
Role of Immune Cells and Receptors in Cancer Treatment: An Immunotherapeutic Approach

 ,
,  ,
,  ,
,  , , , ,
, , , ,  and
and 
Abstract
:1. Cancer and Immunotherapy
2. Dendritic Cells and Cancer
3. Natural Killer (NK) Cells and Cancer
3.1. Autologous NK Cells
3.2. Allogeneic NK Cells
3.3. Genetic Modification of NK Cells
4. B-Cell and Cancer
5. γδT Cell Therapy and Cancer
5.1. Stimulating γδT Cells In Vivo with Phosphoantigens
5.2. Redirecting T Cells to Tumors
6. Regulation of Immune Checkpoints
6.1. Myeloid and Lymphoid Cells in the Tumor Microenvironment
6.2. Mediated by Epigenetics
6.3. Mediated by Gut Microbiota
7. Activatable Cancer Immunotherapy in Response to Internal Stimulus
7.1. Redox-Activated Immunotherapy
7.1.1. GSH-Mediated Activation of Immunological Adjuvants
7.1.2. GSH-Mediated Activation of Antigens
7.1.3. GSH-Mediated Activation of Immunotherapeutic Antibodies
7.2. Enzyme-Activated Immunotherapy
7.2.1. MMP-Activated Immunotherapy
7.2.2. Caspase-Activated Immunotherapy
7.2.3. Hyaluronidase (HAase)-Activated Immunotherapy
8. Immunotherapy for Cancer
8.1. Monoclonal Antibodies
8.2. Bispecific Antibodies
8.3. Cancer Vaccine
8.4. Adoptive Cell Therapy
8.5. Molecular Chaperones and Cancer Immunotherapy
9. Combination Therapy for Cancer
CART Cell Therapy for Cancer
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. (In English) [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. (In English) [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474. (In English) [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Matson, V.; Flood, B.; Spranger, S.; Gajewski, T.F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017, 27, 96–108. (In English) [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer immunotherapy, part 3: Challenges and future trends. Pharm. Ther. 2017, 42, 514. [Google Scholar]
- Couzin-Frankel, J. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef]
- Chevolet, I.; Speeckaert, R.; Schreuer, M.; Neyns, B.; Krysko, O.; Bachert, C.; Hennart, B.; Allorge, D.; van Geel, N.; Van Gele, M. Characterization of the in vivo immune network of IDO, tryptophan metabolism, PD-L1, and CTLA-4 in circulating immune cells in melanoma. Oncoimmunology 2015, 4, e982382. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Chambers, A.; Kundranda, M.; Rao, S.; Mahmoud, F.; Niu, J. Anti-angiogenesis revisited: Combination with immunotherapy in solid tumors. Curr. Oncol. Rep. 2021, 23, 100. [Google Scholar] [CrossRef]
- Boone, C.E.; Wang, L.; Gautam, A.; Newton, I.G.; Steinmetz, N.F. Combining nanomedicine and immune checkpoint therapy for cancer immunotherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1739. [Google Scholar] [CrossRef]
- Gurunathan, S.; Qasim, M.; Kang, M.-H.; Kim, J.-H. Role and therapeutic potential of melatonin in various type of cancers. OncoTargets Ther. 2021, 14, 2019–2052. [Google Scholar] [CrossRef]
- Huppert, L.A.; Green, M.D.; Kim, L.; Chow, C.; Leyfman, Y.; Daud, A.I.; Lee, J.C. Tissue-specific tregs in cancer metastasis: Opportunities for precision immunotherapy. Cell. Mol. Immunol. 2022, 19, 33–45. [Google Scholar] [CrossRef]
- Rahman, M.M.; Mosaddik, A.; Alam, A.K. Traditional foods with their constituent’s antiviral and immune system modulating properties. Heliyon 2021, 7, e05957. [Google Scholar] [CrossRef]
- Bagheri, Y.; Barati, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Yousefi, M. Current progress in cancer immunotherapy based on natural killer cells. Cell Biol. Int. 2021, 45, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Chen, K.; Hu, H.; Gopinath, S.C. Progress in gene therapy treatments for prostate cancer. Biotechnol. Appl. Biochem. 2022, 69, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Sadreddini, S.; Baradaran, B.; Aghebati-Maleki, A.; Sadreddini, S.; Shanehbandi, D.; Fotouhi, A.; Aghebati-Maleki, L. Immune checkpoint blockade opens a new way to cancer immunotherapy. J. Cell. Physiol. 2019, 234, 8541–8549. [Google Scholar] [CrossRef]
- Camelliti, S.; Le Noci, V.; Bianchi, F.; Moscheni, C.; Arnaboldi, F.; Gagliano, N.; Balsari, A.; Garassino, M.C.; Tagliabue, E.; Sfondrini, L. Mechanisms of hyperprogressive disease after immune checkpoint inhibitor therapy: What we (don’t) know. J. Exp. Clin. Cancer Res. 2020, 39, 236. [Google Scholar] [CrossRef] [PubMed]
- Macri, C.; Pang, E.S.; Patton, T.; O’Keeffe, M. Dendritic cell subsets. Semin. Cell Dev. Biol. 2018, 84, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Heath, W.R.; Carbone, F.R. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 2001, 1, 126–134. [Google Scholar] [CrossRef]
- Murphy, T.L.; Murphy, K.M. Dendritic cells in cancer immunology. Cell. Mol. Immunol. 2022, 19, 3–13. [Google Scholar] [CrossRef]
- Gutiérrez-Martínez, E.; Planès, R.; Anselmi, G.; Reynolds, M.; Menezes, S.; Adiko, A.C.; Saveanu, L.; Guermonprez, P. Cross-presentation of cell-associated antigens by MHC class I in dendritic cell subsets. Front. Immunol. 2015, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Dammeijer, F.; Aerts, J.G.; Vroman, H. Current state of dendritic cell-based immunotherapy: Opportunities for in vitro antigen loading of different DC subsets? Front. Immunol. 2018, 9, 2804. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef]
