
CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond

 ,
,  , , , , , , , , , , and
, , , , , , , , , , and
Abstract
:1. Introduction
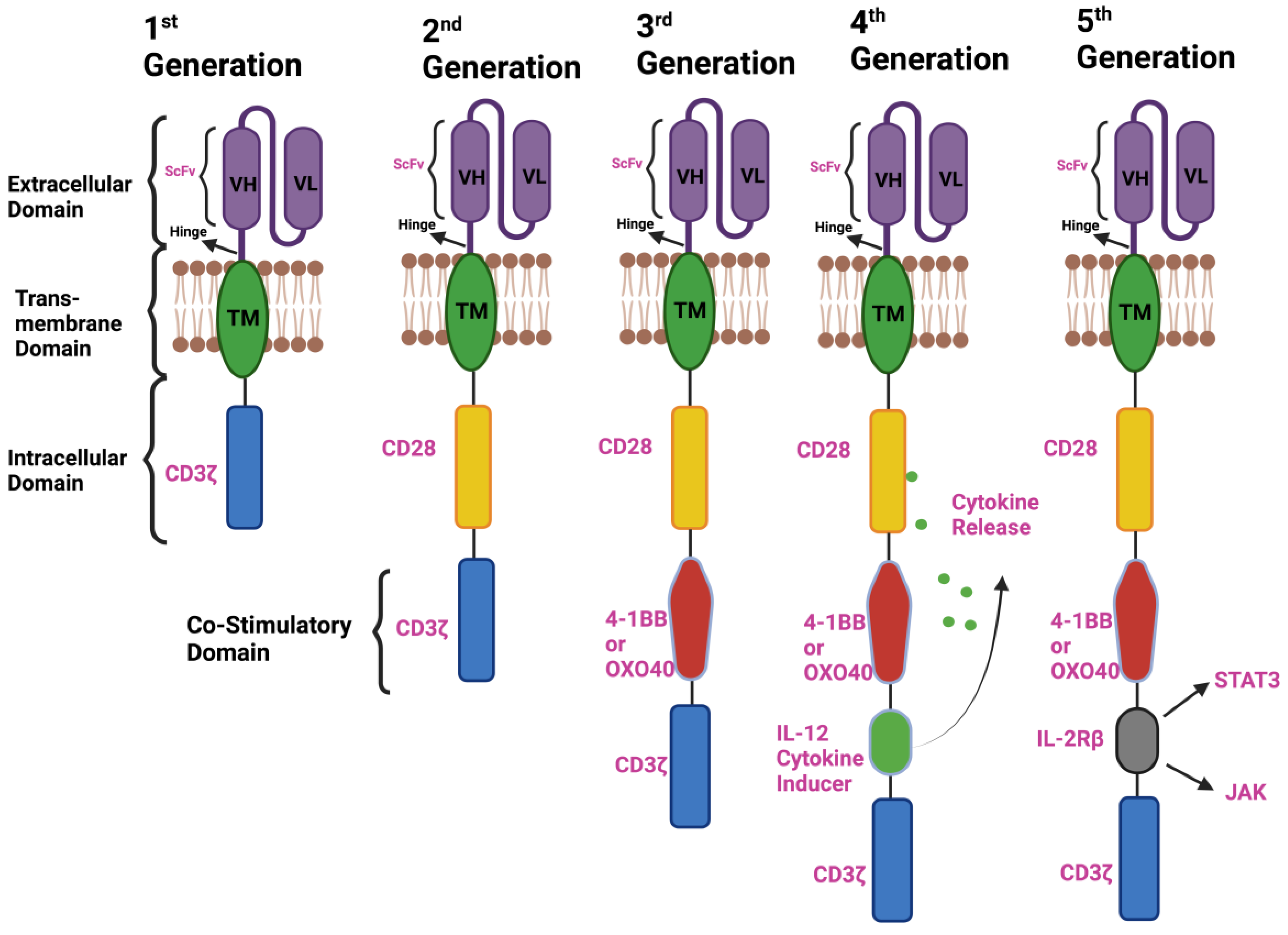
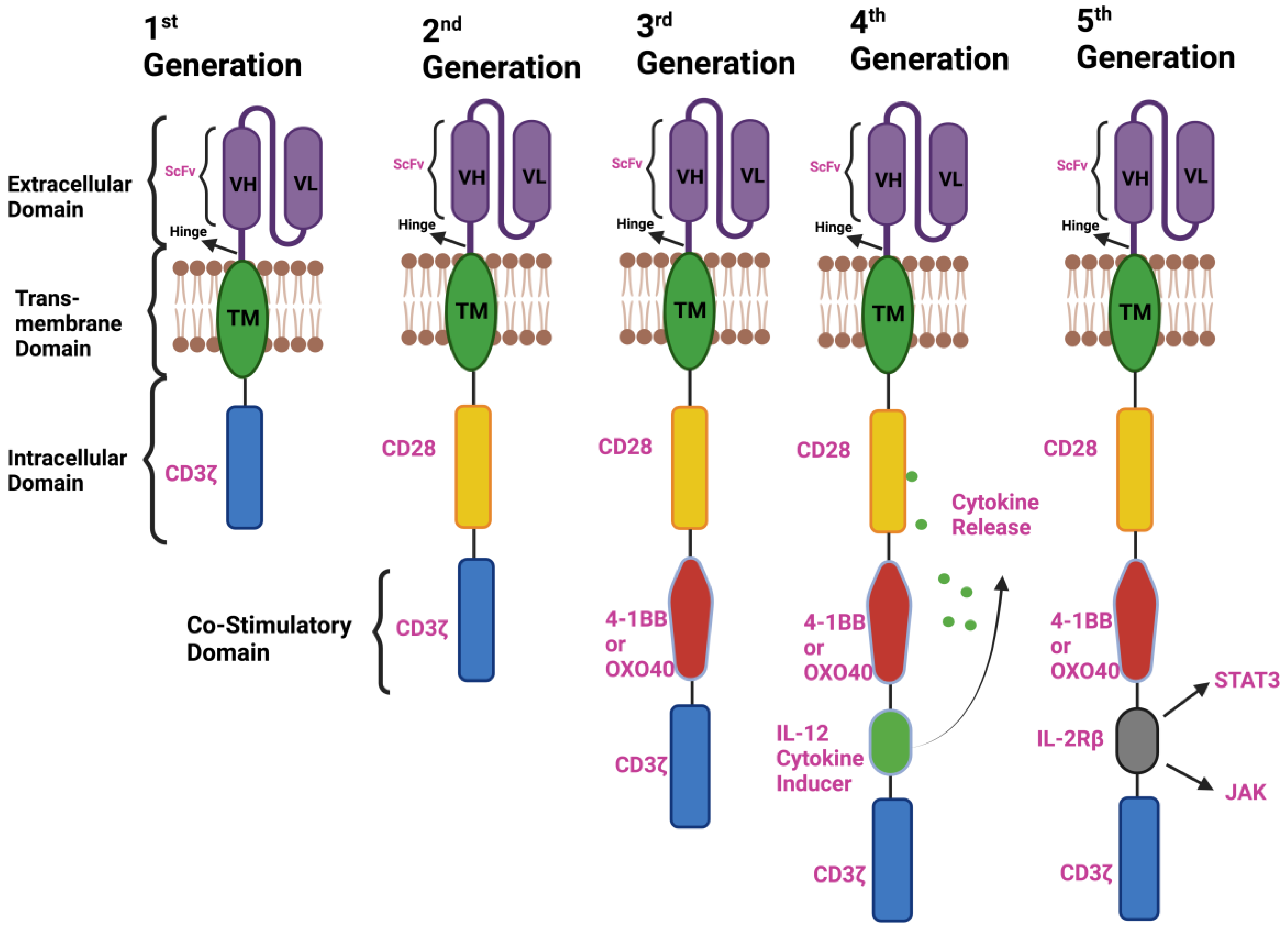
2. Components of CAR-T-Cell Therapy and the Emergence of a Different Generation of CARs
2.1. First-Generation CAR T Cells
2.2. Second-Generation CAR T Cells
2.3. Third-Generation CAR T Cells
2.4. Fourth-Generation CAR T Cells
2.5. Next or Fifth-Generation CAR T Cells
3. Emergence of B-Cell Maturation Antigen (BCMA) as a Promising Target for CAR-T-Cell Therapy in Multiple Myeloma
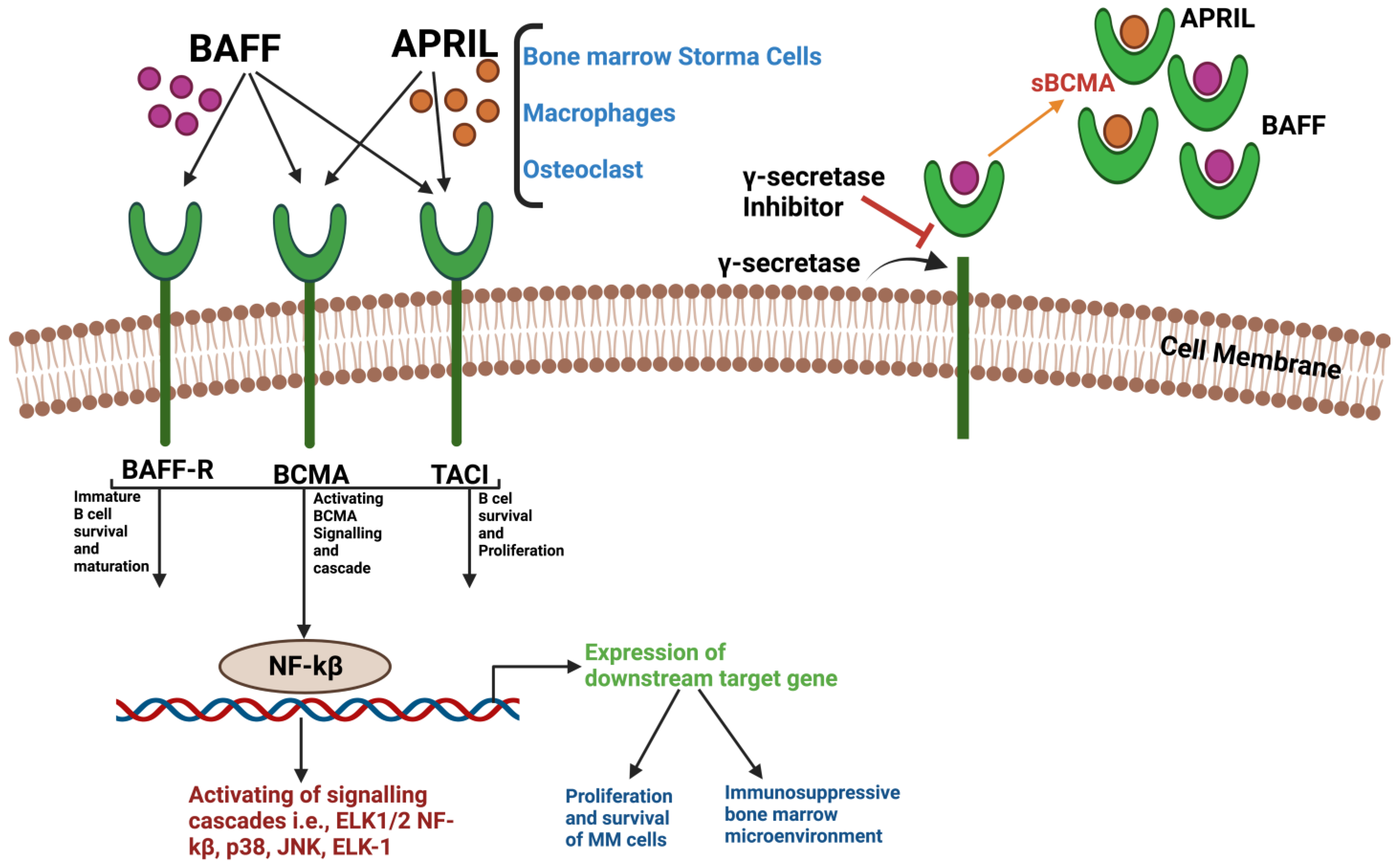
3.1. Structure and Interaction of BCMA
3.2. Idecabtagene Vicleucel (Ide-Cel, bb2121)
3.3. Ciltacabtagene Autoleucel
3.4. JCARH125 (Orva-Cel)
3.5. JNJ-4528 (LCAR-B38M)
4. Non-BCMA CAR-T-Cell Targets
4.1. GPRC5D
