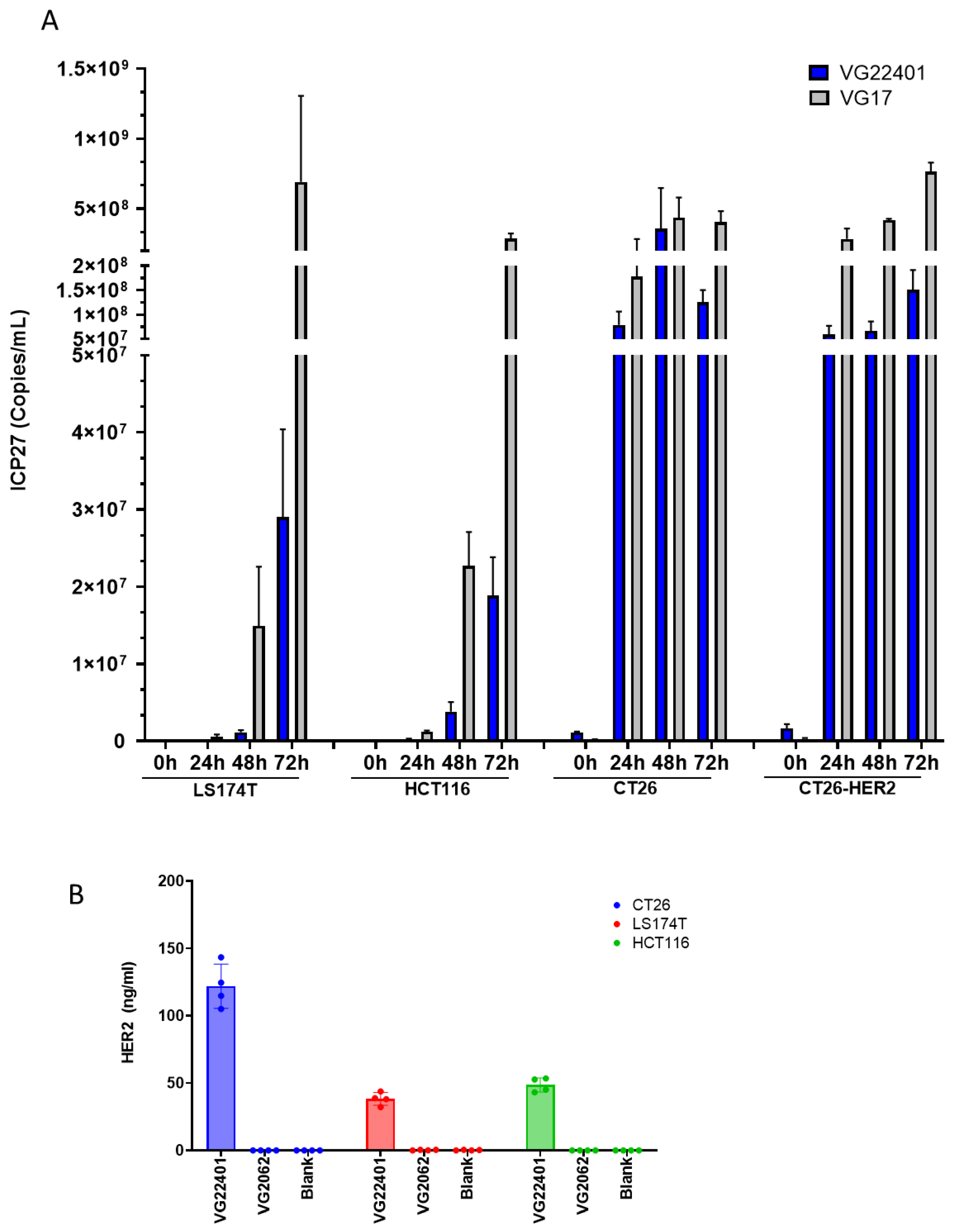
3.3. Replication and Payload Expression of HER2-Expressing HSV-1 in Human and Mouse Colorectal Cancer Cells In Vitro
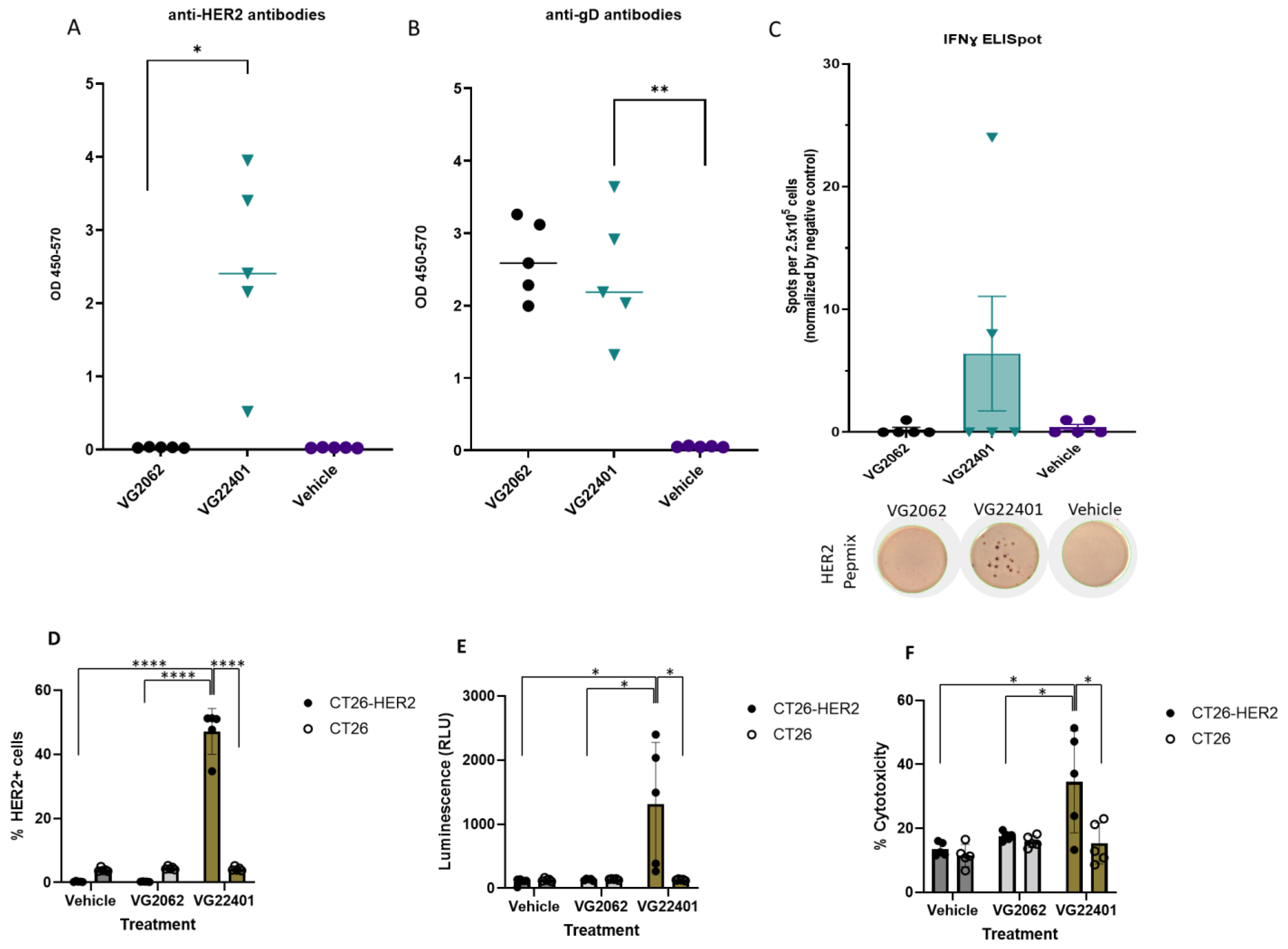
3.4. Immunization with HER2-Expressing oHSV-1 Generates Humoral and Cellular Anti-HER2 Immune Responses
3.5. Mice Vaccinated with oHSV-1 Encoding HER2 Produce Serum Antibodies That Induce Cytotoxicity against HER2-Expressing Tumor Cells
3.6. HER2 Payload Expression Enhances the Antitumor Effectiveness of oHSV-1 in a Bilateral Syngeneic HER2(+) Mouse Tumor Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hemminki, O.; dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Varghese, S.; Rabkin, S.D. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002, 9, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Wakimoto, H. Oncolytic herpes simplex virus-based strategies: Toward a breakthrough in glioblastoma therapy. Front. Microbiol. 2014, 5, 303. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Yin, J.; Markert, J.M.; Leavenworth, J.W. Modulation of the Intratumoral Immune Landscape by Oncolytic Herpes Simplex Virus Virotherapy. Front. Oncol. 2017, 7, 136. [Google Scholar] [CrossRef]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Sorolla, A.; Sorolla, M.A.; Wang, E.; Ceña, V. Peptides, proteins and nanotechnology: A promising synergy for breast cancer targeting and treatment. Expert Opin. Drug Deliv. 2020, 17, 1597–1613. [Google Scholar] [CrossRef]
- You, Z.; Zhou, W.; Weng, J.; Feng, H.; Liang, P.; Li, Y.; Shi, F. Application of HER2 peptide vaccines in patients with breast cancer: A systematic review and meta-analysis. Cancer Cell Int. 2021, 21, 489. [Google Scholar] [CrossRef]
- Riese, D.J.; Stern, D.F. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays 1998, 20, 41–48. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Alrhmoun, S.; Sennikov, S. The Role of Tumor-Associated Antigen HER2/neu in Tumor Development and the Different Approaches for Using It in Treatment: Many Choices and Future Directions. Cancers 2022, 14, 6173. [Google Scholar] [CrossRef]
