
Immunotherapies for Alzheimer’s Disease—A Review
 ,
,  ,
,  and
and 
Abstract
:1. Introduction
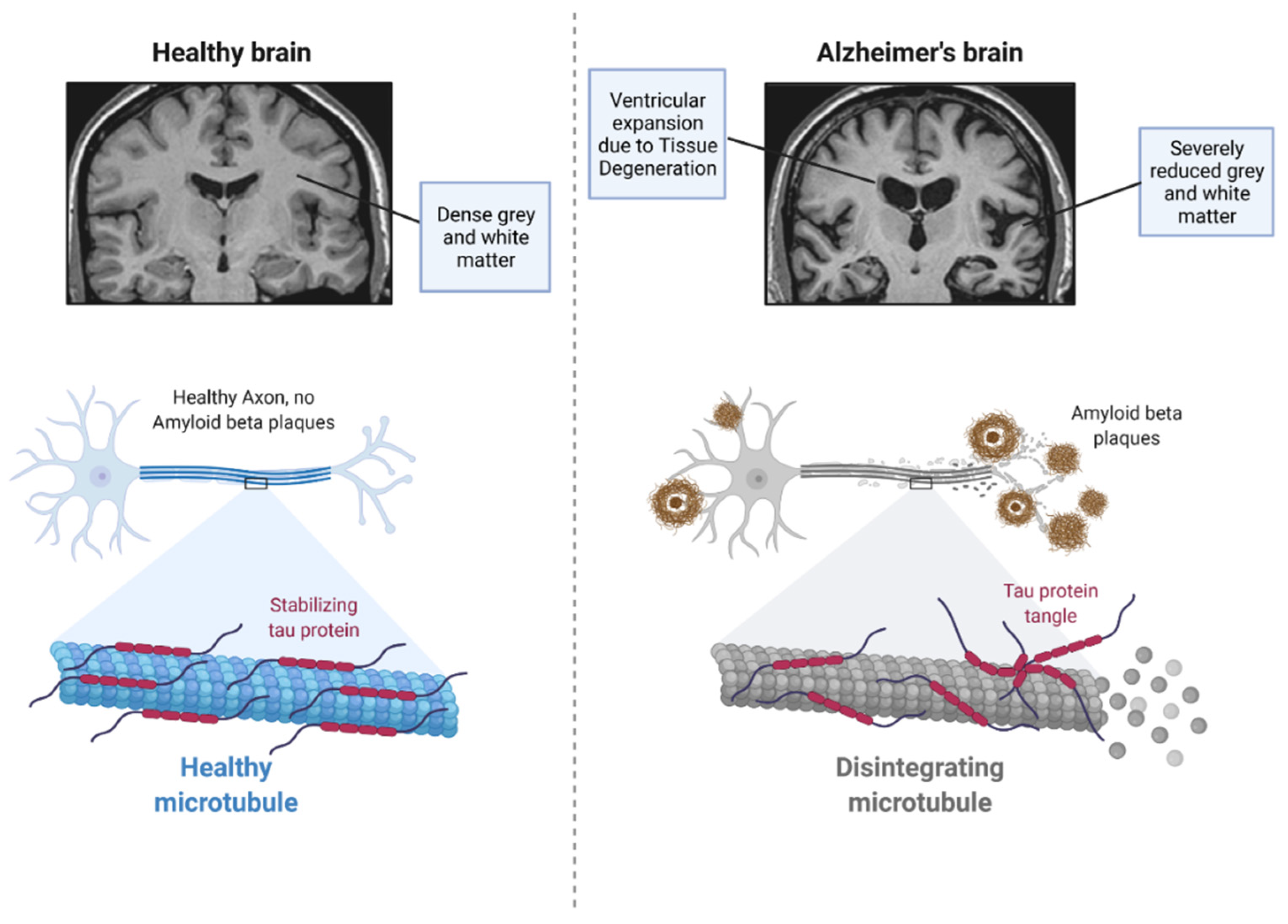
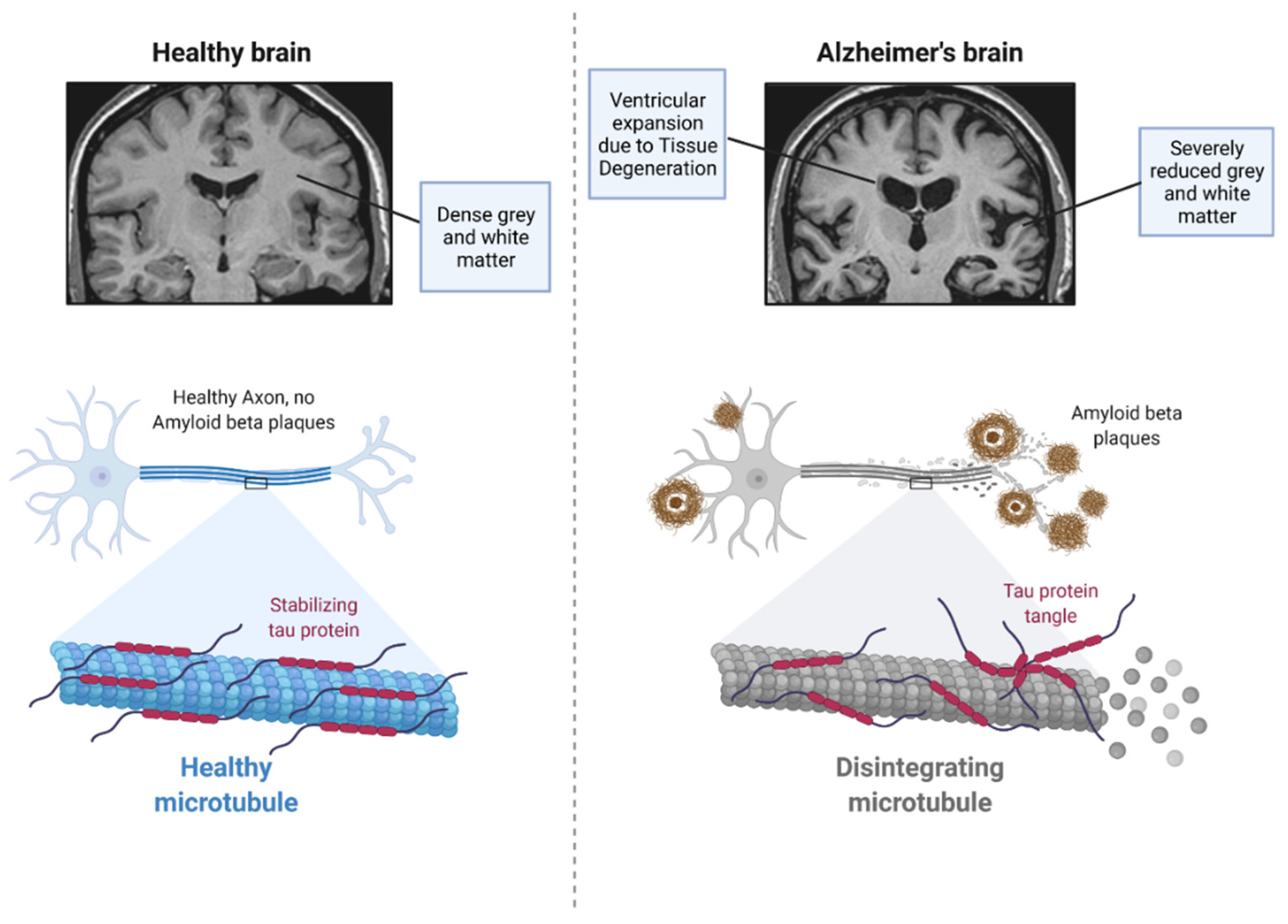
2. Pathophysiology of Alzheimer’s Disease
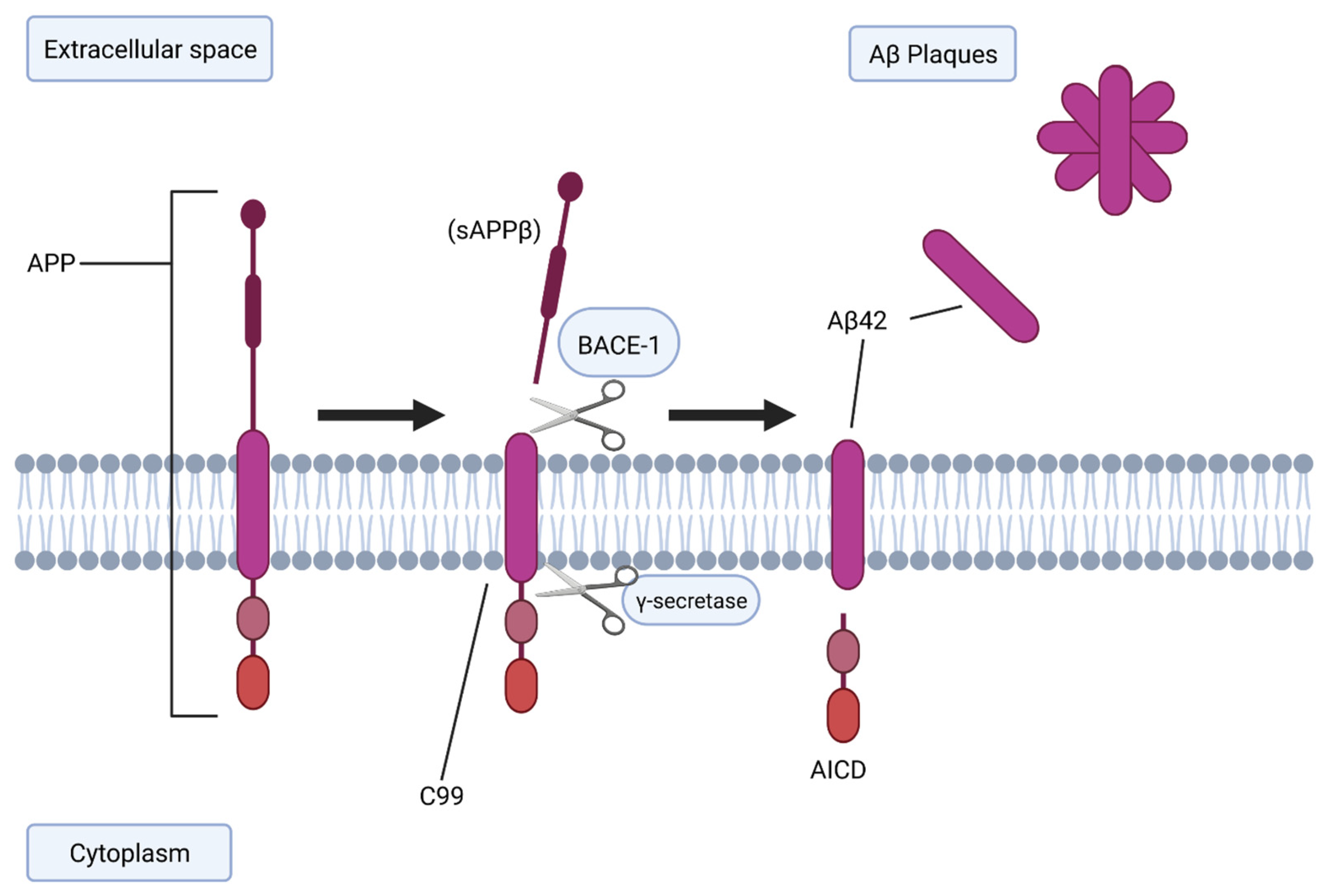
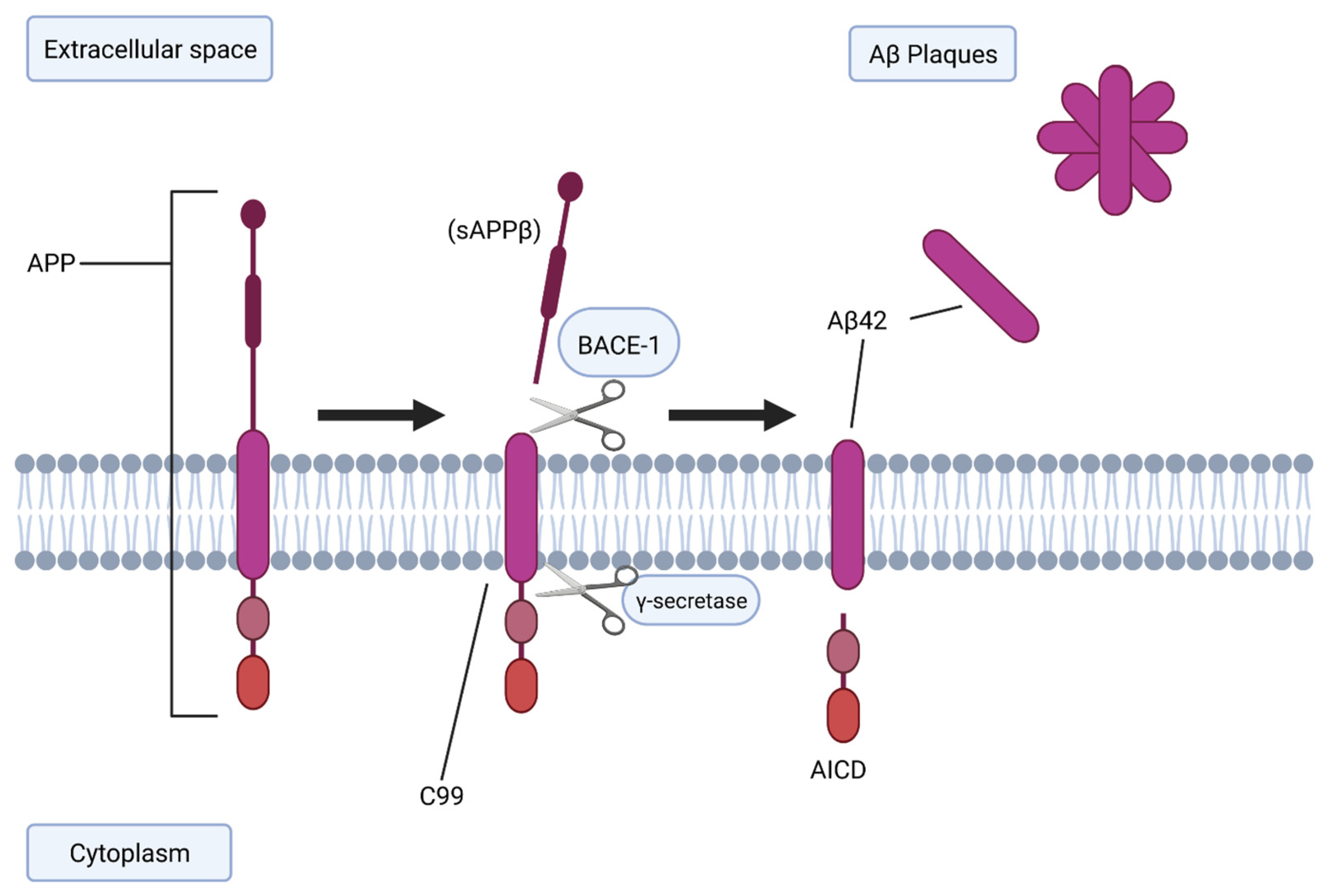
2.1. BACE-1 and Aβ Generation
2.2. Tau Protein
2.3. APOE-ε4
2.4. TREM2
2.5. Other Contributing Factors
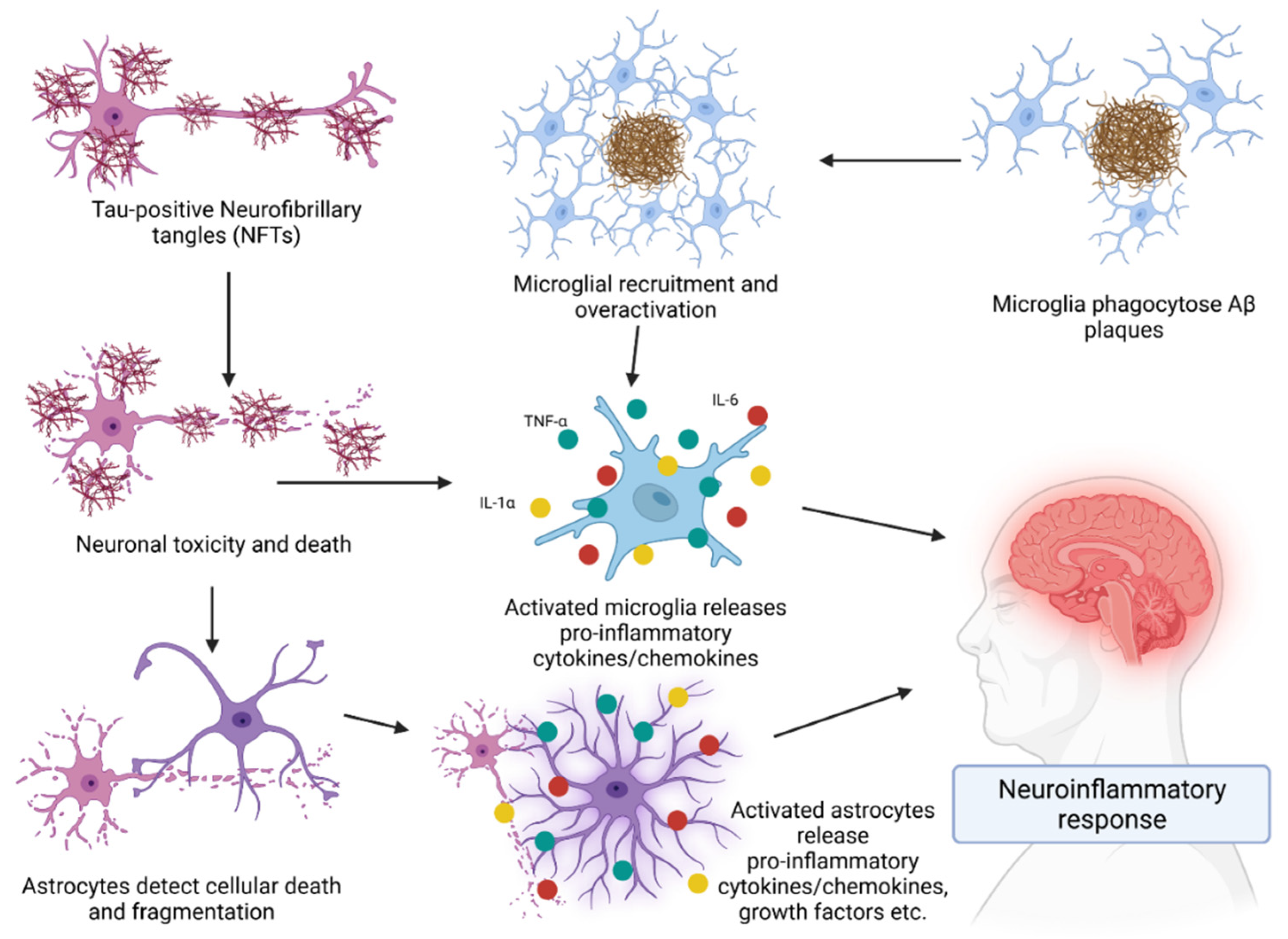
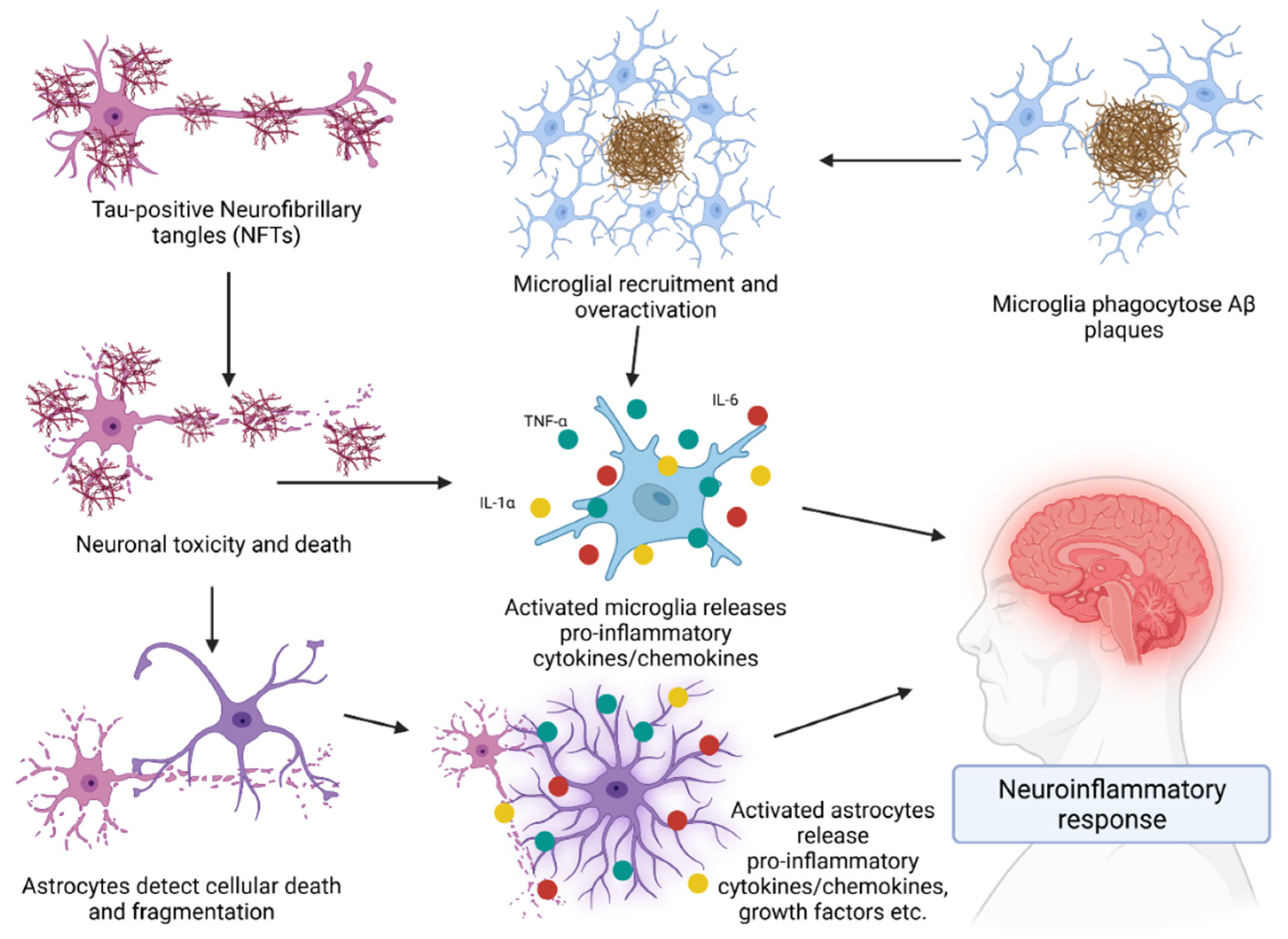
3. Role of the Immune System in AD
Cytokines in AD
4. Immunotherapies for AD
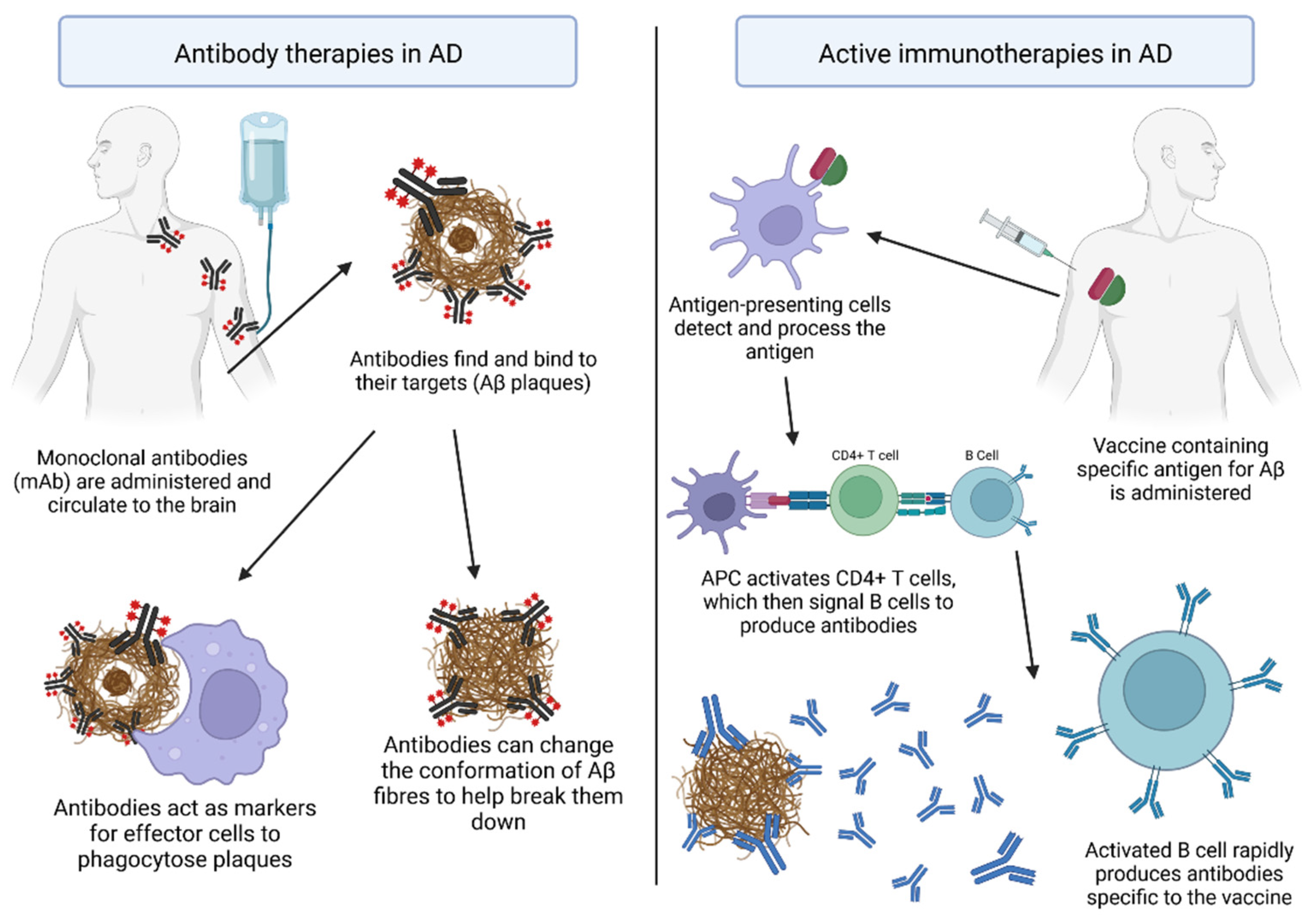
4.1. Antibody Therapies for AD
4.2. Active Vaccinations against AD
4.3. The Limitations and Challenges of Immunotherapies
4.4. Analysis of Immunotherapeutic Efficacy
4.5. Cerebral Amyloid Angiopathy and Amyloid-Related Imaging Abnormalities
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laurent, C.; Buee, L.; Blum, D. Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies? Biomed. J. 2018, 41, 21–33. [Google Scholar] [CrossRef]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Xu, W.; Marseglia, A.; Ferrari, C.; Wang, H.-X. Alzheimer’s Disease: A Clinical Perspective. In Neurodegenerative Diseases; InTech Open: London, UK, 2013; pp. 3–34. [Google Scholar]
- Duong, S.; Patel, T.; Chang, F. Dementia: What pharmacists need to know. Can. Pharm. J. 2017, 150, 118–129. [Google Scholar] [CrossRef]
- Neugroschl, J.; Wang, S. Alzheimer’s disease: Diagnosis and treatment across the spectrum of disease severity. Mt. Sinai J. Med. 2011, 78, 596–612. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Chen, F.; Ghosh, A.; Lin, J.; Zhang, C.; Pan, Y.; Thakur, A.; Singh, K.; Hong, H.; Tang, S. 5-Lipoxygenase pathway and its downstream cysteinyl leukotrienes as potential therapeutic targets for Alzheimer’s disease. Brain Behav. Immun. 2020, 88, 844–855. [Google Scholar] [CrossRef]
- Zhou, F.; He, K.; Guan, Y.; Yang, X.; Chen, Y.; Sun, M.; Qiu, X.; Yan, F.; Huang, H.; Yao, L.; et al. Network pharmacology-based strategy to investigate pharmacological mechanisms of Tinospora sinensis for treatment of Alzheimer’s disease. J. Ethnopharmacol. 2020, 259, 112940. [Google Scholar] [CrossRef]
- Barrera-Ocampo, A.; Lopera, F. Amyloid-Beta immunotherapy: The hope for Alzheimer disease? Colomb. Med. 2016, 47, 203–212. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Jevtic, S.; Sengar, A.S.; Salter, M.W.; McLaurin, J. The role of the immune system in Alzheimer disease: Etiology and treatment. Ageing Res. Rev. 2017, 40, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Korman, D.; Baker, S.L.; Landau, S.M.; Jagust, W.J. Longitudinal Cognitive and Biomarker Measurements Support a Unidirectional Pathway in Alzheimer’s Disease Pathophysiology. Biol. Psychiatry 2020, 89, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Leko, M.B.