
Advancements in Cancer Immunotherapies



Abstract
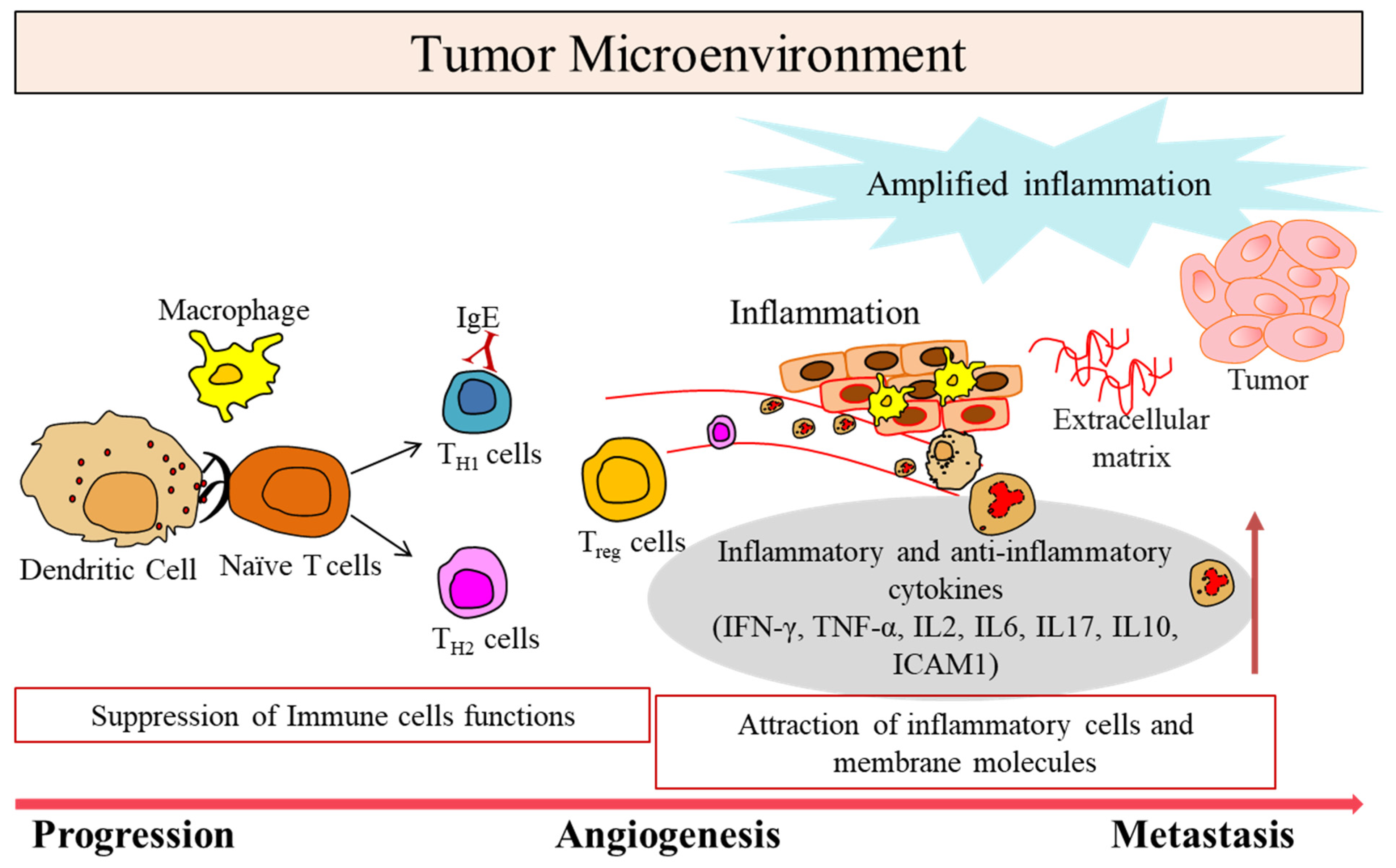
1. Tumor Microenvironment in Cancer
2. Immunomodulatory Roles of Lymphangiogenesis in TME
3. Immune Cells with Specific Phenotypes in TME
4. Cancer Immunotherapy
5. Personalized Recombinant Cancer Vaccines
6. Combination Therapies
7. Immune Checkpoint Inhibitors
8. Monoclonal Antibodies Therapies
9. Immune System Modulators
10. Cytokines Therapy
11. Adoptive Transfer Therapy and Chimeric Antigen Receptor (CAR) T Cell Therapy-Advantages and Disadvantages
12. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Li, M.; Zhou, R.; Zhang, J.; Sun, H.; Shi, M.; Bin, J.; Liao, Y.; Rao, J.; Liao, W. Tumor Microenvironment Characterization in Gastric Cancer Identifies Prognostic and Immunotherapeutically Relevant Gene Signatures. Cancer Immunol. Res. 2019, 7, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Lee, K.; Hwang, H.; Nam, K.T. Immune response and the tumor microenvironment: How they communicate to regulate gastric cancer. Gut Liver 2014, 8, 131–139. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, Q.; Hu, Y.; Li, T.; Yu, J.; Zhao, L.; Ye, G.; Deng, H.; Mou, T.; Cai, S.; et al. ImmunoScore Signature: A Prognostic and Predictive Tool in Gastric Cancer. Ann. Surg. 2018, 267, 504–513. [Google Scholar] [CrossRef]
- Christiansen, A.; Detmar, M. Lymphangiogenesis and cancer. Genes Cancer 2011, 2, 1146–1158. [Google Scholar] [CrossRef]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 1998, 94, 715–725. [Google Scholar] [CrossRef]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. Embo J. 1996, 15, 290–298. [Google Scholar] [CrossRef]
- Orlandini, M.; Marconcini, L.; Ferruzzi, R.; Oliviero, S. Identification of a c-fos-induced gene that is related to the platelet-derived growth factor/vascular endothelial growth factor family. Proc. Natl. Acad. Sci. USA 1996, 93, 11675–11680. [Google Scholar] [CrossRef]
- Yamada, Y.; Nezu, J.; Shimane, M.; Hirata, Y. Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics 1997, 42, 483–488. [Google Scholar] [CrossRef]
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Mäkinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553. [Google Scholar] [CrossRef]
- Favier, B.; Alam, A.; Barron, P.; Bonnin, J.; Laboudie, P.; Fons, P.; Mandron, M.; Herault, J.P.; Neufeld, G.; Savi, P.; et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 2006, 108, 1243–1250. [Google Scholar] [CrossRef]
- Soker, S.; Miao, H.Q.; Nomi, M.; Takashima, S.; Klagsbrun, M. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J. Cell Biochem. 2002, 85, 357–368. [Google Scholar] [CrossRef]
- Pan, Q.; Chanthery, Y.; Liang, W.C.; Stawicki, S.; Mak, J.; Rathore, N.; Tong, R.K.; Kowalski, J.; Yee, S.F.; Pacheco, G.; et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell 2007, 11, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zeng, H.; Wang, P.; Soker, S.; Mukhopadhyay, D. Neuropilin-1-mediated vascular permeability factor/vascular endothelial growth factor-dependent endothelial cell migration. J. Biol. Chem. 2003, 278, 48848–48860. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Varney, M.L.; Backora, M.W.; Cowan, K.; Solheim, J.C.; Talmadge, J.E.; Singh, R.K. Down-regulation of vascular endothelial cell growth factor-C expression using small interfering RNA vectors in mammary tumors inhibits tumor lymphangiogenesis and spontaneous metastasis and enhances survival. Cancer Res. 2005, 65, 9004–9011. [Google Scholar] [CrossRef] [PubMed]
- Kalkunte, S.S.; Mselle, T.F.; Norris, W.E.; Wira, C.R.; Sentman, C.L.; Sharma, S. Vascular endothelial growth factor C facilitates immune tolerance and endovascular activity of human uterine NK cells at the maternal-fetal interface. J. Immunol. 2009, 182, 4085–4092. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Hirakawa, S.; Detmar, M.; Kerjaschki, D.; Nagamatsu, S.; Matsuo, K.; Tanemura, A.; Kamata, N.; Higashikawa, K.; Okazaki, H.; Kameda, K.; et al. Nodal lymphangiogenesis and metastasis: Role of tumor-induced lymphatic vessel activation in extramammary Paget’s disease. Am. J. Pathol. 2009, 175, 2235–2248. [Google Scholar] [CrossRef]