- Brodbeck, T.; Nehmann, N.; Bethge, A.; Wedemann, G.; Schumacher, U. Perforin-dependent direct cytotoxicity in natural killer cells induces considerable knockdown of spontaneous lung metastases and computer modelling-proven tumor cell dormancy in a HT29 human colon cancer xenograft mouse model. Mol. Cancer 2014, 13, 244. [Google Scholar] [CrossRef] [PubMed]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Payer, Á.R.; Gonzalez, S.; López-Soto, A. Mechanisms of apoptosis resistance to NK cell-mediated cytotoxicity in cancer. Int. J. Mol. Sci. 2020, 21, 3726. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, K.; Ambrose, A.R.; Davis, D.M. Escaping death: How cancer cells and infected cells resist cell-mediated cytotoxicity. Front. Immunol. 2022, 13, 867098. [Google Scholar] [CrossRef]
- Beyer, K.; Baukloh, A.-K.; Stoyanova, A.; Kamphues, C.; Sattler, A.; Kotsch, K. Interactions of tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) with the immune system: Implications for inflammation and cancer. Cancers 2019, 11, 1161. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, P.; Sharma, R. Natural killer cells-their role in tumour immunosurveillance. J. Clin. Diagn. Res. JCDR 2017, 11, BE01. [Google Scholar] [CrossRef]
- Lusty, E.; Poznanski, S.M.; Kwofie, K.; Mandur, T.S.; Lee, D.A.; Richards, C.D.; Ashkar, A.A. IL-18/IL-15/IL-12 synergy induces elevated and prolonged IFN-γ production by ex vivo expanded NK cells which is not due to enhanced STAT4 activation. Mol. Immunol. 2017, 88, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Valipour, B.; Velaei, K.; Abedelahi, A.; Karimipour, M.; Darabi, M.; Charoudeh, H.N. NK cells: An attractive candidate for cancer therapy. J. Cell. Physiol. 2019, 234, 19352–19365. [Google Scholar] [CrossRef]
- Woan, K.V.; Miller, J.S. Harnessing natural killer cell antitumor immunity: From the bench to bedside. Cancer Immunol. Res. 2019, 7, 1742–1747. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Hoya, A.; Soto-Cruz, I. NK Cell Regulation in Cervical Cancer and Strategies for Immunotherapy. Cells 2021, 10, 3104. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.A. Cellular therapy: Adoptive immunotherapy with expanded natural killer cells. Immunol. Rev. 2019, 290, 85–99. [Google Scholar] [CrossRef]
- Bashiri Dezfouli, A.; Yazdi, M.; Pockley, A.G.; Khosravi, M.; Kobold, S.; Wagner, E.; Multhoff, G. NK cells armed with chimeric antigen receptors (CAR): Roadblocks to successful development. Cells 2021, 10, 3390. [Google Scholar] [CrossRef]
- Wang, W.-N.; Zhou, G.-Y.; Zhang, W.-L. NK-92 cell, another ideal carrier for chimeric antigen receptor. Immunotherapy 2017, 9, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Zhang, Y.-H.; Guo, X.-H.; Zhang, H.-Y.; Zhang, Y. The double-edge role of B cells in mediating antitumor T-cell immunity: Pharmacological strategies for cancer immunotherapy. Int. Immunopharmacol. 2016, 36, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, Z.; Prinz, I.; Déchanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2020, 19, 169–184. [Google Scholar] [CrossRef]
- Tomogane, M.; Omura, M.; Sano, Y.; Shimizu, D.; Toda, Y.; Hosogi, S.; Kimura, S.; Ashihara, E. Expression level of BTN3A1 on the surface of CD14+ monocytes is a potential predictor of γδ T cell expansion efficiency. Biochem. Biophys. Res. Commun. 2022, 588, 47–54. [Google Scholar] [CrossRef]
- Hoeres, T.; Smetak, M.; Pretscher, D.; Wilhelm, M. Improving the efficiency of Vγ9Vδ2 T-cell immunotherapy in cancer. Front. Immunol. 2018, 9, 800. [Google Scholar] [CrossRef]
- Liu, C.; Skorupinska-Tudek, K.; Eriksson, S.-G.; Parmryd, I. Potentiating Vγ9Vδ2 T cell proliferation and assessing their cytotoxicity towards adherent cancer cells at the single cell level. Biol. Open 2022, 11, bio059049. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Cole, D.K.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Ding, Y.-P.; Tanaka, Y.; Shen, L.-W.; Wei, C.-H.; Minato, N.; Zhang, W. γδ T cells and their potential for immunotherapy. Int. J. Biol. Sci. 2014, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- DeVorkin, L.; Jacobs, T.M.; Tonikian, R.; Hervé, K.; Caldwell, K.; Hwang, Y.; Faralla, C.; Wei, W.; Lam, K.J.; Dhupar, H. Redirecting T cells to tumor targets with functionally diverse CD3-binding antibodies. Cancer Res. 2022, 82, 312. [Google Scholar] [CrossRef]
- Zhao, H.; Xi, X.; Cui, L.; He, W. CDR3δ-grafted γ9δ2T cells mediate effective antitumor reactivity. Cell. Mol. Immunol. 2012, 9, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh, S.; Bayatipoor, H.; Pashangzadeh, S.; Jafarpour, R.; Shojaei, Z.; Motallebnezhad, M. Immune checkpoints and cancer development: Therapeutic implications and future directions. Pathol.-Res. Pract. 2021, 223, 153485. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Panagi, M.; Stylianopoulos, T.; Papageorgis, P. The role of tumor microenvironment in cancer metastasis: Molecular mechanisms and therapeutic opportunities. Cancers 2021, 13, 2053. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhou, J.; Zhu, L.; Hu, W.; Liu, B.; Xie, L. Adoptive tumor infiltrating lymphocytes cell therapy for cervical cancer. Hum. Vaccines Immunother. 2022, 18, 2060019. [Google Scholar] [CrossRef]