4.2. SLAMF7/CS1
4.3. CD38
4.4. CD19
4.5. CD138
4.6. NKG2D
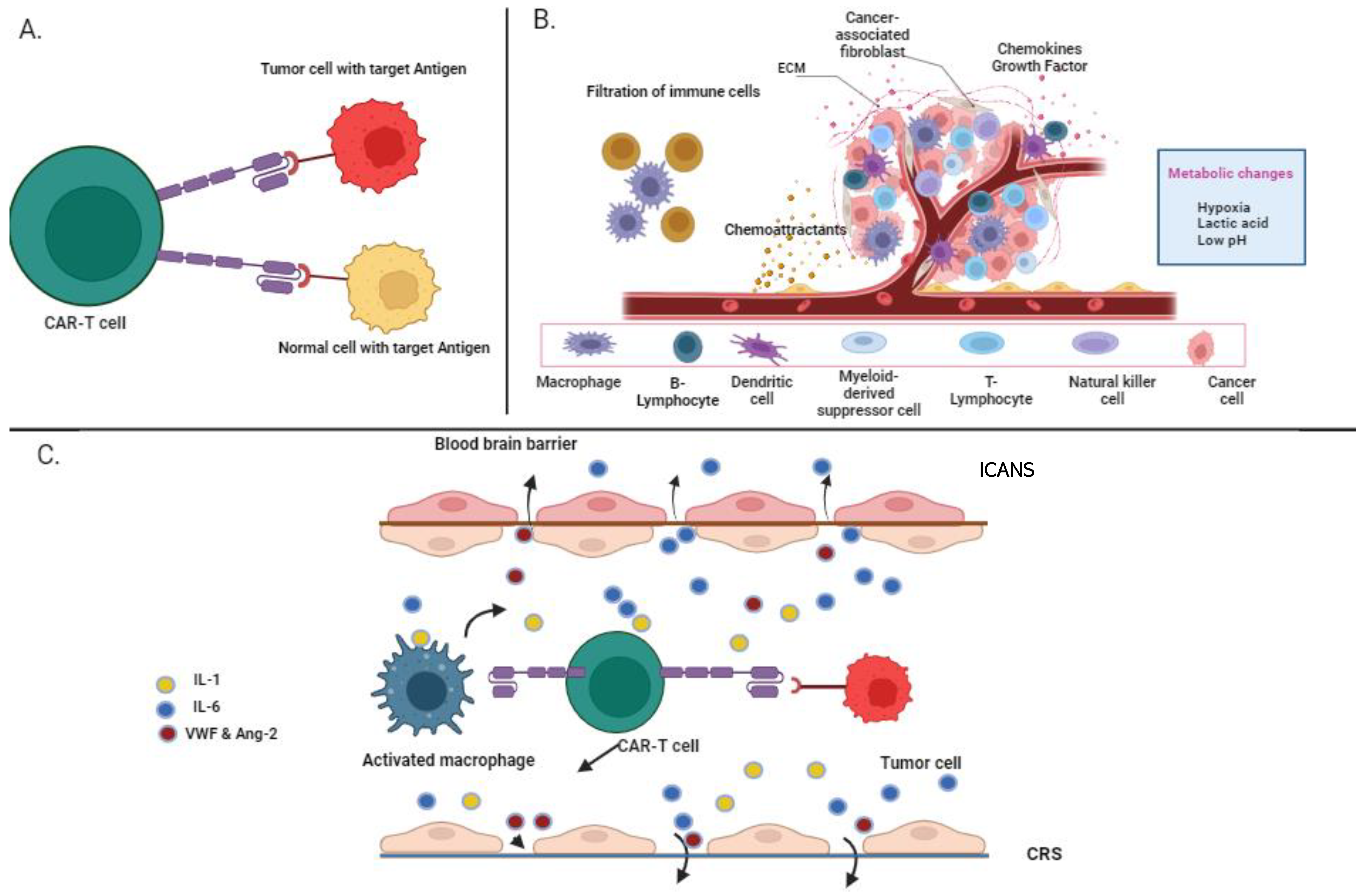
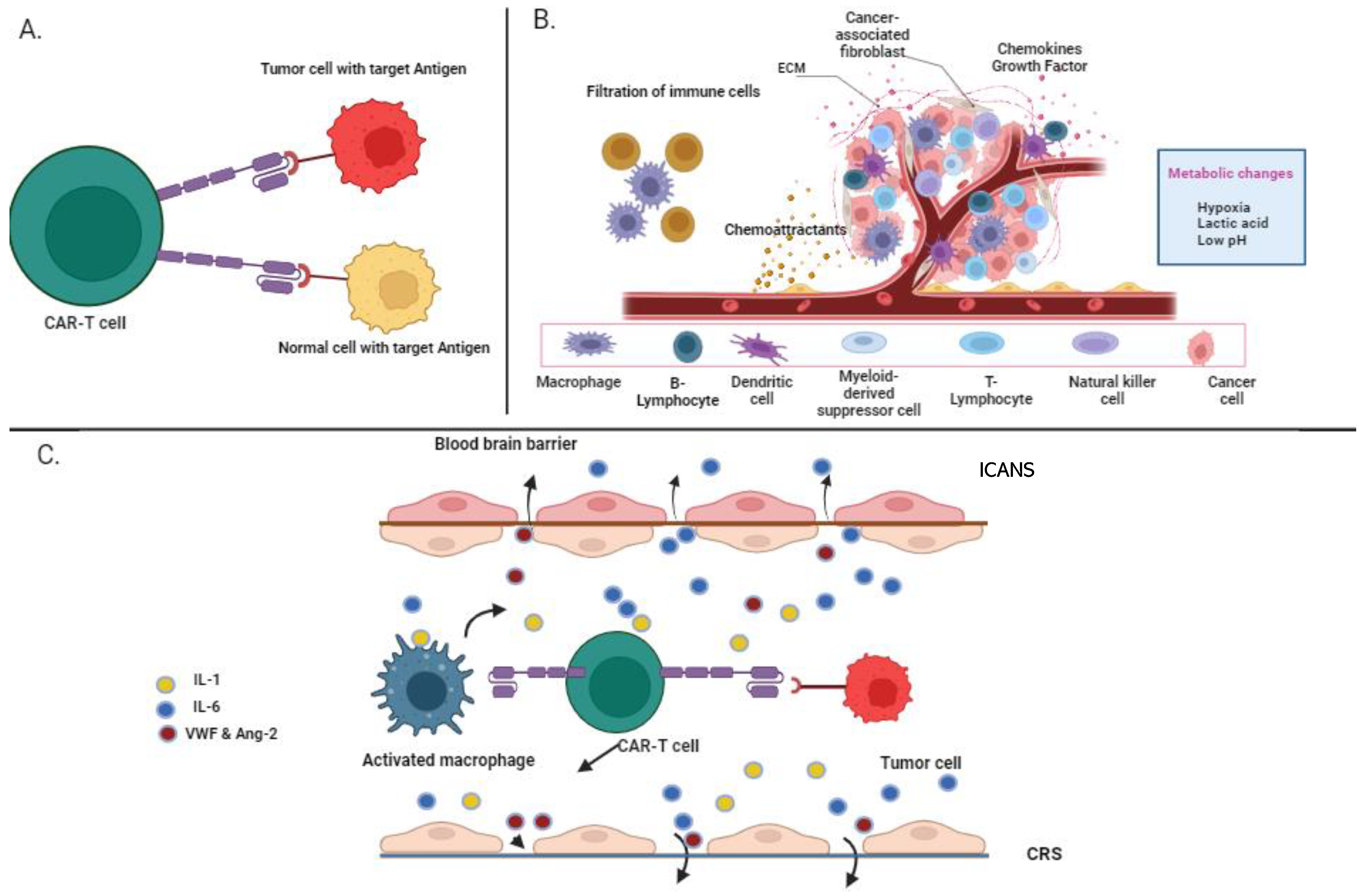
5. Limitations of CAR-T-Cell Therapy in Multiple Myeloma
5.1. On-Target, Off-Tumor
5.2. Cytokine Release Syndrome
5.3. Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)
5.4. Immunosuppressive Microenvironment
6. Recent Advances in BCMA Bispecific Antibodies
6.1. Comparison with BCMA CAR-T Therapy
6.2. Potential Synergies and Complementary Roles
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajkumar, S.V. Multiple Myeloma: 2022 Update on Diagnosis, Risk Stratification, and Management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef]
- Brown, R.D.; Pope, B.; Murray, A.; Esdale, W.; Sze, D.M.; Gibson, J.; Ho, P.J.; Hart, D.; Joshua, D. Dendritic Cells from Patients with Myeloma Are Numerically Normal but