- Zhang, J.P.; Lu, W.G.; Ye, F.; Chen, H.Z.; Zhou, C.Y.; Xie, X. Study on CXCR4/SDF-1alpha axis in lymph node metastasis of cervical squamous cell carcinoma. Int. J. Gynecol. Cancer 2007, 17, 478–483. [Google Scholar] [CrossRef]
- Kaifi, J.T.; Yekebas, E.F.; Schurr, P.; Obonyo, D.; Wachowiak, R.; Busch, P.; Heinecke, A.; Pantel, K.; Izbicki, J.R. Tumor-cell homing to lymph nodes and bone marrow and CXCR4 expression in esophageal cancer. J. Natl. Cancer Inst. 2005, 97, 1840–1847. [Google Scholar] [CrossRef]
- Yoshitake, N.; Fukui, H.; Yamagishi, H.; Sekikawa, A.; Fujii, S.; Tomita, S.; Ichikawa, K.; Imura, J.; Hiraishi, H.; Fujimori, T. Expression of SDF-1 alpha and nuclear CXCR4 predicts lymph node metastasis in colorectal cancer. Br. J. Cancer 2008, 98, 1682–1689. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark Res. 2020, 8, 34. [Google Scholar] [CrossRef]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e1020. [Google Scholar] [CrossRef]
- Gajewski, T.F. Failure at the effector phase: Immune barriers at the level of the melanoma tumor microenvironment. Clin. Cancer Res. 2007, 13, 5256–5261. [Google Scholar] [CrossRef]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Woo, E.Y.; Chu, C.S.; Goletz, T.J.; Schlienger, K.; Yeh, H.; Coukos, G.; Rubin, S.C.; Kaiser, L.R.; June, C.H. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001, 61, 4766–4772. [Google Scholar] [PubMed]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Balatoni, T.; Mohos, A.; Papp, E.; Sebestyén, T.; Liszkay, G.; Oláh, J.; Varga, A.; Lengyel, Z.; Emri, G.; Gaudi, I.; et al. Tumor-infiltrating immune cells as potential biomarkers predicting response to treatment and survival in patients with metastatic melanoma receiving ipilimumab therapy. Cancer Immunol. Immunother. 2018, 67, 141–151. [Google Scholar] [CrossRef]
- Liu, R.B.; Engels, B.; Arina, A.; Schreiber, K.; Hyjek, E.; Schietinger, A.; Binder, D.C.; Butz, E.; Krausz, T.; Rowley, D.A.; et al. Densely granulated murine NK cells eradicate large solid tumors. Cancer Res. 2012, 72, 1964–1974. [Google Scholar] [CrossRef]
- Delahaye, N.F.; Rusakiewicz, S.; Martins, I.; Ménard, C.; Roux, S.; Lyonnet, L.; Paul, P.; Sarabi, M.; Chaput, N.; Semeraro, M.; et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med. 2011, 17, 700–707. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Crist, M.; Yaniv, B.; Palackdharry, S.; Lehn, M.A.; Medvedovic, M.; Stone, T.; Gulati, S.; Karivedu, V.; Borchers, M.; Fuhrman, B.; et al. Metformin increases natural killer cell functions in head and neck squamous cell carcinoma through CXCL1 inhibition. J. Immunother. Cancer 2022, 10, e005632. [Google Scholar] [CrossRef]
- Wang, S.; Lin, Y.; Xiong, X.; Wang, L.; Guo, Y.; Chen, Y.; Chen, S.; Wang, G.; Lin, P.; Chen, H.; et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical Trial. Clin. Cancer Res. 2020, 26, 4921–4932. [Google Scholar] [CrossRef]
- Wei, Z.; Zhang, X.; Yong, T.; Bie, N.; Zhan, G.; Li, X.; Liang, Q.; Li, J.; Yu, J.; Huang, G.; et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat. Commun. 2021, 12, 440. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Z.; Hou, J.; Xiong, W.; Kim, H.; Chen, J.; Zheng, C.; Jiang, X.; Yoon, J.; Shen, J. Tumor Selective Metabolic Reprogramming as a Prospective PD-L1 Depression Strategy to Reactivate Immunotherapy. Adv. Mater. 2022, 34, e2206121. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Song, W.; Jiang, X.; Deng, Z.; Xiong, W.; Shen, J. Metabolic reprogramming mediated PD-L1 depression and hypoxia reversion to reactivate tumor therapy. J. Control. Release 2022, 352, 793–812. [Google Scholar] [CrossRef]