- Singh, B.; Singh, D.K.; Ganatra, S.R.; Escobedo, R.A.; Khader, S.; Schlesinger, L.S.; Kaushal, D.; Mehra, S. Myeloid-derived suppressor cells mediate T cell dysfunction in nonhuman primate TB granulomas. Mbio 2021, 12, e03189-21. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Nisar, S.; Maacha, S.; Carneiro-Lobo, T.C.; Akhtar, S.; Siveen, K.S.; Wani, N.A.; Rizwan, A.; Bagga, P.; Singh, M. Cytokine-chemokine network driven metastasis in esophageal cancer; promising avenue for targeted therapy. Mol. Cancer 2021, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Nava, S.; Lisini, D.; Frigerio, S.; Bersano, A. Dendritic cells and cancer immunotherapy: The adjuvant effect. Int. J. Mol. Sci. 2021, 22, 12339. [Google Scholar] [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Taghiloo, S.; Asgarian-Omran, H. Immune evasion mechanisms in acute myeloid leukemia: A focus on immune checkpoint pathways. Crit. Rev. Oncol. Hematol. 2021, 157, 103164. [Google Scholar] [CrossRef]
- Güç, E.; Pollard, J.W. Redefining macrophage and neutrophil biology in the metastatic cascade. Immunity 2021, 54, 885–902. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Jaillon, S.; Garlanda, C.; Allavena, P. Tumor-associated myeloid cells: Diversity and therapeutic targeting. Cell. Mol. Immunol. 2021, 18, 566–578. [Google Scholar] [CrossRef]
- Katano, I.; Hanazawa, A.; Otsuka, I.; Yamaguchi, T.; Mochizuki, M.; Kawai, K.; Ito, R.; Goto, M.; Kagawa, T.; Takahashi, T. Development of a novel humanized mouse model for improved evaluation of in vivo anticancer effects of anti-PD-1 antibody. Sci. Rep. 2021, 11, 21087. [Google Scholar] [CrossRef] [PubMed]
- Awadasseid, A.; Wu, Y.; Zhang, W. Advance investigation on synthetic small-molecule inhibitors targeting PD-1/PD-L1 signaling pathway. Life Sci. 2021, 282, 119813. [Google Scholar] [CrossRef]
- Wang, Z.; He, L.; Li, W.; Xu, C.; Zhang, J.; Wang, D.; Dou, K.; Zhuang, R.; Jin, B.; Zhang, W. GDF15 induces immunosuppression via CD48 on regulatory T cells in hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, e002787. [Google Scholar] [CrossRef]
- Karpisheh, V.; Mousavi, S.M.; Sheykholeslami, P.N.; Fathi, M.; Saray, M.M.; Aghebati-Maleki, L.; Jafari, R.; Zolbanin, N.M.; Jadidi-Niaragh, F. The role of regulatory T cells in the pathogenesis and treatment of prostate cancer. Life Sci. 2021, 284, 119132. [Google Scholar] [CrossRef] [PubMed]
- Pisibon, C.; Ouertani, A.; Bertolotto, C.; Ballotti, R.; Cheli, Y. Immune Checkpoints in Cancers: From Signaling to the Clinic. Cancers 2021, 13, 4573. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Ma, Y.; Yu, L.; Jiang, J.; Shen, S.; Hou, Y.; Wang, T. Cancer immunotherapy: A focus on the regulation of immune checkpoints. Int. J. Mol. Sci. 2018, 19, 1389. [Google Scholar] [CrossRef]
- Park, H.J.; Park, J.S.; Jeong, Y.H.; Son, J.; Ban, Y.H.; Lee, B.-H.; Chen, L.; Chang, J.; Chung, D.H.; Choi, I. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J. Immunol. 2015, 194, 5801–5811. [Google Scholar] [CrossRef]
- Chen, X.; Du, Y.; Lin, X.; Qian, Y.; Zhou, T.; Huang, Z. CD4+ CD25+ regulatory T cells in tumor immunity. Int. Immunopharmacol. 2016, 34, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 90. [Google Scholar] [CrossRef]
- Villanueva, L.; Álvarez-Errico, D.; Esteller, M. The contribution of epigenetics to cancer immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Deshmukh, S.; Reddy, A.; Lightcap, J.; Hayes, M.; McClellan, S.; Singh, S.; Rabideau, B.; Glover, T.G.; Roberts, B. Cancer immune checkpoint inhibitor therapy and the gut microbiota. Integr. Cancer Ther. 2019, 18, 1534735419846379. [Google Scholar] [CrossRef]
- Routy, B.; Gopalakrishnan, V.; Daillère, R.; Zitvogel, L.; Wargo, J.A.; Kroemer, G. The gut microbiota influences anticancer immunosurveillance and general health. Nat. Rev. Clin. Oncol. 2018, 15, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Won, S.-M.; Park, E.; Jeong, J.-J.; Ganesan, R.; Gupta, H.; Gebru, Y.A.; Sharma, S.; Kim, D.-J.; Suk, K.-T. The gut microbiota-derived immune response in chronic liver disease. Int. J. Mol. Sci. 2021, 22, 8309. [Google Scholar] [CrossRef]
- Lee, S.-H.; Cho, S.-Y.; Yoon, Y.; Park, C.; Sohn, J.; Jeong, J.-J.; Jeon, B.-N.; Jang, M.; An, C.; Lee, S. Bifidobacterium bifidum strains synergize with immune checkpoint inhibitors to reduce tumour burden in mice. Nat. Microbiol. 2021, 6, 277–288. [Google Scholar] [CrossRef]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef]
- Muthusami, S.; Prabakaran, D.; Yu, J.-R.; Park, W.-Y. EGF-induced expression of Fused Toes Homolog (FTS) facilitates epithelial–mesenchymal transition and promotes cell migration in ME180 cervical cancer cells. Cancer Lett. 2014, 351, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, D.; Muthusami, S.; Sivaraman, T.; Yu, J.-R.; Park, W.-Y. Silencing of FTS increases radiosensitivity by blocking radiation-induced Notch1 activation and spheroid formation in cervical cancer cells. Int. J. Biol. Macromol. 2019, 126, 1318–1325. [Google Scholar] [CrossRef]
- DS, P.; Chaturvedi, P.K.; Shimokawa, T.; Kim, K.-H.; Park, W.-Y. Silencing of fused toes homolog (FTS) increases radiosensitivity to carbon-ion through downregulation of Notch signaling in cervical cancer cells. Front. Oncol. 2021, 11, 730607. [Google Scholar]
