

Design Strategies and Precautions for Using Vaccinia Virus in Tumor Virotherapy

Abstract
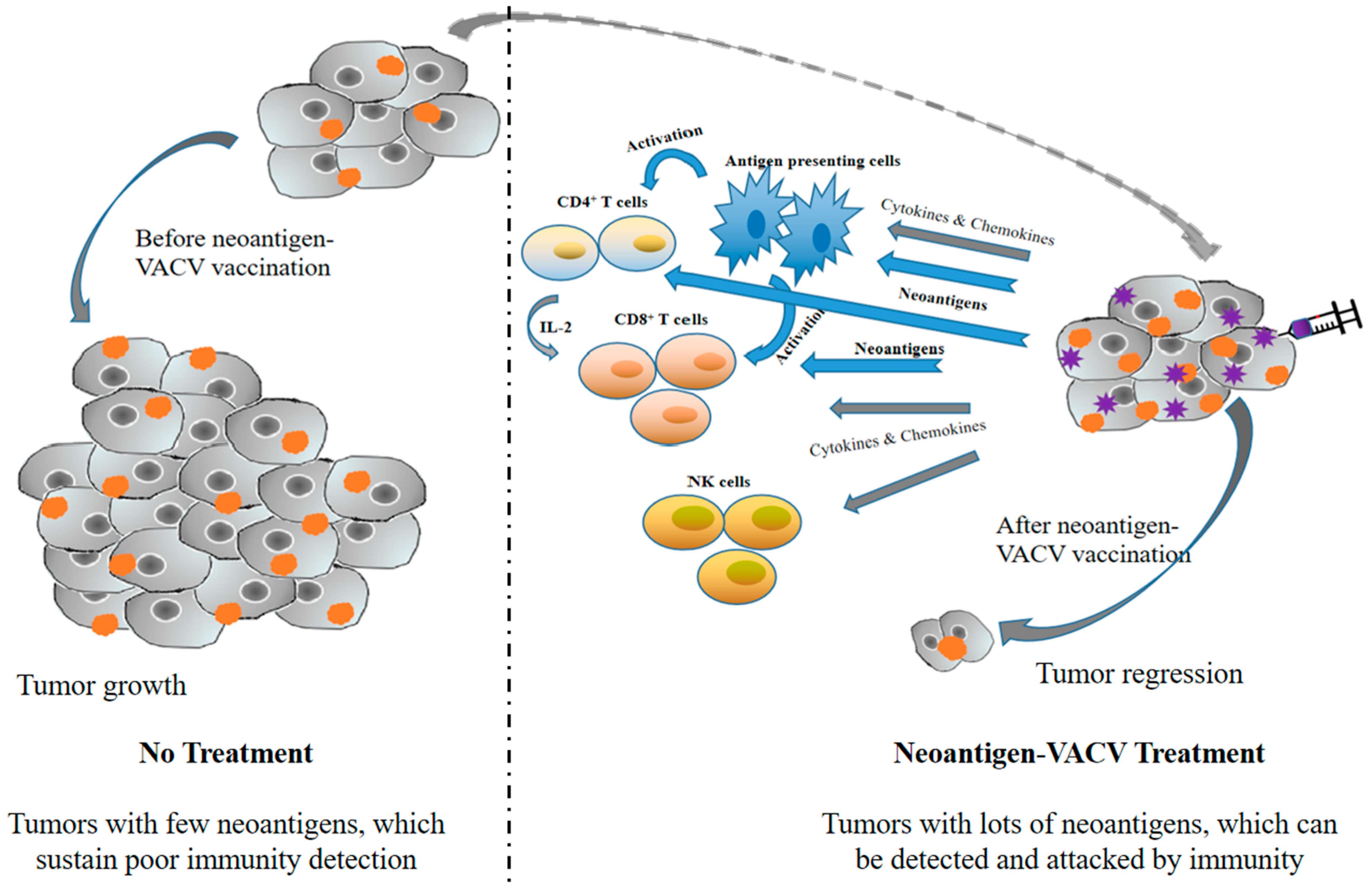
1. Introduction
2. Oncolytic Vaccinia Vectors and Oncolytic Effects
3. Strategies and Approaches to Enhance the Anti-Tumor Capacity of VACV
3.1. Vaccinia Virus Virulence Genes
3.2. Cytokines and Chemokines
3.2.1. Cytokines
3.2.2. Chemokines
3.3. Costimulators
3.4. BiTE
3.5. Tumor Antigens
3.6. Combinations
3.6.1. Combination with Pharmaceutical Drugs
3.6.2. Combination with Anti-Angiogenesis Strategies
3.6.3. Combination with Immune Checkpoint Blockade or CAR-T Cell Therapy
4. Precaution and Risk of VACV in Tumor Therapy
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Kim, M.K.; Breitbach, C.J.; Moon, A.; Heo, J.; Lee, Y.K.; Cho, M.; Lee, J.W.; Kim, S.G.; Kang, D.H.; Bell, J.C.; et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci. Transl. Med. 2013, 15, 185ra63. [Google Scholar] [CrossRef] [PubMed]
- Feola, S.; Russo, S.; Ylosmaki, E.; Cerullo, V. Oncolytic ImmunoViroTherapy: A long history of crosstalk between viruses and immune system for cancer treatment. Pharmacol. Ther. 2021, 23, 108103. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2016, 30, 660. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.Q.; Wang, S.B.; Cai, M.H.; Wang, X.J.; Chen, J.Y.; Tong, X.M.; Chen, X.Y.; Mou, X.Z. Recent advances in oncolytic virus-based cancer therapy. Virus Res. 2019, 270, 197675. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, N.; Kuroda, S.; Kakiuchi, Y.; Kumon, K.; Tsumura, T.; Hashimoto, M.; Morihiro, T.; Kubota, T.; Aoyama, K.; Kikuchi, S.; et al. Immune Modulation by Telomerase-Specific Oncolytic Adenovirus Synergistically Enhances Antitumor Efficacy with Anti-PD1 Antibody. Mol. Ther. 2020, 28, 794–804. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef]
- Mastrangelo, M.J.; Maguire, H.C.; Eisenlohr, L.C.; Laughlin, C.E.; Monken, C.E.; McCue, P.A.; Kovatich, A.J.; Lattime, E.C. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999, 6, 409–422. [Google Scholar] [CrossRef]
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.H.; Sommermann, E.M.; Maruri Avidal, L.; et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef]
- Rojas, J.J.; Thorne, S.H. Theranostic potential of oncolytic vaccinia virus. Theranostics 2012, 2, 363–373. [Google Scholar] [CrossRef]
- Guo, Z.S.; Bartlett, D.L. Vaccinia as a vector for gene delivery. Expert. Opin. Biol. Ther. 2004, 4, 901–917. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Thorne, S.H.; Liang, W.; Sampath, P.; Schmidt, T.; Sikorski, R.; Beilhack, A.; Contag, C.H. Targeting Localized Immune Suppression Within the Tumor Through Repeat Cycles of Immune Cell-oncolytic Virus Combination Therapy. Mol. Ther. 2010, 18, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Crompton, A.M.; Kirn, D.H. From ONYX-015 to armed vaccinia viruses: The education and evolution of oncolytic virus development. Curr. Cancer Drug Targets 2007, 7, 133–139. [Google Scholar] [CrossRef]
- Wittek, R. Vaccinia immune globulin: Current policies, preparedness, and product safety and efficacy. Int. J. Infect. Dis. 2006, 10, 193–201. [Google Scholar] [CrossRef]
- Thorne, S.H.; Hwang, T.H.H.; O’Gorman, W.E.; Bartlett, D.L.; Sei, S.; Kanji, F.; Brown, C.; Werier, J.; Cho, J.H.; Lee, D.E.; et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J. Clin. Investig. 2007, 117, 3350–3358. [Google Scholar] [CrossRef]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar]
- He, Q.; Zou, L.; Zhang, P.A.; Lui, J.X.; Skog, S.; Fornande, T. The clinical significance of thymidine kinase I measurement in serum of breast cancer patients using the anti-TK1 antibody. Int. J. Biol. Markers 2001, 15, 139–146. [Google Scholar] [CrossRef]
- Puhlmann, M.; Gnant, M.; Brown, C.K.; Alexander, H.R.; Bartlett, D.L. Thymidine kinase-deleted vaccinia virus expressing purine nucleoside phosphorylase as a vector for tumor-directed gene therapy. Hum. Gene Ther. 1999, 10, 649–657. [Google Scholar] [CrossRef]