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V. Brain metabolic stress and neuroinflammation at the basis of cognitive impairment in Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 94. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Kapadia, A.; Mirrahimi, A.; Dmytriw, A.A. Intersection between sleep and neurovascular coupling as the driving pathophysiology of Alzheimer’s disease. Med. Hypotheses 2020, 144, 110283. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Zhang, X.; Song, W. The role of APP and BACE1 trafficking in APP processing and amyloid-β generation. Alzheimer’s Res. Ther. 2013, 5, 46. [Google Scholar] [CrossRef]
- Capone, R.; Tiwari, A.; Hadziselimovic, A.; Peskova, Y.; Hutchison, J.M.; Sanders, C.R.; Kenworthy, A.K. The C99 domain of the amyloid precursor protein resides in the disordered membrane phase. J. Biol. Chem. 2021, 296, 100652. [Google Scholar] [CrossRef]
- Sowade, R.F.; Jahn, T.R. Seed-Induced acceleration of amyloid-β mediated neurotoxicity in vivo. Nat. Commun. 2017, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Bolon, B.; Damore, M.A.; Fitzpatrick, D.; Liu, H.; Zhang, J.; Yan, Q.; Vassar, R.; Citron, M. BACE1 (β-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol. Dis. 2003, 14, 81–88. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, H.; Sun, Q.; Yao, H.; Keegan, A.P.; Mullan, M.; Wilson, J.; Lista, S.; Leyhe, T.; Laske, C.; et al. Increased Plasma Beta-Secretase 1 May Predict Conversion to Alzheimer’s Disease Dementia in Individuals with Mild Cognitive Impairment. Biol. Psychiatry 2018, 83, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Vassar, R.; de Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; de Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2020, 89, 745–756. [Google Scholar] [CrossRef]
- Peters, F.; Salihoglu, H.; Rodrigues, E.; Herzog, E.; Blume, T.; Filser, S.; Dorostkar, M.; Shimshek, D.R.; Brose, N.; Neumann, U.; et al. BACE1 inhibition more effectively suppresses initiation than progression of β-amyloid pathology. Acta Neuropathol. 2018, 135, 695–710. [Google Scholar] [CrossRef]
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef]