- Saha, T.; Solomon, J.; Samson, A.O.; Gil-Henn, H. Invasion and metastasis as a central hallmark of breast cancer. J. Clin. Med. 2021, 10, 3498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Pu, K. Molecular and nanoengineering approaches towards activatable cancer immunotherapy. Chem. Soc. Rev. 2020, 49, 4234–4253. [Google Scholar] [CrossRef]
- Ding, Y.; Dai, Y.; Wu, M.; Li, L. Glutathione-mediated nanomedicines for cancer diagnosis and therapy. Chem. Eng. J. 2021, 426, 128880. [Google Scholar] [CrossRef]
- Guo, J.; Huang, L. Membrane-core nanoparticles for cancer nanomedicine. Adv. Drug Deliv. Rev. 2020, 156, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cui, H.; Zhang, R.; Zhang, H.; Huang, W. Nanoparticulation of prodrug into medicines for cancer therapy. Adv. Sci. 2021, 8, 2101454. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Ji, S.; Li, F.; Xu, H. Diselenide-containing hyperbranched polymer with light-induced cytotoxicity. ACS Appl. Mater. Interfaces 2017, 9, 12924–12929. [Google Scholar] [CrossRef] [PubMed]
- Aikins, M.E.; Xu, C.; Moon, J.J. Engineered nanoparticles for cancer vaccination and immunotherapy. Acc. Chem. Res. 2020, 53, 2094–2105. [Google Scholar] [CrossRef]
- Liu, J.; Liew, S.S.; Wang, J.; Pu, K. Bioinspired and biomimetic delivery platforms for cancer vaccines. Adv. Mater. 2022, 34, 2103790. [Google Scholar] [CrossRef]
- Peng, P.; Hu, H.; Liu, P.; Xu, L.X. Neoantigen-specific CD4+ T-cell response is critical for the therapeutic efficacy of cryo-thermal therapy. J. Immunother. Cancer 2020, 8, e000421. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, W.-Q.; Pei, Q.; Lord, M.S.; Yu, H.-J. Engineering nanomedicines through boosting immunogenic cell death for improved cancer immunotherapy. Acta Pharmacol. Sin. 2020, 41, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zheng, Y.; Melo, M.B.; Mabardi, L.; Castaño, A.P.; Xie, Y.-Q.; Li, N.; Kudchodkar, S.B.; Wong, H.C.; Jeng, E.K. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 2018, 36, 707–716. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative stress in cancer cell metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, Y.; Zhang, J.; Li, W.; Shi, M.; Qiao, M.; Zhao, X.; Hu, H.; Chen, D. Laser/GSH-activatable oxaliplatin/phthalocyanine-based coordination polymer nanoparticles combining chemophotodynamic therapy to improve cancer immunotherapy. ACS Appl. Mater. Interfaces 2021, 13, 39934–39948. [Google Scholar] [CrossRef]
- Shim, M.K.; Yang, S.; Sun, I.-C.; Kim, K. Tumor-activated carrier-free prodrug nanoparticles for targeted cancer Immunotherapy: Preclinical evidence for safe and effective drug delivery. Adv. Drug Deliv. Rev. 2022, 183, 114177. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Bai, X.; Shu, Y.; Ahmad, O.; Shen, P. Targeting tumor-associated macrophages as an antitumor strategy. Biochem. Pharmacol. 2021, 183, 114354. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; He, X.; Zhang, Y.; Zhou, M.; Peng, C.; He, Z.; Gui, S.; Li, Z. Smart Nanogatekeepers for Tumor Theranostics. Small 2021, 17, 2103712. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jin, Y.; Liu, X.; Chen, F.; Zheng, X.; Liu, T.; Yang, Y.; Yu, H. Endogenous Stimuli-Activatable Nanomedicine for Immune Theranostics for Cancer. Adv. Funct. Mater. 2021, 31, 2100386. [Google Scholar] [CrossRef]
- Wang, D.; Wang, T.; Yu, H.; Feng, B.; Zhou, L.; Zhou, F.; Hou, B.; Zhang, H.; Luo, M.; Li, Y. Engineering nanoparticles to locally activate T cells in the tumor microenvironment. Sci. Immunol. 2019, 4, eaau6584. [Google Scholar] [CrossRef]
- Li, M.; Zhao, G.; Su, W.-K.; Shuai, Q. Enzyme-responsive nanoparticles for anti-tumor drug delivery. Front. Chem. 2020, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xiao, Z.; An, Y.; Han, S.; Wu, W.; Wang, Y.; Guo, Y.; Shuai, X. Nanodrug with dual-sensitivity to tumor microenvironment for immuno-sonodynamic anticancer therapy. Biomaterials 2021, 269, 120636. [Google Scholar] [CrossRef]
- Kwek, G.; Do, T.C.; Lu, X.; Lin, J.; Xing, B. Scratching the Surface of Unventured Possibilities with In Situ Self-Assembly: Protease-Activated Developments for Imaging and Therapy. ACS Appl. Bio Mater. 2021, 4, 2192–2216. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future SuccessesMatrix Metalloproteinase Inhibitors in Cancer Therapy. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef]
- Voura, E.B.; English, J.L.; Yu, H.-Y.E.; Ho, A.T.; Subarsky, P.; Hill, R.P.; Hojilla, C.V.; Khokha, R. Proteolysis during tumor cell extravasation in vitro: Metalloproteinase involvement across tumor cell types. PLoS ONE 2013, 8, e78413. [Google Scholar] [CrossRef]
- Kapoor, C.; Vaidya, S.; Wadhwan, V.; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. Ther. 2016, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Schegoleva, A.; Khozyainova, A.; Gerashchenko, T.; Zhuikova, L.; Denisov, E.V. Metastasis prevention: Targeting causes and roots. Clin. Exp. Metastasis 2022, 39, 505–519. [Google Scholar] [CrossRef]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and their inhibitors: Potential for the development of new therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef] [PubMed]
- Shilova, O.; Shramova, E.; Proshkina, G.; Deyev, S. Natural and designed toxins for precise therapy: Modern approaches in experimental oncology. Int. J. Mol. Sci. 2021, 22, 4975. [Google Scholar] [CrossRef]