- Puhlmann, M.; Brown, C.K.; Gnant, M.; Huang, J.; Libutti, S.K.; Alexander, H.R.; Bartlett, D.L. Vaccinia as a vector for tumor-directed gene therapy: Biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 2000, 7, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.S.; Falls, T.; Burns, J.; Garcia, V.; et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.M.L.; Chakrabarti, S.; Moss, B.; Fredrickson, T. Cell Proliferative Response to Vaccinia Virus Is Mediated by Vgf. Virology 1988, 164, 182–192. [Google Scholar] [CrossRef]
- Buller, R.M.; Chakrabarti, S.; Cooper, J.A.; Twardzik, D.R.; Moss, B. Deletion of the vaccinia virus growth factor gene reduces virus virulence. J. Virol. 1988, 62, 866–874. [Google Scholar] [CrossRef]
- Mejías-Pérez, E.; CarreO-Fuentes, L.; Esteban, M. Development of a Safe and Effective Vaccinia Virus Oncolytic Vector WR-Δ4 with a Set of Gene Deletions on Several Viral Pathways. Mol. Ther. Oncolytics 2017, 8, 27–40. [Google Scholar] [CrossRef]
- Alcami, A.; Symons, J.A.; Smith, G.L. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J. Virol. 2000, 74, 11230–11239. [Google Scholar] [CrossRef]
- Katsoulidis, E.; Kaur, S.; Platanias, L.C. Deregulation of Interferon Signaling in Malignant Cells. Pharmaceuticals 2010, 3, 406–418. [Google Scholar] [CrossRef]
- Liu, Z. Deletion of C7L and K1L Genes Leads to Significantly Decreased Virulence of Recombinant Vaccinia Virus TianTan. Aids Res. Hum. Retrov. 2013, 29, A153. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.G.; Workenhe, S.T.; Konda, P.; Gujar, S.; Kroemer, G. Cytokines in oncolytic virotherapy. Cytokine Growth Factor Rev. 2020, 56, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Meko, J.B.; Yim, J.H.; Tsung, K.; Norton, J.A. High cytokine production and effective antitumor activity of a recombinant vaccinia virus encoding murine interleukin 12. Cancer Res. 1995, 55, 4765–4770. [Google Scholar] [PubMed]
- van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of dendritic cell development by GM-CSF: Molecular control and implications for immune homeostasis and therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef]
- Chu, Y.; Wang, L.X.; Yang, G.; Ross, H.J.; Urba, W.J.; Prell, R.; Jooss, K.; Xiong, S.; Hu, H.M. Efficacy of GM-CSF-producing tumor vaccine after docetaxel chemotherapy in mice bearing established Lewis lung carcinoma. J. Immunother. 2006, 29, 367–380. [Google Scholar] [CrossRef]
- Malhotra, S.; Kim, T.; Zager, J.; Bennett, J.; Ebright, M.; D’Angelica, M.; Fong, Y. Use of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomas. Surgery 2007, 141, 520–529. [Google Scholar] [CrossRef]
- Ramesh, N.; Ge, Y.; Ennist, D.L.; Zhu, M.; Mina, M.; Ganesh, S.; Reddy, P.S.; Yu, D.C. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor-armed oncolytic adenovirus for the treatment of bladder cancer. Clin. Cancer Res. 2006, 12, 305–313. [Google Scholar] [CrossRef]
- Kohno, S.I.; Luo, C.; Nawa, A.; Fujimoto, Y.; Watanabe, D.; Goshima, F.; Tsurumi, T.; Nishiyama, Y. Oncolytic virotherapy with an HSV amplicon vector expressing granulocyte-macrophage colony-stimulating factor using the replication-competent HSV type 1 mutant HF10 as a helper virus. Cancer Gene Ther. 2007, 14, 918–926. [Google Scholar] [CrossRef]
- Liu, T.C.; Hwang, T.; Park, B.H.; Bell, J.; Kirn, D.H. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol. Ther. 2008, 16, 1637–1642. [Google Scholar] [CrossRef]
- Lee, J.H.; Roh, M.S.; Lee, Y.K.; Kim, M.K.; Han, J.Y.; Park, B.H.; Trown, P.; Kirn, D.H.; Hwang, T.H. Oncolytic and immunostimulatory efficacy of a targeted oncolytic poxvirus expressing human GM-CSF following intravenous administration in a rabbit tumor model. Cancer Gene Ther. 2010, 17, 73–79. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic Vaccinia Virus Disrupts Tumor-Associated Vasculature in Humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- DiPaola, R.S.; Plante, M.; Kaufman, H.; Petrylak, D.P.; Israeli, R.; Lattime, E.; Manson, K.; Schuetz, T. A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J. Transl. Med. 2006, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Grosenbach, D.W.; Aarts, W.M.; Poole, D.J.; Schlom, J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin. Cancer Res. 2003, 9, 1837–1849. [Google Scholar] [PubMed]