- Novak, G.; Streffer, J.R.; Timmers, M.; Henley, D.; Brashear, H.R.; Bogert, J.; Russu, A.; Janssens, L.; Tesseur, I.; Tritsmans, L.; et al. Long-Term safety and tolerability of atabecestat (JNJ-54861911), an oral BACE1 inhibitor, in early Alzheimer’s disease spectrum patients: A randomized, double-blind, placebo-controlled study and a two-period extension study. Alzheimers Res. Ther. 2020, 12, 58. [Google Scholar] [CrossRef]
- Sur, C.; Kost, J.; Scott, D.; Adamczuk, K.; Fox, N.C.; Cummings, J.L.; Tariot, P.N.; Aisen, P.S.; Vellas, B.; Voss, T.; et al. BACE inhibition causes rapid, regional, and non-progressive volume reduction in Alzheimer’s disease brain. Brain 2020, 143, 3816–3826. [Google Scholar] [CrossRef]
- Šimić, G.; Leko, M.B.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-Specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Riphagen, J.M.; Ramakers, I.H.; Freeze, W.M.; Pagen, L.H.; Hanseeuw, B.J.; Verbeek, M.M.; Verhey, F.R.; Jacobs, H.I. Linking APOE-ε4, blood-brain barrier dysfunction, and inflammation to Alzheimer’s pathology. Neurobiol. Aging 2020, 85, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Mentis, A.-F.A.; Dardiotis, E.; Chrousos, G.P. Apolipoprotein E4 and meningeal lymphatics in Alzheimer disease: A conceptual framework. Mol. Psychiatry 2020, 26, 1075–1097. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.; Zhao, L. Human ApoE Isoforms Differentially Modulate Brain Glucose and Ketone Body Metabolism: Implications for Alzheimer’s Disease Risk Reduction and Early Intervention. J. Neurosci. 2018, 38, 6665–6681. [Google Scholar] [CrossRef]
- Fan, J.; Tao, W.; Li, X.; Li, H.; Zhang, J.; Wei, D.; Chen, Y.; Zhang, Z. The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk. Int. J. Mol. Sci. 2019, 20, 1177. [Google Scholar] [CrossRef]
- Pang, S.; Li, J.; Zhang, Y.; Chen, J. Meta-Analysis of the Relationship between the APOE Gene and the Onset of Parkinson’s Disease Dementia. Parkinson’s Dis. 2018, 2018, 9497147. [Google Scholar] [CrossRef]
- Deming, Y.; Li, Z.; Benitez, B.A.; Cruchaga, C. Triggering receptor expressed on myeloid cells 2 (TREM2): A potential therapeutic target for Alzheimer disease? Expert. Opin. Ther. Targets 2018, 22, 587–598. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, X.-F. The Emerging Roles and Therapeutic Potential of Soluble TREM2 in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 328. [Google Scholar] [CrossRef]
- Plotkin, S.S.; Cashman, N.R. Passive immunotherapies targeting Aβ and tau in Alzheimer’s disease. Neurobiol. Dis. 2020, 144, 105010. [Google Scholar] [CrossRef]