Functionally Defective as They Fail to Up-Regulate CD80 (B7-1) Expression after huCD40LT Stimulation Because of Inhibition by Transforming Growth Factor-Beta1 and Interleukin-10. Blood 2001, 98, 2992–2998. [Google Scholar] [CrossRef]
- Brimnes, M.K.; Svane, I.M.; Johnsen, H.E. Impaired Functionality and Phenotypic Profile of Dendritic Cells from Patients with Multiple Myeloma. Clin. Exp. Immunol. 2006, 144, 76–84. [Google Scholar] [CrossRef]
- Urashima, M.; Ogata, A.; Chauhan, D.; Hatziyanni, M.; Vidriales, M.B.; Dedera, D.A.; Schlossman, R.L.; Anderson, K.C. Transforming Growth Factor-Beta1: Differential Effects on Multiple Myeloma versus Normal B Cells. Blood 1996, 87, 1928–1938. [Google Scholar] [CrossRef]
- Kazmi, S.M.; Nusrat, M.; Gunaydin, H.; Cornelison, A.M.; Shah, N.; Kebriaei, P.; Nieto, Y.; Parmar, S.; Popat, U.R.; Oran, B.; et al. Outcomes among High-Risk and Standard-Risk Multiple Myeloma Patients Treated with High-Dose Chemotherapy and Autologous Hematopoietic Stem-Cell Transplantation. Clin. Lymphoma Myeloma Leuk. 2015, 15, 687–693. [Google Scholar] [CrossRef]
- AlDallal, S.M. Yescarta: A New Era for Non-Hodgkin Lymphoma Patients. Cureus 2020, 12, e11504. [Google Scholar] [CrossRef]
- Ali, S.; Kjeken, R.; Niederlaender, C.; Markey, G.; Saunders, T.S.; Opsata, M.; Moltu, K.; Bremnes, B.; Grønevik, E.; Muusse, M.; et al. The European Medicines Agency Review of Kymriah (Tisagenlecleucel) for the Treatment of Acute Lymphoblastic Leukemia and Diffuse Large B-Cell Lymphoma. Oncologist 2020, 25, e321–e327. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research; FDA. Approves Brexucabtagene Autoleucel for Relapsed or Refractory Mantle Cell Lymphoma; FDA: Silver Spring, MD, USA, 2021.