- Downs-Canner, S.; Guo, Z.S.; Ravindranathan, R.; Breitbach, C.J.; O’Malley, M.E.; Jones, H.L.; Moon, A.; McCart, J.A.; Shuai, Y.; Zeh, H.J.; et al. Phase 1 Study of Intravenous Oncolytic Poxvirus (vvDD) in Patients With Advanced Solid Cancers. Mol. Ther. 2016, 24, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lynn, R.C.; Cheng, G.; Alexander, E.; Kapoor, V.; Moon, E.K.; Sun, J.; Fridlender, Z.G.; Isaacs, S.N.; Thorne, S.H.; et al. Treating tumors with a vaccinia virus expressing IFNβ illustrates the complex relationships between oncolytic ability and immunogenicity. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 736. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.H.; Wang, Y.; Le Boeuf, F.; Bell, J.; Thorne, S.H. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007, 4, e353. [Google Scholar] [CrossRef] [PubMed]
- Kowalsky, S.J.; Liu, Z.Q.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef]
- Li, J.; O’Malley, M.; Sampath, P.; Kalinski, P.; Bartlett, D.L.; Thorne, S.H. Expression of CCL19 from Oncolytic Vaccinia Enhances Immunotherapeutic Potential while Maintaining Oncolytic Activity. Neoplasia 2012, 14, 1115–1121. [Google Scholar] [CrossRef]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997, 90, 2541–2548. [Google Scholar]
- Nazari, A.; Ahmadi, Z.; Hassanshahi, G.; Abbasifard, M.; Taghipour, Z.; Falahati-Pour, S.K.; Khorramdelazad, H. Effective Treatments for Bladder Cancer Affecting CXCL9/CXCL10/CXCL11/CXCR3 Axis: A Review. Oman. Med. J. 2020, 35, e103. [Google Scholar] [CrossRef]
- Kistner, L.; Doll, D.; Holtorf, A.; Nitsche, U.; Janssen, K.P. Interferon-inducible CXC-chemokines are crucial immune modulators and survival predictors in colorectal cancer. Oncotarget 2017, 8, 89998–90012. [Google Scholar] [CrossRef] [PubMed]
- Hensbergen, P.J.; Wijnands, P.; Schreurs, M.; Scheper, R.J.; Willemze, R.; Tensen, C.P. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J. Immunother. 2005, 28, 343. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.; Wang, L.C.S.; Bekdache, K.; Lynn, R.C.; Lo, A.; Thorne, S.H.; Albelda, S.M. Intra-Tumoral Delivery of CXCL11 via a Vaccinia Virus, but not by Modified T Cells, Enhances the Efficacy of Adoptive T Cell Therapy and Vaccines. Oncoimmunology 2017, 7, e1395997. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Ravindranathan, R.; Li, J.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology 2016, 5, e1091554. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, E.; Combadière, C.; Iga, M.; Boissonnas, A.; Bonduelle, O.; Maho, M.; Debré, P.; Combadiere, B. Intratumoral CC Chemokine Ligand 5 Overexpression Delays Tumor Growth and Increases Tumor Cell Infiltration. J. Immunol. 2004, 173, 3755. [Google Scholar] [CrossRef] [PubMed]
- Bhat, H.; Zaun, G.; Hamdan, T.A.; Lang, J.; Adomati, T.; Schmitz, R.; Friedrich, S.K.; Bergerhausen, M.; Cham, L.B.; Li, F.; et al. Arenavirus Induced CCL5 Expression Causes NK Cell-Mediated Melanoma Regression. Front. Immunol. 2020, 11, 1849. [Google Scholar] [CrossRef]
- Li, J.; O’Malley, M.; Urban, J.; Sampath, P.; Guo, Z.S.; Kalinski, P.; Thorne, S.H.; Bartlett, D.L. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol. Ther. 2011, 19, 650–657. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D. PD-L1, TMB, and other potential predictors of response to immunotherapy for hepatocellular carcinoma: How can they assist drug clinical trials? Expert Opin. Investig. Drugs 2022, 31, 415–423. [Google Scholar] [CrossRef]
- Yu, F.; Wang, X.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.T. T-cell Engager-armed Oncolytic Vaccinia Virus Significantly Enhances Antitumor Therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Bollard, C.M.; Gottschalk, S.; Leen, A.M.; Weiss, H.; Straathof, K.C.; Carrum, G.; Khalil, M.; Wu, M.F.; Huls, M.H.; Chang, C.C.; et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood 2007, 110, 2838–2845. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Abrams, S.; Schlom, J.; Kantor, J.A. Induction of antitumor immunity by recombinant vaccinia virus expressing B7-1 or B7-2 co-stimulatory molecules. Cancer Res. 1994, 54, 5552–5555. [Google Scholar] [PubMed]