- Xiong, W.; Qi, L.; Jiang, N.; Zhao, Q.; Chen, L.; Jiang, X.; Li, Y.; Zhou, Z.; Shen, J. Metformin Liposome-Mediated PD-L1 Downregulation for Amplifying the Photodynamic Immunotherapy Efficacy. ACS Appl. Mater. Interfaces 2021, 13, 8026–8041. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Z.; Zheng, C.; Liu, Y.; Hao, R.; Ji, X.; Xi, Q.; Shen, J.; Li, Z. Chitosan oligosaccharide regulates AMPK and STAT1 pathways synergistically to mediate PD-L1 expression for cancer chemoimmunotherapy. Carbohydr. Polym. 2022, 277, 118869. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Walker, L.S. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J. Autoimmun. 2013, 45, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S.; Bluestone, J.A. Expression of CTLA-4 and FOXP3 in cis protects from lethal lymphoproliferative disease. Eur. J. Immunol. 2007, 37, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Traversari, C.; van der Bruggen, P.; Luescher, I.F.; Lurquin, C.; Chomez, P.; Van Pel, A.; De Plaen, E.; Amar-Costesec, A.; Boon, T. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J. Exp. Med. 1992, 176, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.G., Jr.; Peters, L.C. Immunotherapy of established micrometastases with Bacillus Calmette-Guérin tumor cell vaccine. Cancer Res. 1978, 38, 204–209. [Google Scholar]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Barbee, M.S.; Ogunniyi, A.; Horvat, T.Z.; Dang, T.O. Current status and future directions of the immune checkpoint inhibitors ipilimumab, pembrolizumab, and nivolumab in oncology. Ann. Pharm. 2015, 49, 907–937. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug. Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef]
- Marrone, K.A.; Brahmer, J.R. Using Immune Checkpoint Inhibitors in Lung Cancer. Oncology 2016, 30, 713–721. [Google Scholar]
- Hatae, R.; Chamoto, K. Immune checkpoint inhibitors targeting programmed cell death-1 (PD-1) in cancer therapy. Rinsho Ketsueki 2016, 57, 2224–2231. [Google Scholar] [CrossRef]
- Swart, M.; Verbrugge, I.; Beltman, J.B. Combination Approaches with Immune-Checkpoint Blockade in Cancer Therapy. Front. Oncol. 2016, 6, 233. [Google Scholar] [CrossRef]
- Olivier, T.; Haslam, A.; Prasad, V. Anticancer Drugs Approved by the US Food and Drug Administration From 2009 to 2020 According to Their Mechanism of Action. JAMA Netw. Open 2021, 4, e2138793. [Google Scholar] [CrossRef]
- Kates, M.; Sopko, N.A.; Matsui, H.; Drake, C.G.; Hahn, N.M.; Bivalacqua, T.J. Immune checkpoint inhibitors: A new frontier in bladder cancer. World J. Urol. 2016, 34, 49–55. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Chen, R.; Chen, B. Brentuximab vedotin for relapsed or refractory Hodgkin’s lymphoma. Drug. Des. Devel. 2015, 9, 1729–1733. [Google Scholar] [CrossRef]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer 2015, 14, 1376–1384. [Google Scholar] [CrossRef]
- Wu, Q.; Qian, W.; Sun, X.; Jiang, S. Small-molecule inhibitors, immune checkpoint inhibitors, and more: FDA-approved novel therapeutic drugs for solid tumors from 1991 to 2021. J. Hematol. Oncol. 2022, 15, 143. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef]
- Duan, F.; Duitama, J.; Al Seesi, S.; Ayres, C.M.; Corcelli, S.A.; Pawashe, A.P.; Blanchard, T.; McMahon, D.; Sidney, J.; Sette, A.; et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med. 2014, 211, 2231–2248. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; LoRusso, P.M.; Khalil, M.; Gartner, E.M.; Khaira, D.; Soulieres, D.; Dorazio, P.; Trosko, J.A.; Rüter, J.; Mariani, G.L.; et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin. Cancer Res. 2010, 16, 3485–3494. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Abdin, S.M.; Zaher, D.M.; Arafa, E.A.; Omar, H.A. Tackling Cancer Resistance by Immunotherapy: Updated Clinical Impact and Safety of PD-1/PD-L1 Inhibitors. Cancers 2018, 10, 32. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Burugu, S.; Dancsok, A.R.; Nielsen, T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2018, 52, 39–52. [Google Scholar] [CrossRef]