- Song, W.; Kuang, J.; Li, C.-X.; Zhang, M.; Zheng, D.; Zeng, X.; Liu, C.; Zhang, X.-Z. Enhanced immunotherapy based on photodynamic therapy for both primary and lung metastasis tumor eradication. ACS Nano 2018, 12, 1978–1989. [Google Scholar] [CrossRef]
- Wang, H.; Picchio, M.L.; Calderón, M. One stone, many birds: Recent advances in functional nanogels for cancer nanotheranostics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2022, 14, e1791. [Google Scholar] [CrossRef]
- Mi, Y.; Hagan IV, C.T.; Vincent, B.G.; Wang, A.Z. Emerging nano-/microapproaches for cancer immunotherapy. Adv. Sci. 2019, 6, 1801847. [Google Scholar] [CrossRef]
- Yang, B.; Gao, J.; Pei, Q.; Xu, H.; Yu, H. Engineering prodrug nanomedicine for cancer immunotherapy. Adv. Sci. 2020, 7, 2002365. [Google Scholar] [CrossRef]
- Duan, X.; Chan, C.; Lin, W. Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew. Chem. Int. Ed. 2019, 58, 670–680. [Google Scholar] [CrossRef]
- Li, L.; Ma, B.; Wang, W. Peptide-based nanomaterials for tumor immunotherapy. Molecules 2020, 26, 132. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Wang, R.; Zhang, L.; Yin, D.; Luo, X.; Solomon, J.; Jiang, R.; Markos, K.; Davidson, W.; Scott, D. Death the Fas way: Regulation and pathophysiology of CD95 and its ligand. Pharmacol. Ther. 2000, 88, 333–347. [Google Scholar] [CrossRef]
- Chhabra, A. Mitochondria-centric activation induced cell death of cytolytic T lymphocytes and its implications for cancer immunotherapy. Vaccine 2010, 28, 4566–4572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nat. Rev. Cancer 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Metkar, S.S.; Wang, B.; Ebbs, M.L.; Kim, J.H.; Lee, Y.J.; Raja, S.M.; Froelich, C.J. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J. Cell Biol. 2003, 160, 875–885. [Google Scholar] [CrossRef]
- Sutton, V.R.; Wowk, M.E.; Cancilla, M.; Trapani, J.A. Caspase activation by granzyme B is indirect, and caspase autoprocessing requires the release of proapoptotic mitochondrial factors. Immunity 2003, 18, 319–329. [Google Scholar] [CrossRef]
- Vogler, M.; Shanmugalingam, S.; Särchen, V.; Reindl, L.M.; Grèze, V.; Buchinger, L.; Kühn, M.; Ullrich, E. Unleashing the power of NK cells in anticancer immunotherapy. J. Mol. Med. 2021, 100, 337–349. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, J.; Yu, S.; Li, Y.; Zhu, J.; Zhang, K.; Zhang, R. Cell pyroptosis in health and inflammatory diseases. Cell Death Discov. 2022, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Du, C.; Xu, M.; Wang, X.; Ley, T.J. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity 2000, 12, 621–632. [Google Scholar] [CrossRef]
- Sharif-Askari, E.; Alam, A.; Rhéaume, E.; Beresford, P.J.; Scotto, C.; Sharma, K.; Lee, D.; DeWolf, W.E.; Nuttall, M.E.; Lieberman, J. Direct cleavage of the human DNA fragmentation factor-45 by granzyme B induces caspase-activated DNase release and DNA fragmentation. EMBO J. 2001, 20, 3101–3113. [Google Scholar] [CrossRef]
- Weigelin, B.; Friedl, P. T cell-mediated additive cytotoxicity–death by multiple bullets. Trends Cancer 2022, 7. [Google Scholar] [CrossRef]
- Cullen, S.; Martin, S. Mechanisms of granule-dependent killing. Cell Death Differ. 2008, 15, 251–262. [Google Scholar] [CrossRef]
- Zhang, C.; Pu, K. Recent Progress on Activatable Nanomedicines for Immunometabolic Combinational Cancer Therapy. Small Struct. 2020, 1, 2000026. [Google Scholar] [CrossRef]
- Peng, S.; Xiao, F.; Chen, M.; Gao, H. Tumor-Microenvironment-Responsive Nanomedicine for Enhanced Cancer Immunotherapy. Adv. Sci. 2022, 9, 2103836. [Google Scholar] [CrossRef]
- Gao, S.; Yang, X.; Xu, J.; Qiu, N.; Zhai, G. Nanotechnology for boosting cancer immunotherapy and remodeling tumor microenvironment: The horizons in cancer treatment. ACS Nano 2021, 15, 12567–12603. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, J.; Yoon, J.; Chen, X. Cancer-associated, stimuli-driven, turn on theranostics for multimodality imaging and therapy. Adv. Mater. 2017, 29, 1606857. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Han, H.; Posner, R.G.; Hostetter, G.; Von Hoff, D.D. Targeting the tumor microenvironment in cancer: Why hyaluronidase deserves a second look. Cancer Discov. 2011, 1, 291–296. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, H.; Gao, N.; Yang, C.; Zhang, R.; Zhang, X. An efficient FRET based theranostic nanoprobe for hyaluronidase detection and cancer therapy in vitro. Sens. Actuators B Chem. 2021, 344, 130201. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Wu, H.X.; Xu, R.H. Advancing to the era of cancer immunotherapy. Cancer Commun. 2021, 41, 803–829. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Ding, J.; Chen, Y. Role of CD8+ T lymphocyte cells: Interplay with stromal cells in tumor microenvironment. Acta Pharm. Sin. B 2021, 11, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, H.; Zuo, Y.; Li, N.; Ding, M.; Li, C. A novel monoclonal antibody KMP1 has potential antitumor activity of bladder cancer by blocking CD44 in vivo and in vitro. Cancer Med. 2018, 7, 2064–2077. (In English) [Google Scholar] [CrossRef]
- Qing, J.; Du, X.; Chen, Y.; Chan, P.; Li, H.; Wu, P.; Marsters, S.; Stawicki, S.; Tien, J.; Totpal, K.; et al. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J. Clin. Investig. 2009, 119, 1216–1229. (In English) [Google Scholar] [CrossRef] [PubMed]