- Zhu, S.-Y.; Yu, K.-D. Breast Cancer Vaccines: Disappointing or Promising? Front. Immunol. 2022, 13, 828386. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Shi, J.; Yang, C. HER2-Based Immunotherapy for Breast Cancer. Cancer Biother. Radiopharm. 2018, 33, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Guthrie, K.A.; Liu, Y.; Coveler, A.L.; Higgins, D.M.; Childs, J.S.; Dang, Y.; Salazar, L.G. Safety and Outcomes of a Plasmid DNA Vaccine Encoding the ERBB2 Intracellular Domain in Patients with Advanced-Stage ERBB2-Positive Breast Cancer: A Phase 1 Nonrandomized Clinical Trial. JAMA Oncol. 2023, 9, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Jahan, N.; Ghouse, S.M.; Martuza, R.L.; Rabkin, S.D. In Situ Cancer Vaccination and Immunovirotherapy Using Oncolytic HSV. Viruses 2021, 13, 1740. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.; Taylor, K.E.; Mossman, K.L. Innate and Adaptive Immune Responses to Herpes Simplex Virus. Viruses 2009, 1, 979–1002. [Google Scholar] [CrossRef]
- Biron, C.A.; Brossay, L. NK cells and NKT cells in innate defense against viral infections. Curr. Opin. Immunol. 2001, 13, 458–464. [Google Scholar] [CrossRef]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Jones, C.M.; Smith, C.M.; Heath, W.R.; Carbone, F.R. Rapid cytotoxic T lymphocyte activation occurs in the draining lymph nodes after cutaneous herpes simplex virus infection as a result of early antigen presentation and not the presence of virus. J. Exp. Med. 2002, 195, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Chouljenko, D.V.; Murad, Y.M.; Lee, I.-F.; Delwar, Z.; Ding, J.; Liu, G.; Liu, X.; Bu, X.; Sun, Y.; Samudio, I.; et al. Targeting carcinoembryonic antigen-expressing tumors using a novel transcriptional and translational dual-regulated oncolytic herpes simplex virus type 1. Mol. Ther. Oncolytics 2023, 28, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Delwar, Z.M.; Liu, G.; Kuo, Y.; Lee, C.; Bu, L.; Rennie, P.S.; Jia, W.W. Tumour-specific triple-regulated oncolytic herpes virus to target glioma. Oncotarget 2016, 7, 28658–28669. [Google Scholar] [CrossRef]
- Lee, C.Y.F.; Rennie, P.S.; Jia, W.W.G. MicroRNA Regulation of Oncolytic Herpes Simplex Virus-1 for Selective Killing of Prostate Cancer Cells. Clin. Cancer Res. 2009, 15, 5126–5135. [Google Scholar] [CrossRef] [PubMed]