- Donini, C.; D’Ambrosio, L.; Grignani, G.; Aglietta, M.; Sangiolo, D. Next generation immune-checkpoints for cancer therapy. J. Thorac Dis. 2018, 10, S1581–S1601. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, E.; Cappello, P.; Sorrentino, C.; D’Antuono, T.; Pellicciotta, A.; Giovarelli, M.; Forni, G.; Musiani, P.; Triebel, F. Immunological mechanisms elicited at the tumour site by lymphocyte activation gene-3 (LAG-3) versus IL-12: Sharing a common Th1 anti-tumour immune pathway. J. Pathol. 2005, 205, 82–91. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Rivard, C.J.; Rozeboom, L.; Yu, H.; Ellison, K.; Kowalewski, A.; Zhou, C.; Hirsch, F.R. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. 2016, 107, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.W.; Mao, L.; Yu, G.T.; Bu, L.L.; Ma, S.R.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology 2016, 5, e1239005. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Yen, H.R.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.; Pardoll, D.M.; Drake, C.G. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J. Immunol. 2009, 182, 6659–6669. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Tu, X.; Qin, B.; Zhang, Y.; Zhang, C.; Kahila, M.; Nowsheen, S.; Yin, P.; Yuan, J.; Pei, H.; Li, H.; et al. PD-L1 (B7-H1) Competes with the RNA Exosome to Regulate the DNA Damage Response and Can Be Targeted to Sensitize to Radiation or Chemotherapy. Mol. Cell 2019, 74, 1215–1226.e4. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaav7431. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Jiang, X.; Zheng, C.; Luo, W.; Xiang, X.; Qi, X.; Shen, J. Metformin modified chitosan as a multi-functional adjuvant to enhance cisplatin-based tumor chemotherapy efficacy. Int. J. Biol. Macromol. 2023, 224, 797–809. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hsu, J.M.; Yang, W.H.; Hung, M.C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol. 2022, 19, 287–305. [Google Scholar] [CrossRef]
- Lucibello, G.; Mograbi, B.; Milano, G.; Hofman, P.; Brest, P. PD-L1 regulation revisited: Impact on immunotherapeutic strategies. Trends Mol. Med. 2021, 27, 868–881. [Google Scholar] [CrossRef]
- Morel, K.L.; Sheahan, A.V.; Burkhart, D.L.; Baca, S.C.; Boufaied, N.; Liu, Y.; Qiu, X.; Cañadas, I.; Roehle, K.; Heckler, M.; et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat. Cancer 2021, 2, 444–456. [Google Scholar] [CrossRef]
- Kim, D.J.; Jang, J.H.; Ham, S.Y.; Choi, S.H.; Park, S.S.; Jeong, S.Y.; Kim, B.C.; Jeon, D.Y.; Lee, B.J.; Ko, B.K.; et al. Doxorubicin inhibits PD-L1 expression by enhancing TTP-mediated decay of PD-L1 mRNA in cancer cells. Biochem. Biophys. Res. Commun. 2020, 522, 402–407. [Google Scholar] [CrossRef]
- Dai, X.; Bu, X.; Gao, Y.; Guo, J.; Hu, J.; Jiang, C.; Zhang, Z.; Xu, K.; Duan, J.; He, S.; et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol. Cell. 2021, 81, 2317–2331.e6. [Google Scholar] [CrossRef]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell. 2018, 71, 606–620.e7. [Google Scholar] [CrossRef]
- Finn, O.J. Human Tumor Antigens Yesterday, Today, and Tomorrow. Cancer Immunol. Res. 2017, 5, 347–354. [Google Scholar] [CrossRef]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef]
- Mendelsohn, J. The epidermal growth factor receptor as a target for therapy with antireceptor monoclonal antibodies. Semin. Cancer Biol. 1990, 1, 339–344. [Google Scholar] [PubMed]
- Grütter, C.; Wilkinson, T.; Turner, R.; Podichetty, S.; Finch, D.; McCourt, M.; Loning, S.; Jermutus, L.; Grütter, M.G. A cytokine-neutralizing antibody as a structural mimetic of 2 receptor interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 20251–20256. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Krupitskaya, Y.; Wakelee, H.A. Ramucirumab, a fully human mAb to the transmembrane signaling tyrosine kinase VEGFR-2 for the potential treatment of cancer. Curr. Opin. Investig. Drugs 2009, 10, 597–605. [Google Scholar] [PubMed]
- Wu, Y.; Zhong, Z.; Huber, J.; Bassi, R.; Finnerty, B.; Corcoran, E.; Li, H.; Navarro, E.; Balderes, P.; Jimenez, X.; et al. Anti-vascular endothelial growth factor receptor-1 antagonist antibody as a therapeutic agent for cancer. Clin. Cancer Res. 2006, 12, 6573–6584. [Google Scholar] [CrossRef]
- Lutterbuese, R.; Raum, T.; Kischel, R.; Hoffmann, P.; Mangold, S.; Rattel, B.; Friedrich, M.; Thomas, O.; Lorenczewski, G.; Rau, D.; et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 12605–12610. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Naidoo, J.; Page, D.B.; Wolchok, J.D. Immune modulation for cancer therapy. Br. J. Cancer 2014, 111, 2214–2219. [Google Scholar] [CrossRef]
- Matsushita, M.; Kawaguchi, M. Immunomodulatory Effects of Drugs for Effective Cancer Immunotherapy. J. Oncol. 2018, 2018, 8653489. [Google Scholar] [CrossRef]