- Inman, B.A.; Longo, T.A.; Ramalingam, S.; Harrison, M.R. Atezolizumab: A PD-L1–Blocking Antibody for Bladder Cancer. Clin. Cancer Res. 2017, 23, 1886–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Ma, J.; Lei, T.; Zhao, M.; Zhang, M. Targeting immunotherapy for bladder cancer by using anti-CD3 × CD155 bispecific antibody. J. Cancer 2019, 10, 5153–5161. (In English) [Google Scholar] [CrossRef]
- Kapoor, R.; Vijjan, V.; Singh, P. Bacillus Calmette-Guerin in the management of superficial bladder cancer. Indian J. Urol. 2008, 24, 72. [Google Scholar] [CrossRef]
- Pang, A.S.D.; Morales, A. Immunoprophylaxis of a Murine Bladder Cancer with High Dose Bcg Immunizations. J. Urol. 1982, 127, 1006–1009. [Google Scholar] [CrossRef]
- Bernard-Marty, C.; Lebrun, F.; Awada, A.; Piccart, M.J. Monoclonal antibody-based targeted therapy in breast cancer: Current status and future directions. Drugs 2006, 66, 1577–1591. (In English) [Google Scholar] [CrossRef] [PubMed]
- Kurai, J.; Chikumi, H.; Hashimoto, K.; Yamaguchi, K.; Yamasaki, A.; Sako, T.; Touge, H.; Makino, H.; Takata, M.; Miyata, M.; et al. Antibody-Dependent Cellular Cytotoxicity Mediated by Cetuximab against Lung Cancer Cell Lines. Clin. Cancer Res. 2007, 13, 1552–1561. [Google Scholar] [CrossRef]
- Russell, P.J.; Ow, K.T.; Tam, P.N.; Juarez, J.; Kingsley, E.A.; Qu, C.F.; Li, Y.; Cozzi, P.J.; Martiniello-Wilks, R. Immunohistochemical characterisation of the monoclonal antibody BLCA-38 for the detection of prostate cancer. Cancer Immunol. Immunother. CII 2004, 53, 995–1004. (In English) [Google Scholar] [CrossRef]
- Muthusami, S.; Prabakaran, D.; Yu, J.-R.; Park, W.-Y. FTS is responsible for radiation-induced nuclear phosphorylation of EGFR and repair of DNA damage in cervical cancer cells. J. Cancer Res. Clin. Oncol. 2015, 141, 203–210. [Google Scholar] [CrossRef]
- Kumar, A.R.; Devan, A.R.; Nair, B.; Vinod, B.S.; Nath, L.R. Harnessing the immune system against cancer: Current immunotherapy approaches and therapeutic targets. Mol. Biol. Rep. 2021, 48, 8075–8095. [Google Scholar] [CrossRef]
- Aghanejad, A.; Bonab, S.F.; Sepehri, M.; Haghighi, F.S.; Tarighatnia, A.; Kreiter, C.; Nader, N.D.; Tohidkia, M.R. A review on targeting tumor microenvironment: The main paradigm shift in the mAb-based immunotherapy of solid tumors. Int. J. Biol. Macromol. 2022, 207, 592–610. [Google Scholar] [CrossRef]
- Lucas, A.T.; Moody, A.; Schorzman, A.N.; Zamboni, W.C. Importance and Considerations of Antibody Engineering in Antibody-Drug Conjugates Development from a Clinical Pharmacologist’s Perspective. Antibodies 2021, 10, 30. [Google Scholar] [CrossRef]
- Le Jeune, C.; Thomas, X. Potential for bispecific T-cell engagers: Role of blinatumomab in acute lymphoblastic leukemia. Drug Des. Dev. Ther. 2016, 10, 757. [Google Scholar] [CrossRef]
- Friend, R.; Bhutani, M.; Voorhees, P.M.; Usmani, S.Z. Clinical potential of SLAMF7 antibodies–focus on elotuzumab in multiple myeloma. Drug Des. Dev. Ther. 2017, 11, 893. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Register, A.C.; Tarighat, S.S.; Lee, H.Y. Bioassay Development for Bispecific Antibodies—Challenges and Opportunities. Int. J. Mol. Sci. 2021, 22, 5350. [Google Scholar] [CrossRef] [PubMed]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune suppression in cancer: Effects on immune cells, mechanisms and future therapeutic intervention. Semin. Cancer Biol. 2006, 16, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Weiner, J.; von Reyn, C.F. Novel approaches to tuberculosis vaccine development. Int. J. Infect. Dis. 2017, 56, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, C.M.; Weiden, J.; Eggermont, L.J.; Figdor, C.G. Dendritic cells in cancer immunotherapy. Nat. Mater. 2018, 17, 474–475. [Google Scholar] [CrossRef]
- Sutherland, S.I.; Ju, X.; Horvath, L.; Clark, G.J. Moving on from sipuleucel-T: New dendritic cell vaccine strategies for prostate cancer. Front. Immunol. 2021, 12, 641307. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Delgado, I.; Calzada-Fraile, D.; Sánchez-Madrid, F. Immune regulation by dendritic cell extracellular vesicles in cancer immunotherapy and vaccines. Cancers 2020, 12, 3558. [Google Scholar] [CrossRef] [PubMed]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: Review of the literature and challenges ahead. J. Immunother. Cancer 2021, 9, e002723. [Google Scholar] [CrossRef]
- Simon, B.; Harrer, D.C.; Thirion, C.; Schuler-Thurner, B.; Schuler, G.; Uslu, U. Enhancing lentiviral transduction to generate melanoma-specific human T cells for cancer immunotherapy. J. Immunol. Methods 2019, 472, 55–64. [Google Scholar] [CrossRef]
- Lu, P.; Hill, H.A.; Navsaria, L.J.; Wang, M.L. CAR-T and other adoptive cell therapies for B cell malignancies. J. Natl. Cancer Cent. 2021, 1, 88–96. [Google Scholar] [CrossRef]
- Novellino, L.; Castelli, C.; Parmiani, G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol. Immunother. 2005, 54, 187–207. [Google Scholar] [CrossRef]
- Brodsky, J.L.; Chiosis, G. Hsp70 molecular chaperones: Emerging roles in human disease and identification of small molecule modulators. Curr. Top. Med. Chem. 2006, 6, 1215–1225. [Google Scholar] [CrossRef]