- Zajac, P.; Schutz, A.; Oertli, D.; Noppen, C.; Schaefer, C.; Heberer, M.; Spagnoli, G.C.; Marti, W.R. Enhanced generation of cytotoxic T lymphocytes using recombinant vaccinia virus expressing human tumor-associated antigens and B7 costimulatory molecules. Cancer Res. 1998, 58, 4567–4571. [Google Scholar] [PubMed]
- Carroll, M.W.; Overwijk, W.W.; Surman, D.R.; Tsung, K.; Moss, B.; Restifo, N.P. Construction and characterization of a triple-recombinant vaccinia virus encoding B7-1, interleukin 12, and a model tumor antigen. J. Natl. Cancer Inst. 1998, 90, 1881–1887. [Google Scholar] [CrossRef]
- Chen, L. Co-inhibitory molecules of the B7–CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Deraffele, G.; Mitcham, J.; Moroziewicz, D.; Cohen, S.M.; Hurst-Wicker, K.S.; Cheung, K.; Lee, D.S.; Divito, J.; Voulo, M.; et al. Targeting the local tumor microenvironment with vaccinia virus expressing B7.1 for the treatment of melanoma. J. Clin. Invest. 2005, 115, 1903–1912. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Koyama, S.; Maruyama, T.; Adachi, S.; Nozue, M. Expression of costimulatory molecules, B7-1 and B7-2 on human gastric carcinoma. J. Cancer Res. Clin. Oncol. 1998, 124, 383–388. [Google Scholar] [CrossRef]
- Feng, X.Y.; Lu, L.; Wang, K.F.; Zhu, B.Y.; Wen, X.Z.; Peng, R.Q.; Ding, Y.; Li, D.D.; Li, J.J.; Li, Y.; et al. Low expression of CD80 predicts for poor prognosis in patients with gastric adenocarcinoma. Future Oncol. 2019, 15, 473–483. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Oertli, D.; Marti, W.R.; Norton, J.A.; Tsung, K. Non-replicating recombinant vaccinia virus encoding murine B-7 molecules elicits effective costimulation of naive CD4+ splenocytes in vitro. J. Gen. Virol. 1996, 77 Pt 12, 3121–3125. [Google Scholar] [CrossRef]
- Haddad, D. Genetically engineered vaccinia viruses As Agents for Cancer Treatment, Imaging, and Transgene Delivery. Front. Oncol. 2017, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 50. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef]
- Scott, E.M.; Duffy, M.R.; Freedman, J.D.; Fisher, K.D.; Seymour, L.W. Solid Tumor Immunotherapy with T Cell Engager-Armed Oncolytic Viruses. Macromol. Biosci. 2018, 18, 1700187. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef]
- Yu, F.; Hong, B.; Song, X. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Transl. Med. 2017, 3, 122–132. [Google Scholar]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Tagliamonte, M.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Antigen-specific vaccines for cancer treatment. Hum. Vaccines Immunother. 2014, 10, 3332–3346. [Google Scholar] [CrossRef] [PubMed]
- Masuelli, L.; Fantini, M.; Benvenuto, M.; Sacchetti, P.; Giganti, M.G.; Tresoldi, I.; Lido, P.; Lista, F.; Cavallo, F.; Nanni, P.; et al. Intratumoral delivery of recombinant vaccinia virus encoding for ErbB2/Neu inhibits the growth of salivary gland carcinoma cells. J. Transl. Med. 2014, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Nesslinger, N.J.; Ng, A.; Tsang, K.-Y.; Ferrara, T.; Schlom, J.; Gulley, J.L.; Nelson, B.H. A viral vaccine encoding PSA induces antigen spreading to a common set of self proteins in prostate cancer patients. Clin. Cancer Res. 2010, 16, 4046. [Google Scholar] [CrossRef]
- Gulley, J.L.; Arlen, P.M.; Madan, R.A.; Tsang, K.Y.; Pazdur, M.P.; Skarupa, L.; Jones, J.L.; Poole, D.J.; Higgins, J.P.; Hodge, J.W.; et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol. Immun. 2010, 59, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.; Chen, A.P.; Dahut, W.; Arlen, P.M.; Bastian, A.; Steinberg, S.M.; Tsang, K.; Panicali, D.; Poole, D.; Schlom, J.; et al. Phase I study of a vaccine using recombinant vaccinia virus expressing PSA (rV-PSA) in patients with metastatic androgen-independent prostate cancer. Prostate 2010, 53, 109–117. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef]
- Bendjama, K.; Quemeneur, E. Modified Vaccinia virus Ankara-based vaccines in the era of personalized immunotherapy of cancer. Hum. Vaccines Immunother. 2017, 13, 1997–2003. [Google Scholar] [CrossRef]