- Booth, F.W.; Roberts, C.K.; Laye, M.J. Lack of exercise is a major cause of chronic diseases. Compr. Physiol. 2012, 2, 1143–1211. [Google Scholar]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Cao, W.; Zheng, H. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Ramos, J.M.P.; Ben-Yehuda, H. A 20-Year Journey from Axonal Injury to Neurodegenerative Diseases and the Prospect of Immunotherapy for Combating Alzheimer’s Disease. J. Immunol. 2020, 204, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Kabir, M.T.; Mamun, A.A.; Barreto, G.E.; Rashid, M.; Perveen, A.; Ashraf, G.M. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int. Immunopharmacol. 2020, 84, 106479. [Google Scholar] [CrossRef]
- Counil, H.; Krantic, S. Synaptic Activity and (Neuro) Inflammation in Alzheimer’s Disease: Could Exosomes be an Additional Link? J. Alzheimer’s Dis. 2020, 74, 1029–1043. [Google Scholar] [CrossRef]
- Papatriantafyllou, M. Immunological bullets against Alzheimer’s disease. Nat. Rev. Immunol. 2013, 13, 3. [Google Scholar] [CrossRef]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef]
- Unger, M.S.; Li, E.; Scharnagl, L.; Poupardin, R.; Altendorfer, B.; Mrowetz, H.; Hutter-Paier, B.; Weiger, T.M.; Heneka, M.T.; Attems, J.; et al. CD8+ T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav. Immun. 2020, 89, 67–86. [Google Scholar] [CrossRef]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou-Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 2015, 6, 7967. [Google Scholar] [CrossRef] [PubMed]
- Monsonego, A.; Nemirovsky, A.; Harpaz, I. CD4 T cells in immunity and immunotherapy of Alzheimer’s disease. Immunology 2013, 139, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.V.; Prokop, S.; Miller, K.R.; Obst, J.; Kälin, R.E.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Scarabino, D.; Peconi, M.; Broggio, E.; Gambina, G.; Maggi, E.; Armeli, F.; Mantuano, E.; Morello, M.; Corbo, R.M.; Businaro, R. Relationship between proinflammatory cytokines (Il-1beta, Il-18) and leukocyte telomere length in mild cognitive impairment and Alzheimer’s disease. Exp. Gerontol. 2020, 136, 110945. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Yee, K.L.; Sumbria, R.K. Tumor necrosis factor α Inhibition for Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2017, 9, 1179573517709278. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Mrak, R.E.; Griffin, W.S. Interleukin-1 and the immunogenetics of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 471–476. [Google Scholar] [CrossRef]
- Gyengesi, E.; Rangel, A.; Ullah, F.; Liang, H.; Niedermayer, G.; Asgarov, R.; Venigalla, M.; Gunawardena, D.; Karl, T.; Münch, G. Chronic Microglial Activation in the GFAP-IL6 Mouse Contributes to Age-Dependent Cerebellar Volume Loss and Impairment in Motor Function. Front. Neurosci. 2019, 13, 303. [Google Scholar] [CrossRef]