- Chouljenko, D.V.; Ding, J.; Lee, I.-F.; Murad, Y.M.; Bu, X.; Liu, G.; Delwar, Z.; Sun, Y.; Yu, S.; Samudio, I.; et al. Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes. Biomedicines 2020, 8, 484. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Randall, J.; Smith, J.; Gilden, D.H.; Dabrowski, C.; van der Keyl, H.; Tal-Singer, R. Analysis of Individual Human Trigeminal Ganglia for Latent Herpes Simplex Virus Type 1 and Varicella-Zoster Virus Nucleic Acids Using Real-Time PCR. J. Virol. 2000, 74, 11464–11471. [Google Scholar] [CrossRef]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Piccart, M.; Procter, M.; Fumagalli, D.; de Azambuja, E.; Clark, E.; Ewer, M.S.; Restuccia, E.; Jerusalem, G.; Dent, S.; Reaby, L.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer in the APHINITY Trial: 6 Years’ Follow-Up. J. Clin. Oncol. 2021, 39, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Zaman, S.; Sharpe, S.; Helenowski, I.; Shaw, C.; Han, H.; Soliman, H.; Czerniecki, B. A brief report of toxicity end points of HER2 vaccines for the treatment of patients with HER2+ breast cancer. Drug Des. Devel. Ther. 2019, 13, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Wallace, D.R.; Gooley, T.A.; Dang, Y.; Slota, M.; Lu, H.; Coveler, A.L.; Childs, J.S.; Higgins, D.M.; Fintak, P.A.; et al. Concurrent Trastuzumab and HER2/neu-Specific Vaccination in Patients with Metastatic Breast Cancer. J. Clin. Oncol. 2009, 27, 4685. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Murad, Y.M.; Sun, Y.; Lee, I.-F.; Samudio, I.; Liu, X.; Jia, W.W.-G.; Zhao, R. Pre-Existing HSV-1 Immunity Enhances Anticancer Efficacy of a Novel Immune-Stimulating Oncolytic Virus. Viruses 2022, 14, 2327. [Google Scholar] [CrossRef] [PubMed]
- Amin, I.; Younas, S.; Afzal, S.; Shahid, M.; Idrees, M. Herpes Simplex Virus Type 1 and Host Antiviral Immune Responses: An Update. Viral. Immunol. 2019, 32, 424–429. [Google Scholar] [CrossRef]
- Chen, A.C.; Xu, R.; Wang, T.; Wei, J.; Yang, X.-Y.; Liu, C.-X.; Lei, G.; Lyerly, H.K.; Heiland, T.; Hartman, Z.C. HER2-LAMP vaccines effectively traffic to endolysosomal compartments and generate enhanced polyfunctional T cell responses that induce complete tumor regression. J. Immunother. Cancer 2020, 8, e000258. [Google Scholar] [CrossRef]
- Crosby, E.J.; Acharya, C.R.; Haddad, A.-F.; Rabiola, C.A.; Lei, G.; Wei, J.-P.; Yang, X.-Y.; Wang, T.; Liu, C.-X.; Wagner, K.U.; et al. Stimulation of Oncogene-Specific Tumor-Infiltrating T Cells through Combined Vaccine and αPD-1 Enable Sustained Antitumor Responses against Established HER2 Breast Cancer. Clin. Cancer Res. 2020, 26, 4670–4681. [Google Scholar] [CrossRef]