- Brun, J.L.; Dalstein, V.; Leveque, J.; Mathevet, P.; Raulic, P.; Baldauf, J.J.; Scholl, S.; Huynh, B.; Douvier, S.; Riethmuller, D.; et al. Regression of high-grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am. J. Obstet. Gynecol. 2011, 204, 169.e1–169.e8. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, B.; Ren, J.; Feng, J.; Pang, Z.; Gao, J.; Zhang, H.; Tan, W.; Tian, H.; Ruan, L. Immunogenicity in mice and rhesus monkeys vaccinated with recombinant vaccinia virus expressing bivalent E7E6 fusion proteins from human papillomavirus types 16 and 18. Virol. J. 2011, 8, 302. [Google Scholar] [CrossRef]
- Ricci, A.D.; Rizzo, A.; Rojas Llimpe, F.L.; Di Fabio, F.; De Biase, D.; Rihawi, K. Novel HER2-Directed Treatments in Advanced Gastric Carcinoma: AnotHER Paradigm Shift? Cancers 2021, 13, 1664. [Google Scholar] [CrossRef] [PubMed]
- de Vries, C.R.; Monken, C.E.; Lattime, E.C. The addition of recombinant vaccinia HER2/neu to oncolytic vaccinia-GMCSF given into the tumor microenvironment overcomes MDSC-mediated immune escape and systemic anergy. Cancer Gene Ther. 2015, 22, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Focaccetti, C.; Izzi, V.; Masuelli, L.; Modesti, A.; Bei, R. Tumor antigens heterogeneity and immune response-targeting neoantigens in breast cancer. Semin. Cancer Biol. 2019, 72, 65–75. [Google Scholar] [CrossRef]
- Heemskerk, B.; Kvistborg, P.; Schumacher, T.N. The cancer antigenome. EMBO J. 2013, 32, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Warren, R.L.; Gibb, E.A.; Martin, S.D.; Spinelli, J.J.; Nelson, B.H.; Holt, R.A. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014, 24, 743–750. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Aldous, A.R.; Dong, J.Z. Personalized neoantigen vaccines: A new approach to cancer immunotherapy. Bioorg. Med. Chem. 2018, 26, 2842–2849. [Google Scholar] [CrossRef]
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R., 3rd; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.D.; Coukos, G.; Holt, R.A.; Nelson, B.H. Targeting the undruggable: Immunotherapy meets personalized oncology in the genomic era. Ann. Oncol. 2015, 26, 2367–2374. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, M.; Reid, T.R.; Larson, C.; Oronsky, B.; Carter, C.; Morris, J.C. Extended treatment with MY-NEOVAX, personalized neoantigen-enhanced oncolytic viruses, for two end-stage cancer patients. Oxf. Med. Case Rep. 2019, 2019, 461–463. [Google Scholar] [CrossRef]
- Kirn, D.H.; Wang, Y.H.; Liang, W.C.; Contag, C.H.; Thorne, S.H. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 2008, 68, 2071–2075. [Google Scholar] [CrossRef]
- Desai, S.J.; Prickril, B.; Rasooly, A. Mechanisms of Phytonutrient Modulation of Cyclooxygenase-2 (COX-2) and Inflammation Related to Cancer. Nutr. Cancer 2018, 70, 350–375. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.P.; Malboeuf, C.M.; Bernard, M.; Rose, R.C.; Phipps, R.P. Cyclooxygenase-2 inhibition attenuates antibody responses against human papillomavirus-like particles. J. Immunol. 2006, 177, 7811–7819. [Google Scholar] [CrossRef]
- Chang, C.L.; Ma, B.; Pang, X.W.; Wu, T.C.; Hung, C.F. Treatment With Cyclooxygenase-2 Inhibitors Enables Repeated Administration of Vaccinia Virus for Control of Ovarian Cancer. Mol. Ther. 2009, 17, 1365–1372. [Google Scholar] [CrossRef]
- Tang, B.; Guo, Z.S.; Bartlett, D.L.; Yan, D.Z.; Schane, C.P.; Thomas, D.L.; Liu, J.; McFadden, G.; Shisler, J.L.; Roy, E.J. Synergistic Combination of Oncolytic Virotherapy and Immunotherapy for Glioma. Clin. Cancer Res. 2020, 26, 2216–2230. [Google Scholar] [CrossRef]
- Hou, W.Z.; Sampath, P.; Rojas, J.J.; Thorne, S.H. Oncolytic Virus-Mediated Targeting of PGE2 in the Tumor Alters the Immune Status and Sensitizes Established and Resistant Tumors to Immunotherapy. Cancer Cell 2016, 30, 108–119. [Google Scholar] [CrossRef]
- Chon, H.J.; Lee, W.S.; Yang, H.; Kong, S.J.; Lee, N.K.; Moon, E.S.; Choi, J.; Han, E.C.; Kim, J.H.; Ahn, J.B.; et al. Tumor Microenvironment Remodeling by Intratumoral Oncolytic Vaccinia Virus Enhances the Efficacy of Immune-Checkpoint Blockade. Clin. Cancer Res. 2019, 25, 1612–1623. [Google Scholar] [CrossRef]