- Balschun, D.; Wetzel, W.; del Rey, A.; Pitossi, F.; Schneider, H.; Zuschratter, W.; Besedovsky, H.O. Interleukin-6: A cytokine to forget. FASEB J. 2004, 18, 1788–1790. [Google Scholar] [CrossRef]
- Wang, J.-C.; Zhu, K.; Zhang, H.-Y.; Wang, G.-Q.; Liu, H.-Y.; Cao, Y.-P. Early active immunization with Aβ(3-10)-KLH vaccine reduces tau phosphorylation in the hippocampus and protects cognition of mice. Neural. Regen. Res. 2020, 15, 519–527. [Google Scholar]
- Kabir, M.; Uddin, M.; Mathew, B.; Das, P.K.; Ashraf, G.M. Emerging Promise of Immunotherapy for Alzheimer’s Disease: A New Hope for the Development of Alzheimer’s Vaccine. Curr. Top. Med. Chem. 2020, 20, 1214–1234. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Frisardi, V.; Solfrizzi, V.; Imbimbo, B.P.; Logroscino, G.; Santamato, A.; Greco, A.; Seripa, D.; Pilotto, A. Immunotherapy for Alzheimer’s disease: From anti-β-amyloid to tau-based immunization strategies. Immunotherapy 2012, 4, 213–238. [Google Scholar] [CrossRef]
- Cacabelos, R. How plausible is an Alzheimer’s disease vaccine? Expert Opin. Drug Discov. 2020, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Drummond, E. Developing therapeutic vaccines against Alzheimer’s disease. Expert Rev. Vaccines 2016, 15, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Herline, K.; Drummond, E.; Wisniewski, T. Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev. Vaccines 2018, 17, 707–721. [Google Scholar] [CrossRef]
- Zipfel, P.; Rochais, C.; Baranger, K.; Rivera, S.; Dallemagne, P. Matrix Metalloproteinases as New Targets in Alzheimer’s Disease: Opportunities and Challenges. J. Med. Chem. 2020, 63, 10705–10725. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Fu, L.; Zhang, Y.; Zhang, X.; Tian, W.; Zhang, W.; Jia, Y.; Zhang, L. Preparation and in vitro activity of single chain antibodies against Alzheimer’s disease. Immunol. Lett. 2020, 227, 1–7. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Deptula, D.; Thurfjell, L.; Barkhof, F.; Bohrmann, B.; Brooks, D.J.; Klunk, W.E.; Ashford, E.; Yoo, K.; Xu, Z.-X. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol. 2012, 69, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef] [PubMed]
- Söllvander, S.; Nikitidou, E.; Gallasch, L.; Zyśk, M.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J. Neuroinflamm. 2018, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Hettmann, T.; Gillies, S.D.; Kleinschmidt, M.; Piechotta, A.; Makioka, K.; Lemere, C.A.; Schilling, S.; Jens-Ulrich, R.; Inge, L. Development of the clinical candidate PBD-C06, a humanized pGlu3-Aβ-specific antibody against Alzheimer’s disease with reduced complement activation. Sci. Rep. 2020, 10, 3294. [Google Scholar] [CrossRef]