- Ansa-Addo, E.A.; Thaxton, J.; Hong, F.; Wu, B.X.; Zhang, Y.; Fugle, C.W.; Metelli, A.; Riesenberg, B.; Williams, K.; Gewirth, D.T. Clients and oncogenic roles of molecular chaperone gp96/grp94. Curr. Top. Med. Chem. 2016, 16, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.H.; Li, C.; Liu, Y.; Shi, L. Mimicking molecular chaperones to regulate protein folding. Adv. Mater. 2020, 32, 1805945. [Google Scholar] [CrossRef]
- Wang, Z.; Shang, Y.; Tan, Z.; Li, X.; Li, G.; Ren, C.; Wang, F.; Yang, Z.; Liu, J. A supramolecular protein chaperone for vaccine delivery. Theranostics 2020, 10, 657. [Google Scholar] [CrossRef]
- Delfi, M.; Sartorius, R.; Ashrafizadeh, M.; Sharifi, E.; Zhang, Y.; De Berardinis, P.; Zarrabi, A.; Varma, R.S.; Tay, F.R.; Smith, B.R. Self-assembled peptide and protein nanostructures for anticancer therapy: Targeted delivery, stimuli-responsive devices and immunotherapy. Nano Today 2021, 38, 101119. [Google Scholar] [CrossRef]
- Doody, A.D.; Kovalchin, J.T.; Mihalyo, M.A.; Hagymasi, A.T.; Drake, C.G.; Adler, A.J. Glycoprotein 96 can chaperone both MHC class I-and class II-restricted epitopes for in vivo presentation, but selectively primes CD8+ T cell effector function. J. Immunol. 2004, 172, 6087–6092. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Tukaj, S. Heat shock protein 70 as a double agent acting inside and outside the cell: Insights into autoimmunity. Int. J. Mol. Sci. 2020, 21, 5298. [Google Scholar] [CrossRef]
- Alberti, G.; Vergilio, G.; Paladino, L.; Barone, R.; Cappello, F.; Conway de Macario, E.; Macario, A.J.; Bucchieri, F.; Rappa, F. The Chaperone System in Breast Cancer: Roles and Therapeutic Prospects of the Molecular Chaperones Hsp27, Hsp60, Hsp70, and Hsp90. Int. J. Mol. Sci. 2022, 23, 7792. [Google Scholar] [CrossRef]
- Sun, B.; Li, G.; Yu, Q.; Liu, D.; Tang, X. HSP60 in cancer: A promising biomarker for diagnosis and a potentially useful target for treatment. J. Drug Target. 2022, 30, 31–45. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat shock proteins 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef]
- Voos, W. Chaperone–protease networks in mitochondrial protein homeostasis. Biochim. Biophys. Acta—Mol. Cell Res. 2013, 1833, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Zgajnar, N.R.; De Leo, S.A.; Lotufo, C.M.; Erlejman, A.G.; Piwien-Pilipuk, G.; Galigniana, M.D. Biological actions of the Hsp90-binding immunophilins FKBP51 and FKBP52. Biomolecules 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Das, J.K.; Xiong, X.; Ren, X.; Yang, J.-M.; Song, J. Heat shock proteins in cancer immunotherapy. J. Oncol. 2019, 2019, 3267207. [Google Scholar] [CrossRef]
- Murshid, A.; Theriault, J.; Gong, J.; Calderwood, S.K. Investigating receptors for extracellular heat shock proteins. In Molecular Chaperones; Springer: New York, NY, USA, 2011; Volume 787, pp. 289–302. [Google Scholar]
- Binder, R.J. Immunosurveillance of cancer and the heat shock protein-CD91 pathway. Cell. Immunol. 2019, 343, 103814. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Cho, Y.B.; Lee, S. Cell surface GRP94 as a novel emerging therapeutic target for monoclonal antibody cancer therapy. Cells 2021, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Iwanowycz, S.; Ngoi, S.; Hill, M.; Zhao, Q.; Liu, B. Molecular chaperone GRP94/GP96 in cancers: Oncogenesis and therapeutic target. Front. Oncol. 2021, 11, 629846. [Google Scholar] [CrossRef]
- Esteva, F.J.; Hubbard-Lucey, V.M.; Tang, J.; Pusztai, L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019, 20, e175–e186. [Google Scholar] [CrossRef]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK cell–cancer cycle: Advances and new challenges in NK cell–based immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef]
- Zaghi, E.; Calvi, M.; Marcenaro, E.; Mavilio, D.; Di Vito, C. Targeting NKG2A to elucidate natural killer cell ontogenesis and to develop novel immune-therapeutic strategies in cancer therapy. J. Leukoc. Biol. 2019, 105, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Xu, Z.; Zhuang, Y.; Ye, Z.; Qian, Q. Current progress in CAR-T cell therapy for hematological malignancies. J. Cancer 2021, 12, 326. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Rui, W.; Zhao, X.; Lin, X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell. Mol. Immunol. 2021, 18, 1085–1095. [Google Scholar] [CrossRef]
- Abrantes, R.; Duarte, H.O.; Gomes, C.; Wälchli, S.; Reis, C.A. CAR-Ts: New perspectives in cancer therapy. FEBS Lett. 2022, 596, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Achkova, D.Y.; Beatson, R.E.; Maher, J. CAR T-Cell Targeting of Macrophage Colony-Stimulating Factor Receptor. Cells 2022, 11, 2190. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, Y.R.; Tung, S.L.; Dudreuilh, C.; Lechler, R.I.; Fruhwirth, G.O.; Lombardi, G. The future of regulatory T cell therapy: Promises and challenges of implementing CAR technology. Front. Immunol. 2020, 11, 1608. [Google Scholar] [CrossRef] [PubMed]
- Roselli, E.; Boucher, J.C.; Li, G.; Kotani, H.; Spitler, K.; Reid, K.; Cervantes, E.V.; Bulliard, Y.; Tu, N.; Lee, S.B. 4-1BB and optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J. Immunother. Cancer 2021, 9, e003354. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef]
- Taefehshokr, N.; Baradaran, B.; Baghbanzadeh, A.; Taefehshokr, S. Promising approaches in cancer immunotherapy. Immunobiology 2020, 225, 151875. [Google Scholar] [CrossRef] [PubMed]