- Draghiciu, O.; Nijman, H.W.; Hoogeboom, B.N.; Meijerhof, T.; Daemen, T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology 2015, 4, e989764. [Google Scholar] [CrossRef] [PubMed]
- Finke, J.H.; Rini, B.; Ireland, J.; Rayman, P.; Richmond, A.; Golshayan, A.; Wood, L.; Elson, P.; Garcia, J.; Dreicer, R.; et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin. Cancer Res. 2008, 14, 6674–6682. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Nitschke, M.; Sennino, B.; Murer, P.; Schriver, B.J.; Bell, A.; Subramanian, A.; McDonald, C.E.; Wang, J.; Cha, H.; et al. Amplification of Oncolytic Vaccinia Virus Widespread Tumor Cell Killing by Sunitinib through Multiple Mechanisms. Cancer Res. 2018, 78, 922–937. [Google Scholar] [CrossRef]
- Farsaci, B.; Higgins, J.P.; Hodge, J.W. Consequence of dose scheduling of sunitinib on host immune response elements and vaccine combination therapy. Int. J. Cancer 2012, 130, 1948–1959. [Google Scholar] [CrossRef]
- Hou, W.; Chen, H.; Rojas, J.; Sampath, P.; Thorne, S.H. Oncolytic vaccinia virus demonstrates antiangiogenic effects mediated by targeting of VEGF. Int. J. Cancer 2014, 135, 1238–1246. [Google Scholar] [CrossRef]
- MacTavish, H.; Diallo, J.S.; Huang, B.; Stanford, M.; Le Boeuf, F.; De Silva, N.; Cox, J.; Simmons, J.G.; Guimond, T.; Falls, T.; et al. Enhancement of vaccinia virus based oncolysis with histone deacetylase inhibitors. PLoS ONE 2010, 5, e14462. [Google Scholar] [CrossRef] [PubMed]
- Francis, L.; Guo, Z.S.; Liu, Z.; Ravindranathan, R.; Urban, J.A.; Sathaiah, M.; Magge, D.; Kalinski, P.; Bartlett, D.L. Modulation of chemokines in the tumor microenvironment enhances oncolytic virotherapy for colorectal cancer. Oncotarget 2016, 7, 22174–22185. [Google Scholar] [CrossRef]
- Peng, J.; Wang, S.; Fan, W.; Li, S.; Wu, Y.; Mou, X.; Wang, J.; Tong, X. Synergistic suppression effect on tumor growth of acute myeloid leukemia by combining cytarabine with an engineered oncolytic vaccinia virus. Onco Targets Ther. 2018, 11, 6887–6900. [Google Scholar] [CrossRef]
- Marme, D. Tumor Angiogenesis: A Key Target for Cancer Therapy. Oncol. Res. Treat. 2018, 41, 164. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37 (Suppl. S4), S9–S15. [Google Scholar] [CrossRef]
- Serrano, M.J.; Alvarez-Cubero, M.J.; De Miguel Perez, D.; Rodríguez-Martínez, A.; Gonzalez-Herrera, L.; Robles-Fernandez, I.; Hernandez, J.E.; Puche, J.L.G.; Lorente, J.A. Significance of EGFR Expression in Circulating Tumor Cells. Adv. Exp. Med. Biol. 2017, 994, 285–296. [Google Scholar]
- De Luca, A.; Carotenuto, A.; Rachiglio, A.; Gallo, M.; Maiello, M.R.; Aldinucci, D.; Pinto, A.; Normanno, N. The role of the EGFR signaling in tumor microenvironment. J. Cell Physiol. 2008, 214, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Moeder, C.B.; Camp, R.L.; Rimm, D.L. Quantitative multiplexed analysis of ErbB family coexpression for primary breast cancer prognosis in a large retrospective cohort. Cancer 2009, 115, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.Q.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Curr. Vasc. Pharmacol. 2017, 15, 339–351. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ema, M. Roles of VEGF-A signalling in development, regeneration, and tumours. J. Biochem. 2014, 156, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Paterson, J.M.; Lemay, C.G.; Falls, T.J.; McGuire, A.; Parato, K.A.; Stojdl, D.F.; Daneshmand, M.; Speth, K.; Kirn, D.; et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007, 15, 1686–1693. [Google Scholar] [CrossRef]
- Gholami, S.; Marano, A.; Chen, N.G.; Aguilar, R.J.; Frentzen, A.; Chen, C.H.; Lou, E.; Fujisawa, S.; Eveno, C.; Belin, L.; et al. A novel vaccinia virus with dual oncolytic and anti-angiogenic therapeutic effects against triple-negative breast cancer. Breast Cancer Res. Treat. 2014, 148, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Adelfinger, M.; Bessler, S.; Frentzen, A.; Cecil, A.; Langbein-Laugwitz, J.; Gentschev, I.; Szalay, A.A. Preclinical Testing Oncolytic Vaccinia Virus Strain GLV-5b451 Expressing an Anti-VEGF Single-Chain Antibody for Canine Cancer Therapy. Viruses 2015, 7, 4075–4092. [Google Scholar] [CrossRef]