- Suryawanshi, R.K.; Patil, C.D.; Agelidis, A.; Koganti, R.; Yadavalli, T.; Ames, J.M.; Borase, H.; Shukla, D. Pathophysiology of reinfection by exogenous HSV-1 is driven by heparanase dysfunction. Sci. Adv. 2023, 9, eadf3977. [Google Scholar] [CrossRef] [PubMed]
- Remeijer, L.; Maertzdorf, J.; Buitenwerf, J.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. Corneal herpes simplex virus type 1 superinfection in patients with recrudescent herpetic keratitis. Invest. Ophthalmol. Vis. Sci. 2002, 43, 358–363. [Google Scholar] [PubMed]
- Blank, H.; Haines, H.G. Experimental human reinfection with herpes simplex virus. J. Invest. Dermatol. 1973, 61, 223–225. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delwar, Z.; Tatsiy, O.; Chouljenko, D.V.; Lee, I.-F.; Liu, G.; Liu, X.; Bu, L.; Ding, J.; Singh, M.; Murad, Y.M.; et al. Prophylactic Vaccination and Intratumoral Boost with HER2-Expressing Oncolytic Herpes Simplex Virus Induces Robust and Persistent Immune Response against HER2-Positive Tumor Cells. Vaccines 2023, 11, 1805. https://doi.org/10.3390/vaccines11121805
Delwar Z, Tatsiy O, Chouljenko DV, Lee I-F, Liu G, Liu X, Bu L, Ding J, Singh M, Murad YM, et al. Prophylactic Vaccination and Intratumoral Boost with HER2-Expressing Oncolytic Herpes Simplex Virus Induces Robust and Persistent Immune Response against HER2-Positive Tumor Cells. Vaccines. 2023; 11(12):1805. https://doi.org/10.3390/vaccines11121805
Chicago/Turabian StyleDelwar, Zahid, Olga Tatsiy, Dmitry V. Chouljenko, I-Fang Lee, Guoyu Liu, Xiaohu Liu, Luke Bu, Jun Ding, Manu Singh, Yanal M. Murad, and et al. 2023. "Prophylactic Vaccination and Intratumoral Boost with HER2-Expressing Oncolytic Herpes Simplex Virus Induces Robust and Persistent Immune Response against HER2-Positive Tumor Cells" Vaccines 11, no. 12: 1805. https://doi.org/10.3390/vaccines11121805
APA StyleDelwar, Z., Tatsiy, O., Chouljenko, D. V., Lee, I.-F., Liu, G., Liu, X., Bu, L., Ding, J., Singh, M., Murad, Y. M., & Jia, W. W.-G. (2023). Prophylactic Vaccination and Intratumoral Boost with HER2-Expressing Oncolytic Herpes Simplex Virus Induces Robust and Persistent Immune Response against HER2-Positive Tumor Cells. Vaccines, 11(12), 1805. https://doi.org/10.3390/vaccines11121805