- Chim, C.S.; Kumar, S.K.; Orlowski, R.Z.; Cook, G.; Richardson, P.G.; Gertz, M.A.; Giralt, S.; Mateos, M.V.; Leleu, X.; Anderson, K.C. Management of relapsed and refractory multiple myeloma: Novel agents, antibodies, immunotherapies and beyond. Leukemia 2018, 32, 252–262. [Google Scholar] [CrossRef]
- Pan, B.; Lentzsch, S. The application and biology of immunomodulatory drugs (IMiDs) in cancer. Pharmacol. Ther. 2012, 136, 56–68. [Google Scholar] [CrossRef]
- Zeldis, J.B.; Knight, R.; Hussein, M.; Chopra, R.; Muller, G. A review of the history, properties, and use of the immunomodulatory compound lenalidomide. Ann. N. Y. Acad. Sci. 2011, 1222, 76–82. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Lu, X.; Ding, Z.C.; Cao, Y.; Liu, C.; Habtetsion, T.; Yu, M.; Lemos, H.; Salman, H.; Xu, H.; Mellor, A.L.; et al. Alkylating agent melphalan augments the efficacy of adoptive immunotherapy using tumor-specific CD4+ T cells. J. Immunol. 2015, 194, 2011–2021. [Google Scholar] [CrossRef]
- Derissen, E.J.; Beijnen, J.H.; Schellens, J.H. Concise drug review: Azacitidine and decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef]
- Fucikova, J.; Truxova, I.; Hensler, M.; Becht, E.; Kasikova, L.; Moserova, I.; Vosahlikova, S.; Klouckova, J.; Church, S.E.; Cremer, I.; et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 2016, 128, 3113–3124. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Atallah-Yunes, S.A.; Robertson, M.J. Cytokine Based Immunotherapy for Cancer and Lymphoma: Biology, Challenges and Future Perspectives. Front. Immunol. 2022, 13, 872010. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Su, M.; Liu, L.; Tang, Y.; Pan, Y.; Sun, J. Clinical Application of Cytokines in Cancer Immunotherapy. Drug. Des. Devel. 2021, 15, 2269–2287. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Duvic, M.; Martin, A.; Sterry, W.; Assaf, C.; Sun, Y.; Straus, D.; Acosta, M.; Negro-Vilar, A. Phase III placebo-controlled trial of denileukin diftitox for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Eisenbeis, C.F.; Grainger, A.; Fischer, B.; Baiocchi, R.A.; Carrodeguas, L.; Roychowdhury, S.; Chen, L.; Banks, A.L.; Davis, T.; Young, D.; et al. Combination immunotherapy of B-cell non-Hodgkin’s lymphoma with rituximab and interleukin-2: A preclinical and phase I study. Clin. Cancer Res. 2004, 10, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Gluck, W.L.; Hurst, D.; Yuen, A.; Levine, A.M.; Dayton, M.A.; Gockerman, J.P.; Lucas, J.; Denis-Mize, K.; Tong, B.; Navis, D.; et al. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin. Cancer Res. 2004, 10, 2253–2264. [Google Scholar] [CrossRef]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef]
- West, E.E.; Jin, H.T.; Rasheed, A.U.; Penaloza-Macmaster, P.; Ha, S.J.; Tan, W.G.; Youngblood, B.; Freeman, G.J.; Smith, K.A.; Ahmed, R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Investig. 2013, 123, 2604–2615. [Google Scholar] [CrossRef]
- Hansen, R.M.; Borden, E.C. Current status of interferons in the treatment of cancer. Oncology 1992, 6, 19–24; discussion 26, 29. [Google Scholar]
- Ziegler-Heitbrock, H.W.; Thiel, E. Recombinant IFN-alpha in lymphomas. J. Invest. Derm. 1990, 95, 213s–215s. [Google Scholar] [CrossRef]
- Zhang, L.; Tai, Y.T.; Ho, M.Z.G.; Qiu, L.; Anderson, K.C. Interferon-alpha-based immunotherapies in the treatment of B cell-derived hematologic neoplasms in today’s treat-to-target era. Exp. Hematol. Oncol. 2017, 6, 20. [Google Scholar] [CrossRef]
- Müller, L.; Aigner, P.; Stoiber, D. Type I Interferons and Natural Killer Cell Regulation in Cancer. Front. Immunol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Santegoets, S.J.; Reyners, A.K.; Goedemans, R.; Wouters, M.C.; Kenter, G.G.; van Erkel, A.R.; van Poelgeest, M.I.; Nijman, H.W.; van der Hoeven, J.J.; et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2015, 26, 2141–2149. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- CAR T Cells: Engineering Patients’ Immune Cells to Treat Their Cancers. Available online: https://www.cancer.gov/about-cancer/treatment/research/car-t-cells (accessed on 10 March 2022).