- Patil, S.S.; Gentschev, I.; Adelfinger, M.; Donat, U.; Hess, M.; Weibel, S.; Nolte, I.; Frentzen, A.; Szalay, A.A. Virotherapy of canine tumors with oncolytic vaccinia virus GLV-1h109 expressing an anti-VEGF single-chain antibody. PLoS ONE 2012, 7, e47472. [Google Scholar] [CrossRef]
- Huang, T.; Wang, H.Q.; Chen, N.G.; Frentzen, A.; Minev, B.; Szalay, A.A. Expression of anti-VEGF antibody together with anti-EGFR or anti-FAP enhances tumor regression as a result of vaccinia virotherapy. Mol. Ther.Oncolytics 2015, 2, 15003. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Rioth, M.J.; Horn, L. Immune Checkpoint Inhibitors in NSCLC. Curr. Treat Option. Oncol. 2014, 15, 658–669. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bondarenko, I.; Luft, A. Ipilimumab in Combination With Paclitaxel and Carboplatin As First-Line Treatment in Stage IIIB/IV Non-Small-Cell Lung Cancer: Results From a Randomized, Double-Blind, Multicenter Phase II Study. J. Clin. Oncol. 2012, 30, 3654. [Google Scholar] [CrossRef]
- Hodi, F.S. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 1290. [Google Scholar] [CrossRef]
- Woller, N.; Gurlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.C.; Jing, S.S.; Wang, B.; Wu, K.; Shenglin, M.A.; Zhang, S. Anti-PD-1/PD-L1 Therapy as a Promising Option for Non-Small Cell Lung Cancer: A Single arm Meta-Analysis. Pathol. Oncol. Res. 2016, 22, 331–339. [Google Scholar] [CrossRef]
- Remy-Ziller, C.; Thioudellet, C.; Hortelano, J.; Gantzer, M.; Nourtier, V.; Claudepierre, M.C.; Sansas, B.; Préville, X.; Bendjama, K.; Quemeneur, E.; et al. Sequential administration of MVA-based vaccines and PD-1/PD-L1-blocking antibodies confers measurable benefits on tumor growth and survival: Preclinical studies with MVA-Gal and MVA-MUC1 (TG4010) in a murine tumor model. Hum. Vaccinia Immunother. 2018, 14, 140–145. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Adamow, M.; Ginsberg, B.A.; Rasalan, T.S.; Ritter, E.; Gallardo, H.F.; Xu, Y.; Pogoriler, E.; Terzulli, S.L.; Kuk, D.; et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc. Natl. Acad. Sci. USA 2011, 108, 16723–16728. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Foy, S.P.; Mandl, S.J.; dela Cruz, T.; Cote, J.J.; Gordon, E.J.; Trent, E.; Delcayre, A.; Breitmeyer, J.; Franzusoff, A.; Rountree, R.B. Poxvirus-based active immunotherapy synergizes with CTLA-4 blockade to increase survival in a murine tumor model by improving the magnitude and quality of cytotoxic T cells. Cancer Immunol. Immunother. 2016, 65, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.G.; Mansfield, D.; Roulstone, V.; Kyula-Currie, J.N.; McLaughlin, M.; Patel, R.R.; Bergerhoff, K.F.; Paget, J.T.; Dillon, M.T.; Khan, A.; et al. PD-1 Blockade Following Isolated Limb Perfusion with Vaccinia Virus Prevents Local and Distant Relapse of Soft-tissue Sarcoma. Clin. Cancer Res. 2019, 25, 3443–3454. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Lu, J.; Kim, S.I.; Warner, S.G.; Von Hoff, D.; Fong, Y. Novel Chimeric Immuno-Oncolytic Virus CF33-hNIS-antiPDL1 for the Treatment of Pancreatic Cancer. J. Am. Coll Surg. 2020, 230, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, R.; Esmaeili Gouvarchin Ghaleh, H.; Farzanehpour, M.; Dorostkar, R.; Ranjbar, R.; Bolandian, M.; Mirzaei Nodooshan, M.; Ghorbani Alvanegh, A. Combination therapy with CAR T cells and oncolytic viruses: A new era in cancer immunotherapy. Cancer Gene Ther. 2022, 29, 647–660. [Google Scholar] [CrossRef]
- Watanabe, N.; McKenna, M.K.; Rosewell Shaw, A.; Suzuki, M. Clinical CAR-T Cell and Oncolytic Virotherapy for Cancer Treatment. Mol. Ther. 2021, 29, 505–520. [Google Scholar] [CrossRef]
- Park, A.K.; Fong, Y.; Kim, S.I.; Yang, J.; Murad, J.P.; Lu, J.; Jeang, B.; Chang, W.C.; Chen, N.G.; Thomas, S.H.; et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020, 12, eaaz1863. [Google Scholar] [CrossRef]
- Pelner, L.; Fowler, G.A.; Nauts, H.C. Effects of concurrent infections and their toxins on the course of leukemia. Acta Med. Scand. Suppl. 1958, 338, 1–47. [Google Scholar] [CrossRef]