- Bourbon, E.; Ghesquières, H.; Bachy, E. CAR-T Cells, from Principle to Clinical Applications. Bull. Cancer 2021, 108, S4–S17. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Ramos, C.A.; Dotti, G. Chimeric Antigen Receptor (CAR)-Engineered Lymphocytes for Cancer Therapy. Expert. Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T Cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Non-Signaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for in Vivo Antitumor Activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef]
- Chandran, S.S.; Klebanoff, C.A. T Cell Receptor-Based Cancer Immunotherapy: Emerging Efficacy and Pathways of Resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Romeo, C.; Seed, B. Cellular Immunity to HIV Activated by CD4 Fused to T Cell or Fc Receptor Polypeptides. Cell 1991, 64, 1037–1046. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-Cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically Targeted T Cells Eradicate Systemic Acute Lymphoblastic Leukemia Xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef]
- Hwu, P.; Yang, J.C.; Cowherd, R.; Treisman, J.; Shafer, G.E.; Eshhar, Z.; Rosenberg, S.A. In Vivo Antitumor Activity of T Cells Redirected with Chimeric Antibody/T-Cell Receptor Genes. Cancer Res. 1995, 55, 3369–3373. [Google Scholar]
- Moritz, D.; Wels, W.; Mattern, J.; Groner, B. Cytotoxic T Lymphocytes with a Grafted Recognition Specificity for ERBB2-Expressing Tumor Cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4318–4322. [Google Scholar] [CrossRef]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-Mediated Signalling Co-Stimulates Murine T Cells and Prevents Induction of Anergy in T-Cell Clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric Receptors Providing Both Primary and Costimulatory Signaling in T Cells from a Single Gene Product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [CrossRef]
- Valiullina, A.K.; Zmievskaya, E.A.; Ganeeva, I.A.; Zhuravleva, M.N.; Garanina, E.E.; Rizvanov, A.A.; Petukhov, A.V.; Bulatov, E.R. Evaluation of CAR-T Cells’ Cytotoxicity against Modified Solid Tumor Cell Lines. Biomedicines 2023, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-Specific T Cell Activation by Recombinant Immunoreceptors: CD3 Zeta Signaling and CD28 Costimulation Are Simultaneously Required for Efficient IL-2 Secretion and Can Be Integrated into One Combined CD28/CD3 Zeta Signaling Receptor Molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric Receptors with 4-1BB Signaling Capacity Provoke Potent Cytotoxicity against Acute Lymphoblastic Leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.W.C.J.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-Tuned Chimeric Antigen Receptor-Engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered with an Inducible Cytokine to Modulate the Tumor Stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef]
- Zhang, Q.; Nowak, I.; Vonderheid, E.C.; Rook, A.H.; Kadin, M.E.; Nowell, P.C.; Shaw, L.M.; Wasik, M.A. Activation of Jak/STAT Proteins Involved in Signal Transduction Pathway Mediated by Receptor for Interleukin 2 in Malignant T Lymphocytes Derived from Cutaneous Anaplastic Large T-Cell Lymphoma and Sezary Syndrome. Proc. Natl. Acad. Sci. USA 1996, 93, 9148–9153. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.-L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Anderson, K.C. Targeting B-Cell Maturation Antigen in Multiple Myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef]
- Ma, T.; Shi, J.; Xiao, Y.; Bian, T.; Wang, J.; Hui, L.; Wang, M.; Liu, H. Study on the Relationship Between the Expression of B Cell Mature Antigen and the Classification, Stage, and Prognostic Factors of Multiple Myeloma. Front. Immunol. 2021, 12, 724411. [Google Scholar] [CrossRef]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T Cells Expressing an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Multiple Myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-F.; Anderson, K.C.; Tai, Y.-T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-Cell Maturation Antigen Is a Promising Target for Adoptive T-Cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA Promote Human Multiple Myeloma Growth and Immunosuppression in the Bone Marrow Microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase Inhibition Increases Efficacy of BCMA-Specific Chimeric Antigen Receptor T Cells in Multiple Myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef]
- Celgene. CRB-401 A Phase 1 Study of Bb2121 in BCMA-Expressing Multiple Myeloma; clinicaltrials.gov: Bethesda, MD, USA, 2023.
- Sanoyan, D.A.; Seipel, K.; Bacher, U.; Kronig, M.-N.; Porret, N.; Wiedemann, G.; Daskalakis, M.; Pabst, T. Real-Life Experiences with CAR T-Cell Therapy with Idecabtagene Vicleucel (Ide-Cel) for Triple-Class Exposed Relapsed/Refractory Multiple Myeloma Patients. BMC Cancer 2023, 23, 345. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; Siegel, D.S.D.; Bensinger, W.; Cota, M.; et al. Orvacabtagene Autoleucel (Orva-Cel), a B-Cell Maturation Antigen (BCMA)-Directed CAR T Cell Therapy for Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Update of the Phase 1/2 EVOLVE Study (NCT03430011). J. Clin. Oncol. 2020, 38, 8504. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Liu, J.; Wang, B.-Y.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. A Phase 1, Open-Label Study of LCAR-B38M, a Chimeric Antigen Receptor T Cell Therapy Directed against B Cell Maturation Antigen, in Patients with Relapsed or Refractory Multiple Myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Wang, B.-Y.; Chen, L.-J.; Fu, W.-J.; Xu, J.; Liu, J.; Jin, S.-W.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; et al. Four-Year Follow-up of LCAR-B38M in Relapsed or Refractory Multiple Myeloma: A Phase 1, Single-Arm, Open-Label, Multicenter Study in China (LEGEND-2). J. Hematol. Oncol. 2022, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mahendravada, A.; Ballard, B.; Kale, B.; Ramos, C.; West, J.; Maguire, T.; McKay, K.; Lichtman, E.; Tuchman, S.; et al. Safety and Efficacy of Targeting CD138 with a Chimeric Antigen Receptor for the Treatment of Multiple Myeloma. Oncotarget 2019, 10, 2369–2383. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D Is a Target for the Immunotherapy of Multiple Myeloma with Rationally Designed CAR T Cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Jiang, D.; Huang, H.; Qin, H.; Tang, K.; Shi, X.; Zhu, T.; Gao, Y.; Zhang, Y.; Tian, X.; Fu, J.; et al. Chimeric Antigen Receptor T Cells Targeting FcRH5 Provide Robust Tumour-Specific Responses in Murine Xenograft Models of Multiple Myeloma. Nat. Commun. 2023, 14, 3642. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, M.; Usmani, S.Z. GPRC5D: The Next Frontier for Immunotherapy in Multiple Myeloma. Hematologist 2023, 20. [Google Scholar] [CrossRef]
- Mailankody, S.; Devlin, S.M.; Landa, J.; Nath, K.; Diamonte, C.; Carstens, E.J.; Russo, D.; Auclair, R.; Fitzgerald, L.; Cadzin, B.; et al. GPRC5D-Targeted CAR T Cells for Myeloma. N. Engl. J. Med. 2022, 387, 1196–1206. [Google Scholar] [CrossRef]
- Bal, S.; Kocoglu, M.H.; Nadeem, O.; Htut, M.; Gregory, T.; Anderson, L.D., Jr.; Costa, L.J.; Buchholz, T.J.; Ziyad, S.; Li, M.; et al. Clinical Activity of BMS-986393 (CC-95266), a G Protein-Coupled Receptor Class C Group 5 Member D (GPRC5D)-Targeted Chimeric Antigen Receptor (CAR) T Cell Therapy, in Patients with Relapsed and/or Refractory (R/R) Multiple Myeloma (MM): First Results from a Phase 1, Multicenter, Open-Label Study. Blood 2022, 140, 883–885. [Google Scholar] [CrossRef]
- Xia, J.; Li, H.; Yan, Z.; Zhou, D.; Wang, Y.; Qi, Y.; Cao, J.; Li, D.; Cheng, H.; Sang, W.; et al. Anti–G Protein–Coupled Receptor, Class C Group 5 Member D Chimeric Antigen Receptor T Cells in Patients with Relapsed or Refractory Multiple Myeloma: A Single-Arm, Phase Ⅱ Trial. J. Clin. Oncol. 2023, 41, 2583–2593. [Google Scholar] [CrossRef]
- Zhang, M.; Wei, G.; Zhou, L.; Zhou, J.; Chen, S.; Zhang, W.; Wang, D.; Luo, X.; Cui, J.; Huang, S.; et al. GPRC5D CAR T Cells (OriCAR-017) in Patients with Relapsed or Refractory Multiple Myeloma (POLARIS): A First-in-Human, Single-Centre, Single-Arm, Phase 1 Trial. Lancet Haematol. 2023, 10, e107–e116. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Safety and Efficacy of Anti-BCMA/GPRC5D CAR-T Cell Therapy in Treating Relapsed and Refractory Multiple Myeloma(Rr/MM). Available online: https://clinicaltrials.gov/ct2/show/NCT05509530 (accessed on 26 June 2023).