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- FDA. FDA Approves Tisagenlecleucel for B-Cell ALL and Tocilizumab for Cytokine Release Syndrome. FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome (accessed on 7 September 2017).
- FDA. FDA Approves Axicabtagene Ciloleucel for Large B-Cell Lymphoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-axicabtagene-ciloleucel-large-b-cell-lymphoma (accessed on 25 October 2017).
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Murad, J.P.; Kozlowska, A.K.; Lee, H.J.; Ramamurthy, M.; Chang, W.C.; Yazaki, P.; Colcher, D.; Shively, J.; Cristea, M.; Forman, S.J.; et al. Effective Targeting of TAG72(+) Peritoneal Ovarian Tumors via Regional Delivery of CAR-Engineered T Cells. Front. Immunol. 2018, 9, 2268. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Target | Drug INN (Brand Name) | Cancer Type | Current Status | Ref. |
|---|---|---|---|---|
| Antibodies Based | ||||
| CTLA-4 | Ipilimumab (Yervoy) | Melanoma (2011) and Renal cell carcinoma (2018) | FDA approved | [67,68,69] |
| Multiple cancers | Phase I-III | [69] | ||
| Tremelimumab (Imjudo) | Antineoplastic; liver cancer | FDA approved | [69,70] | |
| PD-1 | Nivolumab (Opdivo) | Melanoma (2014), NSCLC (2015), and Renal (2018) cancers, Hodgkin lymphoma | FDA approved | [69,71,72] |
| Multiple cancers | Phase I-III | [69] | ||
| Pembrolizumab (Keytruda) | Melanoma (2014), Various (2015) | FDA approved | [73] | |
| Multiple cancers | Phase I-III | [69] | ||
| MED10680 | Multiple cancers | Phase I | [69] | |
| AMP-224 | Multiple cancers | Phase I | [69] | |
| Pidilizumab | Multiple cancers | Phase I-II | [69] | |
| Cemiplimab (Libtayo) | Cutaneous squamous-cell carcinoma (2018) | FDA approved | [69,74] | |
| PD-L1 | Atezolizumab (Tecentriq) | Bladder, NSCLC (2016), and TNBC (2019), hepatocellular carcinoma, HCC (2020) | FDA approved | [69] [74,75] |
| Avelumab (Bavencio) | Urothelial Carcinoma (2017), Merkel Cell Carcinoma (2017), Renal carcinoma (2019) | FDA approved | [74] | |
| MED14736 | Multiple cancers | Phase III | [69] | |
| Avelumab (Bavencio) | Merkel cell carcinoma (2017), Rena (2019), Urothelial carcinoma (2020) | FDA approved | [74] | |
| BMS-936559 | Multiple cancers | Phase I | [69] | |
| Durvalumab (IMFINZI) | Bladder Cancer (2017), NSCLC (2018) | FDA approved | [69,74] | |
| LAG-3 | IMP321 | Multiple cancers | Phase I | [69] |
| BMS-986016 | Multiple cancers | Phase I | [69] | |
| Relatlimab (Opdualag) | Melanoma (2022) | FDA approved | [70] | |
| VEGF | Bevacizumab | Colorectal (2004), NSCLC (2006, 2018), Renal (2009), Cervical (2014), Glioblastoma (2009), and Ovarian (2018) Cancers | FDA approved | [74,76] |
| VEGF-A, Ang-2 | Faricimab (Vabysmo) | wAMD, DME | FDA approved | [70] |
| VEGFR2 | Ramucirumab (Cyramza) | Gastric cancer (2014), NSCLC (2020), HCC (2019) | FDA approved | [74,76,77] |
| EGFR | Cetuximab | Colorectal cancer (CRC) (2004, 2012) and Head and neck squamous cell carcinoma (2006, 2011) | FDA approved | [74,76] |
| Necitumumab (Portrazza) | NSCLC (2015) | FDA approved | [74,76,77] | |
| Panitumumab (Vectibix) | Colorectal Cancer (2006) | FDA approved | [76] | |
| PDGFRα | Olaratumab (Lartruvo) | Soft-tissue sarcoma (2016) | FDA approved | [76] |
| HER2 | Pertuzumab (Perjeta) | HER2-positive Breast cancer (2012) | FDA approved | [74,76] |
| Trastuzumab (Herceptin) | HER2-positive Breast cancer (1998) | FDA approved | [74,76] | |
| Ado-trastuzumab emtansine (Kadcyla) | HER2-Breast cancer (2013) | FDA approved | [76] | |
| Fam-trastuzumab deruxtecan (Enhertu) | HER2-positive Breast cancer (2019) | FDA approved | [76] | |
| Trastuzumab tucatinib | HER2-positive Breast cancer (2020) | FDA approved | [74] | |