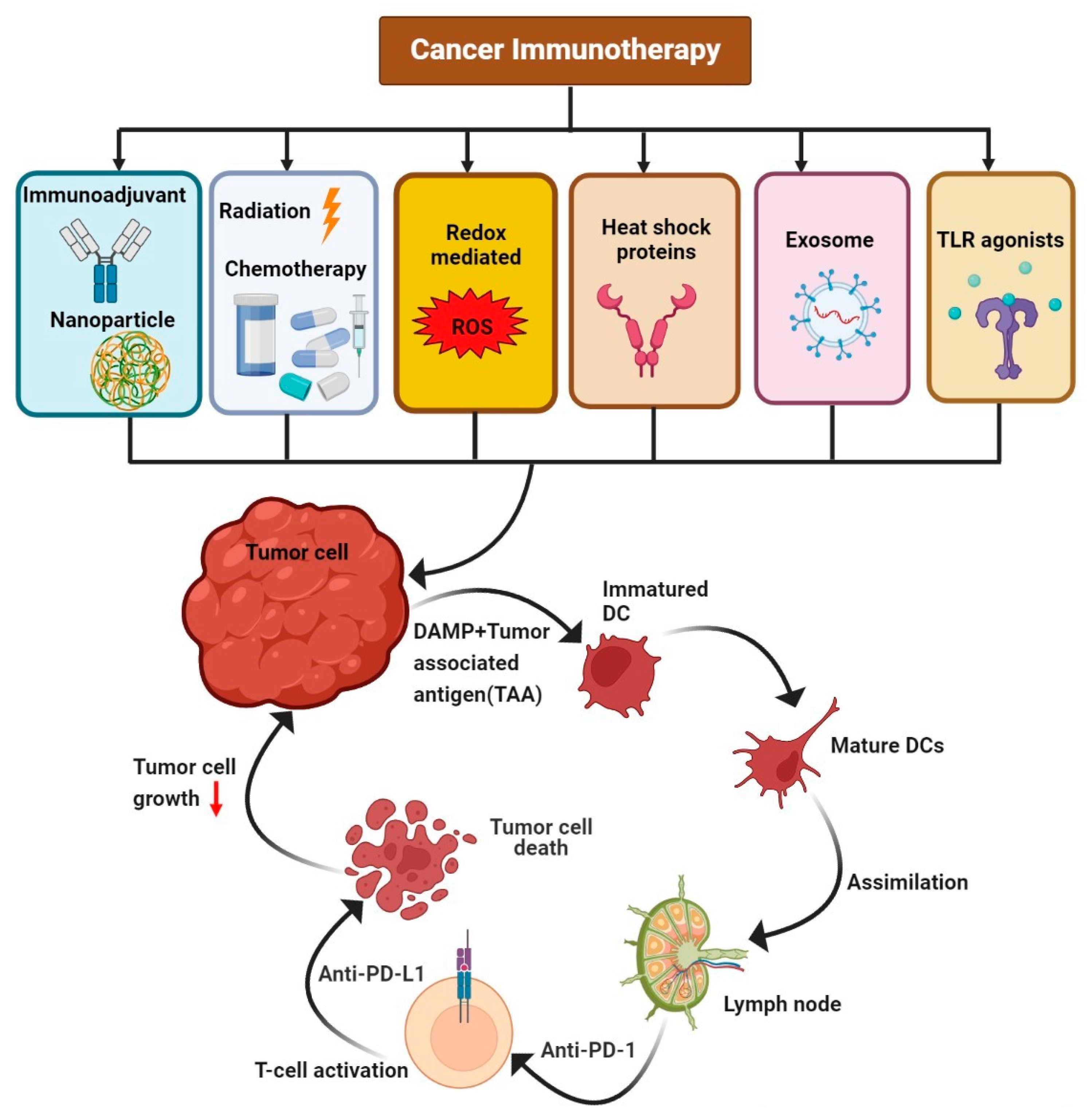

{kind=link}
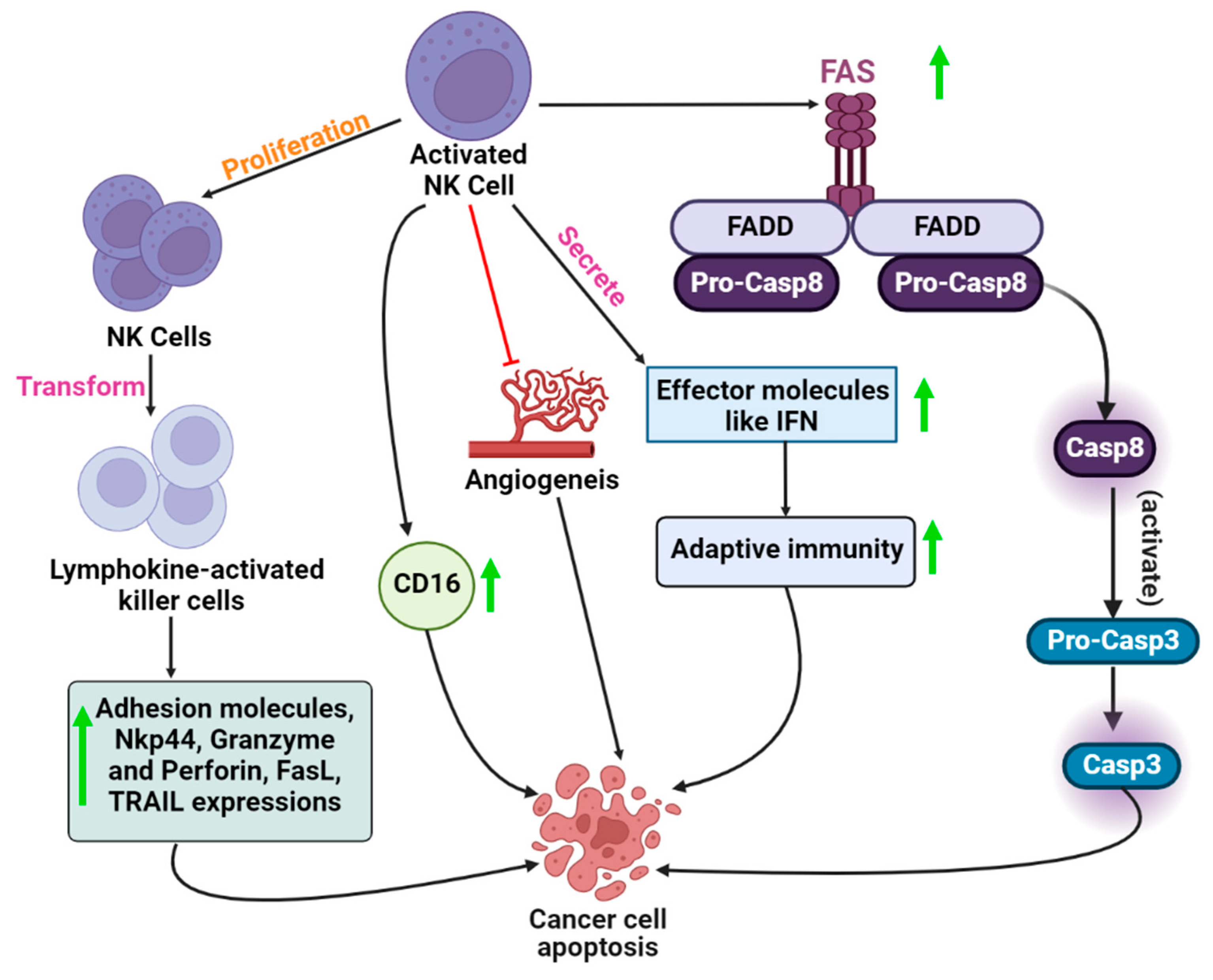
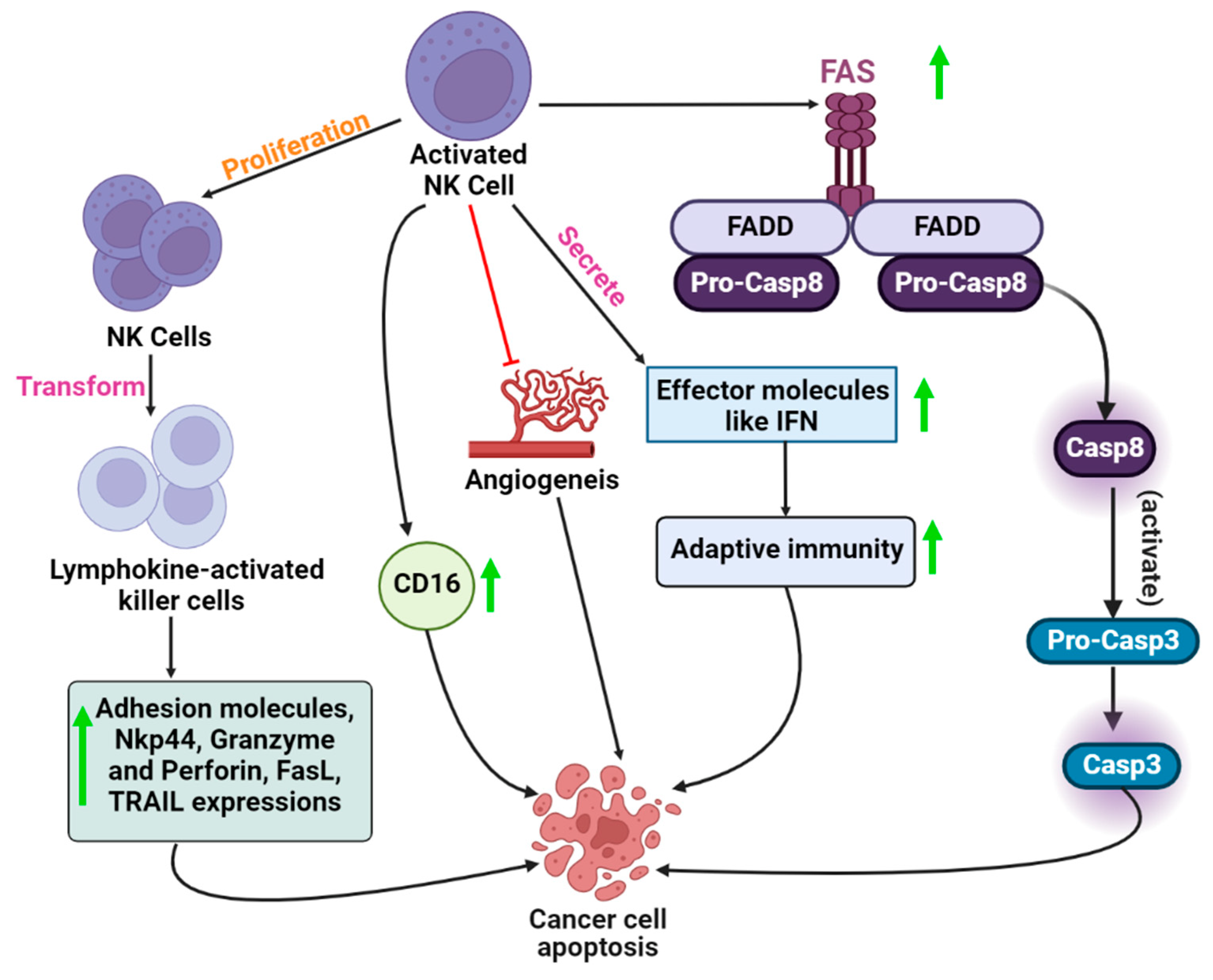{kind=link}
| Cancer Type | Immunotherapy Approach | Targets/Mechanism | Study Type | Cell Lines Used | Animals Used | Technologies Used/Assays Performed | Results | Reference |
|---|---|---|---|---|---|---|---|---|
| Bladder cancer | Monoclonal antibodies | |||||||
| (i) KMP1 mAb | KMP1 binds to CD44 and blocks its function | In vivo and in vitro | Human bladder cancer cell lines EJ, BIU-87, and T24; normal human bladder cell line HCV29; human liver cancer cell line HepG; human cervical cancer cell lines HeLa | BALB/c normal mice and nude mice | Mass spectrometry and antigen affinity to determine KMP1 mechanism; RNA interference technology to knockdown CD44 expression | Bladder cancer clinical severity and prognosis were consistent with the expression of KMP1 epitope | [130] | |
| (ii) R3 mAb/ vofatamab | Inhibits proliferation and FGFR3 signaling by binding to wild-type FGFR3 and FGFR3 mutants | In vivo and in vitro | RT112, RT4, OPM2, Ba/F3, and UMUC-14 | Female nu/nu mice or CB17 SCID mice | Cell proliferation assay, FACS assay, clonal growth assay, and FACS assay | Bladder cancer development was reduced in vivo by induced shRNA knockdown of FGFR3. | [131] | |
| Immune checkpoint inhibitors | ||||||||
| (i) Atezolizumab | Blocks immune checkpoint PD-L1/PD-1; reduces immunosuppressive signals; increases T-cell-mediated immunity against tumors | In vivo In vivo and in vitro | Human bladder cancer cell line pumc-91 | In comparison to historical controls treated with conventional second-line regimens, individuals with advanced bladder cancer treated with atezolizumab exhibited a significantly improved response rate and survival | [132] | |||
| Bispecific antibody anti-CD3 x anti-CD155 | CD155Bi-Ab-armed ATCs secrete more IFN-γ and TNF-α, which increases cytokines and activates endogenous immune cells in vivo, inducing an immune response against tumor cells | MBT-2 cell line | Flow cytometry and ELISA | For CD155-positive bladder cancer, CD155 is a useful target. Additionally, CD155Bi-Ab-armed ATCs show promise concerning developing a novel approach to the present treatment of CD155-positive bladder cancer. | [133] | |||
| BCG vaccines | Prominent infiltration of the bladder wall by immunocompetent cells and the release of cytokines into the urine | In vivo | Female C3H/HeN mice | Vaccine increased NK cell activity | [134,135] | |||
| Breast cancer | Monoclonal antibodies | |||||||
| Trastuzumab | Inhibits intracellular signaling by binding to the extracellular domain of the receptor | In vitro | SK-BR-3 cell line | NK cells killed trastuzumab-coated erbB2-overexpressing cells through an ADCC mechanism mediated by the FcRIII receptor (CD16) | [136] | |||
| Lung cancer | Monoclonal antibodies | |||||||
| Cetuximab | Binds to the extracellular domain of EGFR and blocks EGFR-mediated signal transduction | In vitro | LK-1, EBC-1, A549, LK87, Lu99, N417, Ms1, and LU65; epidermoid carcinoma cell line (A431) | Flow cytometry and immunohistochemistry | A correlation was observed between EGFR molecules on the cell, exerting cytotoxicity against lung cancer cell lines. | [137] | ||
| Monoclonal antibodies | ||||||||
| Prostate cancer | BLCA-38 | In patients with prostate cancer, the BLCA-38 antibody binds primarily to prostate cancer cells but not to normal cells and may be useful in targeting novel therapies | In vivo and In vitro | PZ-HPV-7 prostate cells; LNCaP, DU145, and PC-3; LNCaP-C4 and LNCaP-C4–2; LNCaP-LN3, PC3-M, and PC3-M-MM2; MDA PCa 2a and MDA PCa 2b; LAPC4 cells. | Male 6–8-week-old athymic nude mice, BALB/c (nu/ nu) | Flow cytometry | Prostate cancer lines PC-3, PC-3 M, PC-3 M-MM2, and DU-145 all expressed cell surface BLCA-38 antigen, whereas LNCaP, MDA PCa 2a, and MDA PCa 2b did not. | [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, A.G.; Wanjari, U.R.; Namachivayam, A.; Murali, R.; Prabakaran, D.S.; Ganesan, R.; Renu, K.; Dey, A.; Vellingiri, B.; Ramanathan, G.; et al. Role of Immune Cells and Receptors in Cancer Treatment: An Immunotherapeutic Approach. Vaccines 2022, 10, 1493. https://doi.org/10.3390/vaccines10091493
Mukherjee AG, Wanjari UR, Namachivayam A, Murali R, Prabakaran DS, Ganesan R, Renu K, Dey A, Vellingiri B, Ramanathan G, et al. Role of Immune Cells and Receptors in Cancer Treatment: An Immunotherapeutic Approach. Vaccines. 2022; 10(9):1493. https://doi.org/10.3390/vaccines10091493
Chicago/Turabian StyleMukherjee, Anirban Goutam, Uddesh Ramesh Wanjari, Arunraj Namachivayam, Reshma Murali, D. S. Prabakaran, Raja Ganesan, Kaviyarasi Renu, Abhijit Dey, Balachandar Vellingiri, Gnanasambandan Ramanathan, and et al. 2022. "Role of Immune Cells and Receptors in Cancer Treatment: An Immunotherapeutic Approach" Vaccines 10, no. 9: 1493. https://doi.org/10.3390/vaccines10091493
APA StyleMukherjee, A. G., Wanjari, U. R., Namachivayam, A., Murali, R., Prabakaran, D. S., Ganesan, R., Renu, K., Dey, A., Vellingiri, B., Ramanathan, G., Doss C., G. P., & Gopalakrishnan, A. V. (2022). Role of Immune Cells and Receptors in Cancer Treatment: An Immunotherapeutic Approach. Vaccines, 10(9), 1493. https://doi.org/10.3390/vaccines10091493