- ClinicalTrials.Gov. A Study of CAR-T Cells Targeting Both BCMA and GPRC5D in Treatment of Relapsed or Refractory Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT05325801 (accessed on 26 June 2023).
- ClinicalTrials.Gov. A Study of MCARH109 and MCARH125 in People with Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT05431608 (accessed on 26 June 2023).
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM Family Receptors and SAP Adaptors in Immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Granell, M.; Oriol, A.; Martinez-Lopez, J.; Blade, J.; Hernandez, M.T.; Martín, J.; Gironella, M.; Lynch, M.; Bleickardt, E.; et al. Elotuzumab in Combination with Thalidomide and Low-Dose Dexamethasone: A Phase 2 Single-Arm Safety Study in Patients with Relapsed/Refractory Multiple Myeloma. Br. J. Haematol. 2016, 175, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T Cells Eliminate Myeloma and Confer Selective Fratricide of SLAMF7+ Normal Lymphocytes. Blood 2017, 130, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-Specific Chimeric Antigen Receptor (CAR)-Engineered Natural Killer Cells Enhance in Vitro and in Vivo Antitumor Activity against Human Multiple Myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2021, 29, 702–717. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Off-the-Shelf CAR T Cells for Multiple Myeloma. Nat. Med. 2023, 29, 303–304. [Google Scholar] [CrossRef]
- Korst, C.L.B.M.; Bruins, W.S.C.; Cosovic, M.; Verkleij, C.P.M.; Twickler, I.; Le Clerre, D.; Chion-Sotinel, I.; Zweegman, S.; Galetto, R.; Mutis, T.; et al. Preclinical Activity of Allogeneic CS1-Specific CAR T-Cells (UCARTCS1) in Multiple Myeloma. Blood 2022, 140, 4215–4216. [Google Scholar] [CrossRef]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically Optimized BCMA/CS1 Bispecific CAR-T Cells Robustly Control Heterogeneous Multiple Myeloma. Nat. Commun. 2020, 11, 2283. [Google Scholar] [CrossRef]
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A First-in-Human Clinical Trial with SLAMF7 CAR-T Cells Prepared by Virus-Free Sleeping Beauty Gene Transfer to Treat Multiple Myeloma. Gene Ther. 2021, 28, 560–571. [Google Scholar] [CrossRef]
- Leleu, X.; Martin, T.; Weisel, K.; Schjesvold, F.; Iida, S.; Malavasi, F.; Manier, S.; Min, C.-K.; Ocio, E.M.; Pawlyn, C.; et al. Anti-CD38 Antibody Therapy for Patients with Relapsed/Refractory Multiple Myeloma: Differential Mechanisms of Action and Recent Clinical Trial Outcomes. Ann. Hematol. 2022, 101, 2123–2137. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Nahi, H.; Plesner, T.; Weiss, B.M.; Bahlis, N.J.; Belch, A.; Voorhees, P.M.; Laubach, J.P.; van de Donk, N.W.; Ahmadi, T.; et al. Daratumumab Monotherapy in Patients with Heavily Pretreated Relapsed or Refractory Multiple Myeloma: Final Results from the Phase 2 GEN501 and SIRIUS Trials. Lancet Haematol. 2020, 76, e447–e455. Available online: https://pubmed.ncbi.nlm.nih.gov/32470437/ (accessed on 13 October 2023). [CrossRef]
- Ghosh, A.; Mailankody, S.; Giralt, S.A.; Landgren, C.O.; Smith, E.L.; Brentjens, R.J. CAR T Cell Therapy for Multiple Myeloma: Where Are We Now and Where Are We Headed? Leuk. Lymphoma 2018, 59, 2056–2067. [Google Scholar] [CrossRef]
- Drent, E.; Groen, R.; Noort, W.; van Bueren, J.L.; Parren, P.; Kuball, J.; Sebestyen, Z.; van de Donk, N.; Martens, A.; Lokhorst, H.; et al. Preclinical Evaluation of CD38 Chimeric Antigen Receptor Engineered T Cells for the Treatment of Multiple Myeloma. J. ImmunoTherapy Cancer 2015, 3, P13. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Study to Evaluate the Safety and Efficacy of Anti-CD38 CAR-T in Relapsed or Refractory Multiple Myeloma Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03464916 (accessed on 5 July 2023).