| CCR4 | Mogamulizumab (Poteligeo) | Cutaneous T cell lymphoma (2018) | FDA approved | [76] |
| CD20 | Obinutuzumab (Gazyva) | Chronic lymphocytic leukemia (2013), follicular lymphoma (2017) | FDA approved | [74,76] |
| Ofatumumab (Arzerra) | Chronic lymphocytic leukemia (2014) | FDA approved | [74,76] | |
| Rituximab (MabThera, Rituxan) | B-Cell Lymphoma (1997) | FDA approved | [76] | |
| Ibritumomab tiuxetan (Zevalin) | NHL (2004) | FDA approved | [70,76] | |
| tositumomab Iodine-131 (Bexxar) | NHL (2003) | FDA approved | [76] | |
| Ublituximab | Chronic lymphocytic leukemia, CLL, non-Hodgkin’s lymphoma) and non-cancer (multiple sclerosis) | Phase III | [70,77] | |
| CD33 | Gemtuzumab ozogamicin (Mylotarg) | Acute myeloid leukemia (2000) | FDA approved | [74,76] |
| CD30 | Brentuximab vedotin (Adcetris) | Hodgkin’s lymphoma and Anaplastic large-cell lymphoma (2011) | FDA approved | [76,78,79] |
| CD79B | Polatuzumab vedotin (Polivy) | Diffuse large B-cell lymphoma (2019) | FDA approved | [74,76] |
| CD22 | Inotuzumab ozogamicin (BESPONSA) | Acute lymphoblastic leukemia (2017) | FDA approved | [74,76] |
| Moxetumomab pasudotox (Lumoxiti) | Hairy-cell leukemia (2018) | FDA approved | [74,76] | |
| CD19 | Inebilizumab (Uplizna) | Neuromyelitis optica and neuromyelitis optica spectrum disorders (2022) | FDA approved | [70] |
| CD19, CD3 | Blinatumomab (Blincyto) | Acute lymphoblastic leukemia (2014) | FDA approved | [74,76] |
| TROP2 | Sacituzumab govitecan (Trodelvy) | TNBC (2020) | FDA approved | [70,74,76] |
| CD3 | Muromonab-CD3 (Orthoclone Okt3) | Reversal of kidney transplant rejection (1986) | FDA approved | [77] |
| CD3, BCMA | Teclistamab (TECVAYLI) | Multiple myeloma (2022) | FDA approved | [70] |
| gp100, CD3 | Tebentafusp (KIMMTRAK) | Metastatic uveal melanoma (2022) | FDA approved | [70] |
| CD30, CD3 | Mosunetuzumab (Lunsumio) | Follicular lymphoma (2022) | FDA Review | [70] |
| CD38 | Daratumumab (Darzalex) | Multiple Myeloma (2015) | FDA approved | [74,76] |
| Isatuximab (Sarclisa) | Multiple Myeloma (2020) | FDA approved | [74,76] | |
| GD2 | Dinutuximab (Qarziba; Unituxin) | Neuroblastoma (2015) | FDA approved | [74,76] |
| Nectin-4 | Enfortumab Vedotin (Padcev) | Bladder cancer (2019), Urothelial cancer (2022) | FDA approved | [70,74,76] |
| Small Drugs Based | ||||
|---|---|---|---|---|
| Target | Drug INN (Brand Name) | Cancer Type | Current Status | Ref. |
| EGFR | Gefitinib | NSCLC (2015) | FDA approved | [74,80] |
| Erlotinib HCl (Tarceva) | NSCLC (2004) | FDA approved | [80] | |
| Osimertinib mesylate | NSCLC (2020) | FDA approved | [74,80] | |
| Dacomitinib (Vizimpro) | EGFR-mutated NSCLC (2018) | FDA approved | [74,80] | |
| Mobocertinib succinate (Exkivity) | EGFR exon 20-mutated NSCLC (2021) | FDA approved | [80] | |
| HER2 | Tucatinib (Tukysa) | HER2-positive breast cancer (2020) | FDA approved | [74,80] |
| EGFR, HER2, and HER4 | Neratinib maleate (Nerlynx) | HER2-overexpressed breast cancer (2017) | FDA approved | [80] |
| Afatinib dimaleate (Gilotrif) | Metastatic NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutation (2013) | FDA approved | [80] | |
| PARP | Olaparib (Lynparza) | Advanced BRCA-mutated ovarian cancer (2020) | FDA approved | [74,80] |
| Rucaparib camsylate (Rubraca) | BRCA-positive ovarian cancer (2016) | FDA approved | [74,80] | |
| Niraparib tosylate (Zejula) | Epithelial ovarian, fallopian tube, or primary peritoneal cancer (2017) | FDA approved | [80] | |
| PDGFRα | Avapritinib (Ayvakit) | metastatic gastrointestinal stromal tumor (GIST) with platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutations (2020) | FDA approved | [74,80] |