- Lee, W.; Jiang, Z.; Liu, J.; Haverty, P.M.; Guan, Y.; Stinson, J.; Yue, P.; Zhang, Y.; Pant, K.P.; Bhatt, D.; et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2010, 465, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Auer, R.; Bell, J.C. Oncolytic viruses: Smart therapeutics for smart cancers. Future Oncol. 2012, 8, 1–4. [Google Scholar] [CrossRef]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. Adv. Virus Res. 2017, 97, 187–243. [Google Scholar] [PubMed]
- Chiuppesi, F.; Salazar, M.D.; Contreras, H.; Nguyen, V.H.; Martinez, J.; Park, Y.; Nguyen, J.; Kha, M.; Iniguez, A.; Zhou, Q.; et al. Development of a multi-antigenic SARS-CoV-2 vaccine candidate using a synthetic poxvirus platform. Nat. Commun. 2020, 11, 6121. [Google Scholar] [CrossRef] [PubMed]
- Rihawi, K.; Ricci, A.D.; Rizzo, A.; Brocchi, S.; Marasco, G.; Pastore, L.V.; Llimpe, F.L.R.; Golfieri, R.; Renzulli, M. Tumor-Associated Macrophages and Inflammatory Microenvironment in Gastric Cancer: Novel Translational Implications. Int. J. Mol. Sci. 2021, 22, 3805. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name | Description | Delivery Route | Cancer | Co-Therapy | Phase | Status | Reference |
|---|---|---|---|---|---|---|---|
| JX-594 | Wyeth Strain; Deletion:
| i.t. (intrathecal) | Solid tumors |
| 1 | Recruiting | NCT02977156 |
| i.v. (intravenous) | Refractory colorectal cancer |
| 1/2 | Active, not recruiting | NCT03206073 | ||
| i.v., i.t. | Renal cell carcinoma | REGN2810 (Anti-PD-1) | 1b | Recruiting | NCT03294083 | ||
| i.t. | Hepatocellular carcinoma | Sorafenib | 3 | Completed | NCT02562755 | ||
| GL-ONC1 (GLV-1h68) | Lister strain; Deletions:
| i.v. | Head and neck carcinoma | - | 1 | Completed | NCT01584284 |
| i.p. (intraperitoneal) | Ovarian cancer | bevacizumab | 1b/2 | Recruiting | NCT02759588 | ||
| i.p. | Peritoneal carcinomatosis | - | 1/2 | Completed | NCT01443260 | ||
| i.v. | Solid tumors | - | 1 | Completed | NCT00794131 | ||
| PROSTVAC-V (Vaccinia-PSA-TRICOM) | Strain: partially attenuated version of the virus used for smallpox immunization [42]; Transgenes:
| s.c. (subcutaneous) | Prostate cancer |
| 2 | Completed | NCT00450619 |
| s.c. | Prostate Cancer |
| 3 | Completed | NCT01322490 | ||
| Vaccinia-CEA-TRICOM | Wyeth strain; Transgenes:
| s.c. | Breast Cancer |
| 2 | Completed | NCT00052351 |
| p53MVA | Ankara strain; Transgenes:
| i.v. |
| Pembrolizumab | 2 | Recruiting | NCT03113487 |
| s.c. |
| - | 1 | Completed | NCT01191684 | ||
| TG6002 | Copenhagen strain; Deletions:
| i.v. | Glioblastoma | 5-flucytosine (5-FC) | 1/2 | Recruiting | NCT03294486 |
| Intrahepatic arterial (IHA) administration | Colorectal cancer | 5-FC | 1/2 | Recruiting | NCT04194034 | ||
| i.v. | Gastro-intestinal tumors | 5-FC | 1/2 | Recruiting | NCT03724071 | ||
| MVA-brachyury- TRICOM | Ankara strain; Transgenes:
| s.c. |
| - | 1 | Completed | NCT02179515 |
| TG4010 | Ankara strain; Transgenes:
| s.c. | Non-small cell lung cancer | Nivolumab | 2 | Active, not recruiting | NCT02823990 |
| - | MUC-1 positive advanced cancer | - | Completed | NCT00004881 | |||
| s.c. | Non-small cell lung cancer |
| 2 | Active, not recruiting | NCT03353675 | ||
| vvDD-CDSR | Western Reserve strain; Deletion:
| i.t. i.v. |
| - | 1 | Completed | NCT00574977 |
| MVA-5T4 (TroVax®®) | Ankara strain; Transgenes:
| i.m. (intramuscular) |
| - | 2 | Completed | NCT01556841 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Zhao, J.; Li, X.; Lao, F.; Fang, M. Design Strategies and Precautions for Using Vaccinia Virus in Tumor Virotherapy. Vaccines 2022, 10, 1552. https://doi.org/10.3390/vaccines10091552
Liu X, Zhao J, Li X, Lao F, Fang M. Design Strategies and Precautions for Using Vaccinia Virus in Tumor Virotherapy. Vaccines. 2022; 10(9):1552. https://doi.org/10.3390/vaccines10091552
Chicago/Turabian StyleLiu, Xinjun, Jian Zhao, Xiaopeng Li, Fengxue Lao, and Min Fang. 2022. "Design Strategies and Precautions for Using Vaccinia Virus in Tumor Virotherapy" Vaccines 10, no. 9: 1552. https://doi.org/10.3390/vaccines10091552
APA StyleLiu, X., Zhao, J., Li, X., Lao, F., & Fang, M. (2022). Design Strategies and Precautions for Using Vaccinia Virus in Tumor Virotherapy. Vaccines, 10(9), 1552. https://doi.org/10.3390/vaccines10091552