- Feng, Y.; Liu, X.; Li, X.; Zhou, Y.; Song, Z.; Zhang, J.; Shi, B.; Wang, J. Novel BCMA-OR-CD38 Tandem-Dual Chimeric Antigen Receptor T Cells Robustly Control Multiple Myeloma. OncoImmunology 2021, 10, 1959102. [Google Scholar] [CrossRef]
- Tang, Y.; Yin, H.; Zhao, X.; Jin, D.; Liang, Y.; Xiong, T.; Li, L.; Tang, W.; Zhang, J.; Liu, M.; et al. High Efficacy and Safety of CD38 and BCMA Bispecific CAR-T in Relapsed or Refractory Multiple Myeloma. J. Exp. Clin. Cancer Res. 2022, 41, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, M.; Xiao, X.; Lv, H.; Jiang, Y.; Li, X.; Yuan, T.; Zhao, M. A Combination of Humanized Anti-BCMA and Murine Anti-CD38 CAR-T Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma. Leuk. Lymphoma 2022, 63, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.W.C.J.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Timmers, M.; Roex, G.; Wang, Y.; Campillo-Davo, D.; Van Tendeloo, V.F.I.; Chu, Y.; Berneman, Z.N.; Luo, F.; Van Acker, H.H.; Anguille, S. Chimeric Antigen Receptor-Modified T Cell Therapy in Multiple Myeloma: Beyond B Cell Maturation Antigen. Front. Immunol. 2019, 10, 1613. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Stadtmauer, E.A.; Hwang, W.-T.; Lacey, S.F.; Melenhorst, J.J.; Krevvata, M.; Carroll, M.P.; Matsui, W.H.; Wang, Q.; Dhodapkar, M.V.; et al. Anti-CD19 CAR T Cells with High-Dose Melphalan and Autologous Stem Cell Transplantation for Refractory Multiple Myeloma. JCI Insight 2018, 3, e120505. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.; Gu, W.; Shi, M.; Lan, J.; Yan, Z.; Jin, L.; Xia, J.; Ma, S.; Liu, Y.; et al. Long-Term Follow-Up of Combination of B-Cell Maturation Antigen and CD19 Chimeric Antigen Receptor T Cells in Multiple Myeloma. J. Clin. Oncol. 2022, 40, 2246–2256. [Google Scholar] [CrossRef]
- Shi, X.; Yan, L.; Shang, J.; Kang, L.; Yan, Z.; Jin, S.; Zhu, M.; Chang, H.; Gong, F.; Zhou, J.; et al. Anti-CD19 and Anti-BCMA CAR T Cell Therapy Followed by Lenalidomide Maintenance after Autologous Stem-Cell Transplantation for High-Risk Newly Diagnosed Multiple Myeloma. Am. J. Hematol. 2022, 97, 537–547. [Google Scholar] [CrossRef]
- Gouard, S.; Pallardy, A.; Gaschet, J.; Faivre-Chauvet, A.; Bruchertseifer, F.; Morgenstern, A.; Maurel, C.; Matous, E.; Kraeber-Bodéré, F.; Davodeau, F.; et al. Comparative Analysis of Multiple Myeloma Treatment by CD138 Antigen Targeting with Bismuth-213 and Melphalan Chemotherapy. Nucl. Med. Biol. 2014, 41, e30–e35. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Treatment of Chemotherapy Refractory Multiple Myeloma by CART-138. Available online: https://clinicaltrials.gov/ct2/show/NCT01886976 (accessed on 5 July 2023).
- Guo, B.; Chen, M.; Han, Q.; Hui, F.; Dai, H.; Zhang, W.; Zhang, Y.; Wang, Y.; Zhu, H.; Han, W. CD138-Directed Adoptive Immunotherapy of Chimeric Antigen Receptor (CAR)-Modified T Cells for Multiple Myeloma. J. Cell. Immunother. 2016, 2, 28–35. [Google Scholar] [CrossRef]
- Leivas, A.; Valeri, A.; Córdoba, L.; García-Ortiz, A.; Ortiz, A.; Sánchez-Vega, L.; Graña-Castro, O.; Fernández, L.; Carreño-Tarragona, G.; Pérez, M.; et al. NKG2D-CAR-Transduced Natural Killer Cells Efficiently Target Multiple Myeloma. Blood Cancer J. 2021, 11, 146. [Google Scholar] [CrossRef]
- Spear, P.; Barber, A.; Rynda-Apple, A.; Sentman, C.L. NKG2D CAR T-Cell Therapy Inhibits the Growth of NKG2D Ligand Heterogeneous Tumors. Immunol. Cell Biol. 2013, 91, 435–440. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase 1 Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1– and CTLA-4–Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin—Specific Chimeric Antibody Receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book. 2019, 39, 433–444. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef]
- Pehlivan, K.C.; Duncan, B.B.; Lee, D.W. CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Curr. Hematol. Malig. Rep. 2018, 13, 396–406. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Czajka-Francuz, P.; Prendes, M.J.; Mankan, A.; Quintana, Á.; Pabla, S.; Ramkissoon, S.; Jensen, T.J.; Peiró, S.; Severson, E.A.; Achyut, B.R.; et al. Mechanisms of Immune Modulation in the Tumor Microenvironment and Implications for Targeted Therapy. Front. Oncol. 2023, 13, 1200646. [Google Scholar] [CrossRef]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Cipkar, C.; Chen, C.; Trudel, S. Antibodies and Bispecifics for Multiple Myeloma: Effective Effector Therapy. Hematol. Am. Soc. Hematol. Educ. Program. 2022, 2022, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Li, C.; Jiang, H.; Zhao, X.; Huang, Z.; Jin, D.; Guo, T.; Kou, H.; Liu, L.; Tang, L.; et al. A Bispecific CAR-T Cell Therapy Targeting BCMA and CD38 in Relapsed or Refractory Multiple Myeloma. J. Hematol. Oncol. 2021, 14, 161. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| (A) BCMA Targeted CAR-T for MM | |||||
| Clinical Trial Name | Antigen Target | CAR Name | Sponsor | Phase | Current Status |