- Liu, D.-q.; Lu, S.; Zhang, L.; Huang, Y.-r.; Ji, M.; Sun, X.-y.; Liu, X.-g.; Liu, R.-t. Yeast-Based Aβ1-15 Vaccine Elicits Strong Immunogenicity and Attenuates Neuropathology and Cognitive Deficits in Alzheimer’s Disease Transgenic Mice. Vaccines 2020, 8, 351. [Google Scholar] [CrossRef]
- Petrushina, I.; Hovakimyan, A.; Harahap, I.; Davtyan, H.; Antonyan, T.; Chailyan, G.; Kazarian, K.; Antonenko, M.; Jullienne, A.; Hamer, M.M. Characterization and preclinical evaluation of the cGMP grade DNA based vaccine, AV-1959D to enter the first-in-human clinical trials. Neurobiol. Dis. 2020, 139, 104823. [Google Scholar] [CrossRef]
- Oluwasanmi Adeloye, O.; Babatunde David, O.; Idowu, O. Amyloid-β Immunotherapy on Alzheimer disease: Prevention and Therapeutic Target. J. Dermatol. Res. Rev. Rep. 2020, 1, 2. [Google Scholar]
- Cao, W.; Kim, J.H.; Reber, A.J.; Hoelscher, M.; Belser, J.A.; Lu, X.; Katz, J.M.; Gangappa, S.; Plante, M.; Burt, D.S.; et al. Nasal delivery of Protollin-adjuvanted H5N1 vaccine induces enhanced systemic as well as mucosal immunity in mice. Vaccine 2017, 35, 3318–3325. [Google Scholar] [CrossRef]
- Frenkel, D.; Maron, R.; Burt, D.S.; Weiner, H.L. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears beta-amyloid in a mouse model of Alzheimer disease. J. Clin. Investig. 2005, 115, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Puckett, L.; Petrovic, S.; Xia, W.; Chen, G.; Vega, J.; Dembinsky-Vaknin, A.; Shen, J.; Plante, M.; Burt, D.S.; et al. A nasal proteosome adjuvant activates microglia and prevents amyloid deposition. Ann. Neurol. 2008, 63, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Ishiura, S.; Yoshida, T. Plant-Based vaccines for Alzheimer’s disease. Proc. Jpn. Acad. Ser. B 2019, 95, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Denham, N.; Holmes, C.; Nicoll, J.A.R. Neuropathology after active Aβ42 immunotherapy: Implications for Alzheimer’s disease pathogenesis. Acta Neuropathol. 2010, 120, 369–384. [Google Scholar] [CrossRef]
- Takeda, S. Therapeutic Vaccines Targeting Alzheimer’s Disease. In Therapeutic Vaccines as Novel Immunotherapy: Biological and Clinical Concepts; Nakagami, H., Ed.; Springer: Singapore, 2019; pp. 9–20. [Google Scholar]
- Saresella, M.; Calabrese, E.; Marventano, I.; Piancone, F.; Gatti, A.; Farina, E.; Alberoni, M.; Clerici, M. A potential role for the PD1/PD-L1 pathway in the neuroinflammation of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 624e.11–624e.22. [Google Scholar] [CrossRef]