| Multitarget TKI (VEGFRs, PDGFRα/β, CSF1R, KIT, and FLT3) | Sunitinib malate (Sutent) | Imatinib-resistant GIST and advanced RCC (2013) | FDA approved | [74,80] |
| Multitarget TKI (RET, VEGFRs, KIT, PDGFRα/β, FGFR1/2, RAF1, BRAF, and BRAFV600E) | Regorafenib (Stivarga) | Metastatic colorectal cancer (2012) | FDA approved | [74,80] |
| Multitarget TKI (VEGFR2/3, PDGFRβ, FLT3, KIT, RAF1, and BRAF) | Sorafenib toylate (Nexavar) | Advanced RCC (2005) | FDA approved | [80] |
| Multitarget TKI (VEGFRs, PDGFRα/β, FGFR1/2, KIT) | Pazopanib HCl (Votrient) | Metastatic RCC (2009) | FDA approved | [80] |
| Multitarget TKI (VEGFRs, FGFRs, PDGFRα, RET, and KIT) | Lenvatinib mesylate (Lenvima) | Thyroid cancer (2015) | FDA approved | [80] |
| Multitarget TKI (VEGFR2/3, EGFR, and RET) | Vandetanib (Caprelsa) | Unresectable or metastatic medullary thyroid cancer (2011) | FDA approved | [74,80] |
| Multitarget TKI (VEGFRs, MET, RET, FLT3, KIT, TIE2, and AXL) | Cabozantinib S-malate (Cometriq) | Progressive, metastatic medullary thyroid cancer (2012) | [80] | |
| VEGFRs | Axitinib (Inlyta) | Advanced RCC (2012) | FDA approved | [74,80] |
| Tivozanib HCl (Fotivda) | Advanced RCC (2021) | FDA approved | [80] | |
| mTOR | Everolimus (Afinitor) | Advanced RCC (2009), HER2-negative breast cancer after failure of treatment with letrozole or anastrozole (2012), nonfunctional neuroendocrine tumors of gastrointestinal or lung origin with unresectable, locally advanced, or metastatic disease (2016) | FDA approved | [74,80] |
| Temsirolimus (Torisel) | Advanced RCC (2007) | FDA approved | [80] | |
| Target | Drug | Cancer Type | Modulate PDL1 | Ref. |
|---|---|---|---|---|
| Histone methyltransferase EZH2 | Tazemetostat and DZNep | Prostate cancer | Transcriptional upregulation of PD-L1 | [114] |
| Tazemetostat (Tazverik) | Epithelioid sarcoma (2020) | Transcriptional upregulation of PD-L1 via decreased H3K27me3, FDA approved | [80] | |
| Histone deacetylase inhibitor | Vorinostat (Zolinza) | Cutaneous T cell lymphoma (2006) | Transcriptional upregulation of PD-L1, FDA approved | [80,114] |
| DNA methyltransferases | Decitabine (Dacogen) | Myelodysplastic syndrome (2006) | Transcriptional upregulation of PD-L1 via decreased DNA methylation in the PD-L1 promoter region, FDA approved | [80,114] |
| EGFR | Gefitinib (Iressa) | NSCLC (2004), HNSCC and TNBC | Transcriptional downregulation of PD-L1, FDA approved | [80,114] |
| Osimertinib mesylate (Tagrisso) | NSCLC with EGFRT790M mutations (2015) | |||
| Erlotinib HCL (Tarceva) | NSCLC (2003) | |||
| JAK | Ruxolitinib phosphate (Jakafi) | Intermediate or high-risk myelofibrosis (2011) | Downregulation of PD-L1, FDA approved | [80,114] |
| Fedratinib HCl (Inrebic) | Myelofibrosis (2019) | Downregulation of PD-L1, FDA approved | [80,114] | |
| ZFP36 (Tristetraprolin) | Doxorubicin | NSCLC and breast cancers | Downregulates translation of PD-L1 | [80,114,115] |
| AMPK | Metformin and A-769662 | Multiple cancers | Increased PD-L1 phosphorylation and degradation | [80,116,117] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, R.; Singh, S.K.; Misra, S. Advancements in Cancer Immunotherapies. Vaccines 2023, 11, 59. https://doi.org/10.3390/vaccines11010059
Roy R, Singh SK, Misra S. Advancements in Cancer Immunotherapies. Vaccines. 2023; 11(1):59. https://doi.org/10.3390/vaccines11010059
Chicago/Turabian StyleRoy, Ruchi, Sunil Kumar Singh, and Sweta Misra. 2023. "Advancements in Cancer Immunotherapies" Vaccines 11, no. 1: 59. https://doi.org/10.3390/vaccines11010059
APA StyleRoy, R., Singh, S. K., & Misra, S. (2023). Advancements in Cancer Immunotherapies. Vaccines, 11(1), 59. https://doi.org/10.3390/vaccines11010059