| NCT05393804 | BCMA | Idecabtagene Vicleucel (ide-cel, bb2121) | Memorial Sloan Kettering Cancer Cente | 2 | Recruiting |
| NCT05347485 | BCMA | Ciltacabtagene Autoleucel | Janssen Scientific Affairs, LLC | 2 | Recruiting |
| NCT04181827 | BCMA | JCARH125 | Janseen Research & Development | 3 | Active/Non-recruiting |
| NCT03090659 | BCMA | LEGEND-2 | Nanjing Legend Biotech Co. Second Affiliated Hospital of Xi’an Jiaotong University Ruijin Hospital Jiangsu Provincial People’s Hospital Shanghai Changzheng Hospital | 1/2 | Active, not recruiting |
| NCT03548207 | BCMA | CARTITUDE-1 | Janssen Research & Development, LLC | 1/2 | Completed |
| NCT02954445 | BCMA | NA | Shiqi Li, Southwest Hospital, China (Responsible Party) Southwest Hospital, China | 1/2 | Unknown |
| NCT05860036 | BCMA | NA | Institute of Hematology & Blood Diseases Hospital Gang An, Institute of Hematology & Blood Diseases Hospital (Responsible Party) | 1 | Recruiting |
| NCT05740891 | BCMA | NA | Zhejiang University He Huang, Zhejiang University | 1 | Recruiting |
| NCT03716856 | BCMA | NA | First Affiliated Hospital of Zhejiang University | 1 | Unknown |
| NCT05594797 | BCMA | NA | Hrain Biotechnology Co., Ltd. | 2 | Recruiting |
| NCT04003168 | BCMA | NA | Hrain Biotechnology Co., Ltd. | 1 | Recruiting |
| NCT03338972 | BCMA | NA | Fred Hutchinson Cancer Center | 1 | Completed |
| NCT04727008 | BCMA | CXCR4 | Sichuan University Ting Niu, West China Hospital | Early Phase 1 | Not Yet Recruiting |
| NCT05698303 | BCMA | NA | Nanjing IASO Biotherapeutics Co., Ltd. | 1 | Not Yet Recruiting |
| NCT04637269 | Xinqiao Hospital of Chongqing Xi Zhang, MD, Xinqiao Hospital of Chongqing | Early Phase 1 | Recruiting | ||
| NCT04322292 | BCMA | C-CAR088 | Institute of Hematology & Blood Diseases Hospital AnGang, Institute of Hematology & Blood Diseases Hospital | 1 | Unknown |
| (B) Non-BCMA Targeted CAR-T for MM | |||||
| Clinical Trial Name | Antigen Target | CAR Name | Sponsor | Phase | Current Status |
| NCT04674813 | GPRC5D | CC-95266 | Juno Therapeutics, a Subsidiary of Celgene | 1 | Active, Recruiting |
| NCT05016778 | GPRC5D | POLARIS | Zhejiang University & OriCell Therapeutics Co., Ltd. | Early Phase 1 | Active, not recruiting |
| NCT05431608 | GPRC5D | NA | Memorial Sloan Kettering Cancer Center | 1 | Active, Recruiting |
| NCT05325801 | GPRC5D & BCMA | OriC321 | Zhejiang University | 1 | Recruiting |
| NCT05509530 | GPRC5D & BCMA | NA | Xuzhou Medical University | 2 | Recruiting |
| NCT03958656 | SLAMF7 | NA | National Cancer Institute (NCI) | 1 | Completed |
| NCT03710421 | CS1 | NA | City of Hope Medical Center | 1 | Active, Recruiting |
| NCT04499339 | SLAMF7 | CARAMBA-1 | European Commission and Wuerzburg University Hospital | 1/2 | Active, Recruiting |
| NCT04142619 | SLAMF7 | UCARTCS1 | Cellectis S.A | 1 | Active, Recruiting |
| NCT03464916 | CD38 | CAR2 Anti-CD38 A2 | Sorrento Therapeutics, Inc. | 1 | Completed |
| NCT03767751 | CD38 | NA | Chinese PLA General Hospital | 1/2 | Unknown |
| NCT02135406 | CD19 | CTL019/tisagenlecleucel | University of Pennsylvania | 1 | Completed |
| NCT03672318 | CD138 | ATLCAR.CD138 | UNC Lineberger Comprehensive Cancer Center | 1 | Active, Recruiting |
| NCT01886976 | CD138 | CART-138 | Chinese PLA General Hospital | 1/2 | Unknown |
| NCT04182581 | BCMA/CD19 | BCMA/CD19 Dual-Target | Xijing Hospital | Early Phase1 | Unknown |
| NCT03706547 | CD19 | NA | Peng Liu Peng Liu, Shanghai Zhongshan Hospital | 1 | Unknown |
| NCT04714827 | CD19 | CD19-CD22 | Shanxi Province Cancer Hospital | 1/2 | Recruiting |
| NCT04541368 | SLAMF7 | CS1 | Zhejiang University He Huang, Zhejiang University | Early Phase 1 | Not Yet Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.K.; Gupta, A.; Dagar, G.; Das, D.; Chakraborty, A.; Haque, S.; Prasad, C.P.; Singh, A.; Bhat, A.A.; Macha, M.A.; et al. CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond. Vaccines 2023, 11, 1721. https://doi.org/10.3390/vaccines11111721
Mishra AK, Gupta A, Dagar G, Das D, Chakraborty A, Haque S, Prasad CP, Singh A, Bhat AA, Macha MA, et al. CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond. Vaccines. 2023; 11(11):1721. https://doi.org/10.3390/vaccines11111721
Chicago/Turabian StyleMishra, Abhinava K., Ashna Gupta, Gunjan Dagar, Dayasagar Das, Abhijit Chakraborty, Shabirul Haque, Chandra Prakash Prasad, Archana Singh, Ajaz A. Bhat, Muzafar A. Macha, and et al. 2023. "CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond" Vaccines 11, no. 11: 1721. https://doi.org/10.3390/vaccines11111721
APA StyleMishra, A. K., Gupta, A., Dagar, G., Das, D., Chakraborty, A., Haque, S., Prasad, C. P., Singh, A., Bhat, A. A., Macha, M. A., Benali, M., Saini, K. S., Previs, R. A., Saini, D., Saha, D., Dutta, P., Bhatnagar, A. R., Darswal, M., Shankar, A., & Singh, M. (2023). CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond. Vaccines, 11(11), 1721. https://doi.org/10.3390/vaccines11111721