- Xing, Z.; Zuo, Z.; Hu, D.; Zheng, X.; Wang, X.; Yuan, L.; Zhou, L.; Qi, F.; Yao, Z. Influenza vaccine combined with moderate-dose PD1 blockade reduces amyloid-β accumulation and improves cognition in APP/PS1 mice. Brain Behav. Immun. 2020, 91, 128–141. [Google Scholar] [CrossRef]
- Brigham and Women’s Hospital. Brigham and Women’s Hospital Launches Clinical Trial of Nasal Vaccine for Alzheimer’s Disease; Brigham and Women’s Hospital: Boston, MA, USA, 2021. [Google Scholar]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Uyemura, K.; Castle, S.C.; Makinodan, T. The frail elderly: Role of dendritic cells in the susceptibility of infection. Mech. Ageing Dev. 2002, 123, 955–962. [Google Scholar] [CrossRef]
- Kotb, A.; Ismail, S.; Kimito, I.; Mohamed, W.; Salman, A.; Mohammed, A.A. Increased CD5+ B-cells are associated with autoimmune phenomena in lepromatous leprosy patients. J. Infect. Public Health 2019, 12, 656–659. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B. Aging affects human B cell responses. J. Clin. Immunol. 2011, 31, 430–435. [Google Scholar] [CrossRef]
- Crooke, S.N.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Immunosenescence and human vaccine immune responses. Immun. Ageing 2019, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Pawelec, G.; Cohen, A.A.; Provost, G.; Khalil, A.; Lacombe, G.; Rodrigues, S.; Desroches, M.; Hirokawa, K.; et al. Immunosenescence and Altered Vaccine Efficiency in Older Subjects: A Myth Difficult to Change. Vaccines 2022, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Wattmo, C.; Blennow, K.; Hansson, O. Cerebro-Spinal fluid biomarker levels: Phosphorylated tau (T) and total tau (N) as markers for rate of progression in Alzheimer’s disease. BMC Neurol. 2020, 20, 10. [Google Scholar] [CrossRef] [PubMed]
- Casaletto, K.B.; Elahi, F.M.; Bettcher, B.M.; Neuhaus, J.; Bendlin, B.B.; Asthana, S.; Johnson, S.C.; Yaffe, K.; Carlsson, C.; Blennow, K.; et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology 2017, 89, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Brenowitz, W.D.; Nelson, P.T.; Besser, L.M.; Heller, K.B.; Kukull, W.A. Cerebral amyloid angiopathy and its co-occurrence with Alzheimer’s disease and other cerebrovascular neuropathologic changes. Neurobiol. Aging 2015, 36, 2702–2708. [Google Scholar] [CrossRef] [PubMed]
- Hemonnot, A.-L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef]
- Kwan, P.; Konno, H.; Chan, K.Y.; Baum, L. Rationale for the development of an Alzheimer’s disease vaccine. Hum. Vaccines Immunother. 2020, 16, 645–653. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Compound | Phase | Target | Type | Participants | Findings |
|---|---|---|---|---|---|---|
| [68] | Aducanumab | Ib | Aβ | mAb | 197 | Reduced Aβ, did not improve cognition. |
| [70,78] | Bapineuzumab | III | Aβ | mAb | 1121, 1331 | Did not improve cognition, did not reduce Aβ deposition. |
| [71] | Solanezumab | III | Aβ | mAb | 2052 | Did not improve cognition, Levels of Aβ40 decreased, Aβ42 did not change. |
| [73] | Gantenerumab | III | Aβ | mAb | 799 | Study halted due to no effect on cognition or Aβ deposition. |
| [74] | Crenezumab | II | Aβ | mAb | 448 | No effect on cognition, elevated CSF levels of Aβ were associated with treatment. |
| [76] | Lecanemab | II | Aβ | mAb | 854 | Treatment showed a reduction in Aβ and a reduction in cognitive decline over an 18-month period, missing 12-month primary endpoints. |
| Ref. | Compound | Phase | Target | Type | Participants | Findings |
|---|---|---|---|---|---|---|
| [79] | AV-1959D | Pre-clinical | Aβ | DNA Vaccine | 60 | No short- or long-term toxicities demonstrated. The vaccine elicited an immune response in the form of antibody production specific to Aβ42 |
| [78] | Y-5a15 | Pre-clinical | Aβ | Vaccine | N/A | Treatment elicited significant levels of Aβ antibodies, reduced levels of Aβ, and improved cognitive function in mice. |
| [10,80] | AN1792 | IIa | Aβ | Vaccine | 375 | Reduced Aβ load in the brain, terminated due to development of adverse events resulting from the treatment. |
| [79,81,82,83] | Protollin | Pre-clinical | Aβ | Vaccine | N/A | Significant reduction in Aβ in mice, cognitive function improved following treatment. Adjuvant was not observed in brain tissue. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valiukas, Z.; Ephraim, R.; Tangalakis, K.; Davidson, M.; Apostolopoulos, V.; Feehan, J. Immunotherapies for Alzheimer’s Disease—A Review. Vaccines 2022, 10, 1527. https://doi.org/10.3390/vaccines10091527
Valiukas Z, Ephraim R, Tangalakis K, Davidson M, Apostolopoulos V, Feehan J. Immunotherapies for Alzheimer’s Disease—A Review. Vaccines. 2022; 10(9):1527. https://doi.org/10.3390/vaccines10091527
Chicago/Turabian StyleValiukas, Zachary, Ramya Ephraim, Kathy Tangalakis, Majid Davidson, Vasso Apostolopoulos, and Jack Feehan. 2022. "Immunotherapies for Alzheimer’s Disease—A Review" Vaccines 10, no. 9: 1527. https://doi.org/10.3390/vaccines10091527
APA StyleValiukas, Z., Ephraim, R., Tangalakis, K., Davidson, M., Apostolopoulos, V., & Feehan, J. (2022). Immunotherapies for Alzheimer’s Disease—A Review. Vaccines, 10(9), 1527. https://doi.org/10.3390/vaccines10091527