
An In-Silico Investigation to Design a Multi-Epitopes Vaccine against Multi-Drug Resistant Hafnia alvei

 , , and
, , and 
Abstract
1. Introduction
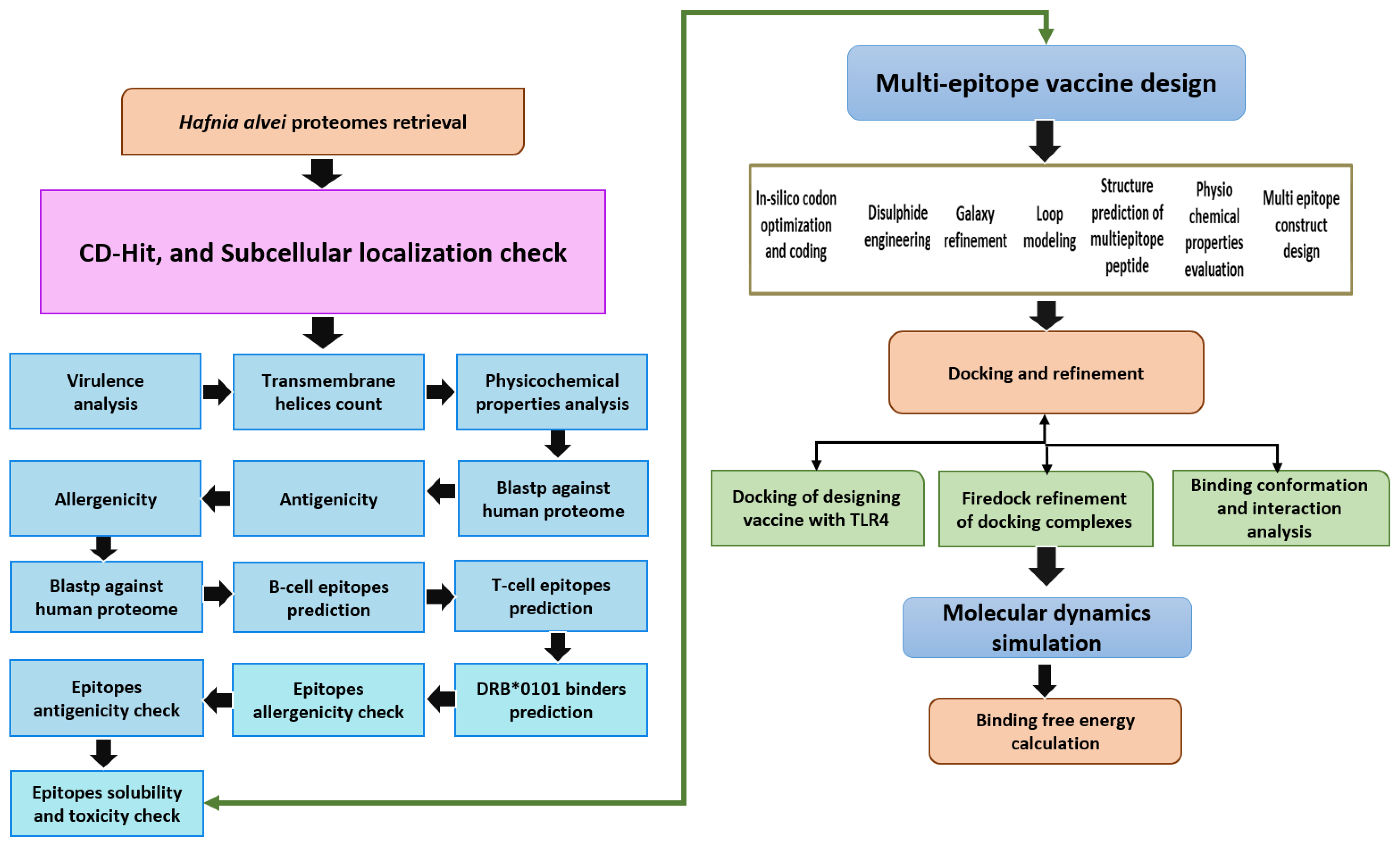
2. Methodology
2.1. Subtractive Proteomics and Reverse Vaccinology
2.2. Pre-Selection Stage
2.3. CD-Hit Analysis
2.4. Subcellular Localization Phase
2.5. Vaccine Candidate’s Prioritization Phase
2.6. Analysis of Potential Transmembrane Helices
2.7. Physiochemical Properties Analysis
2.8. Homology Check with Human and Normal Flora
2.9. Prediction of Immune Cell Epitopes
2.10. MHCPred 2.0 Analysis
2.11. Antigenicity, Allergenicity, Solubility and Toxicity Prediction
2.12. Multi-Epitopes Peptide Designing
2.13. Codon Optimization
2.14. Docking and Refinement
2.15. Molecular Dynamics (MD) Simulation Assay
2.16. Free Energy of TLR4 and Vaccine Design
3. Results
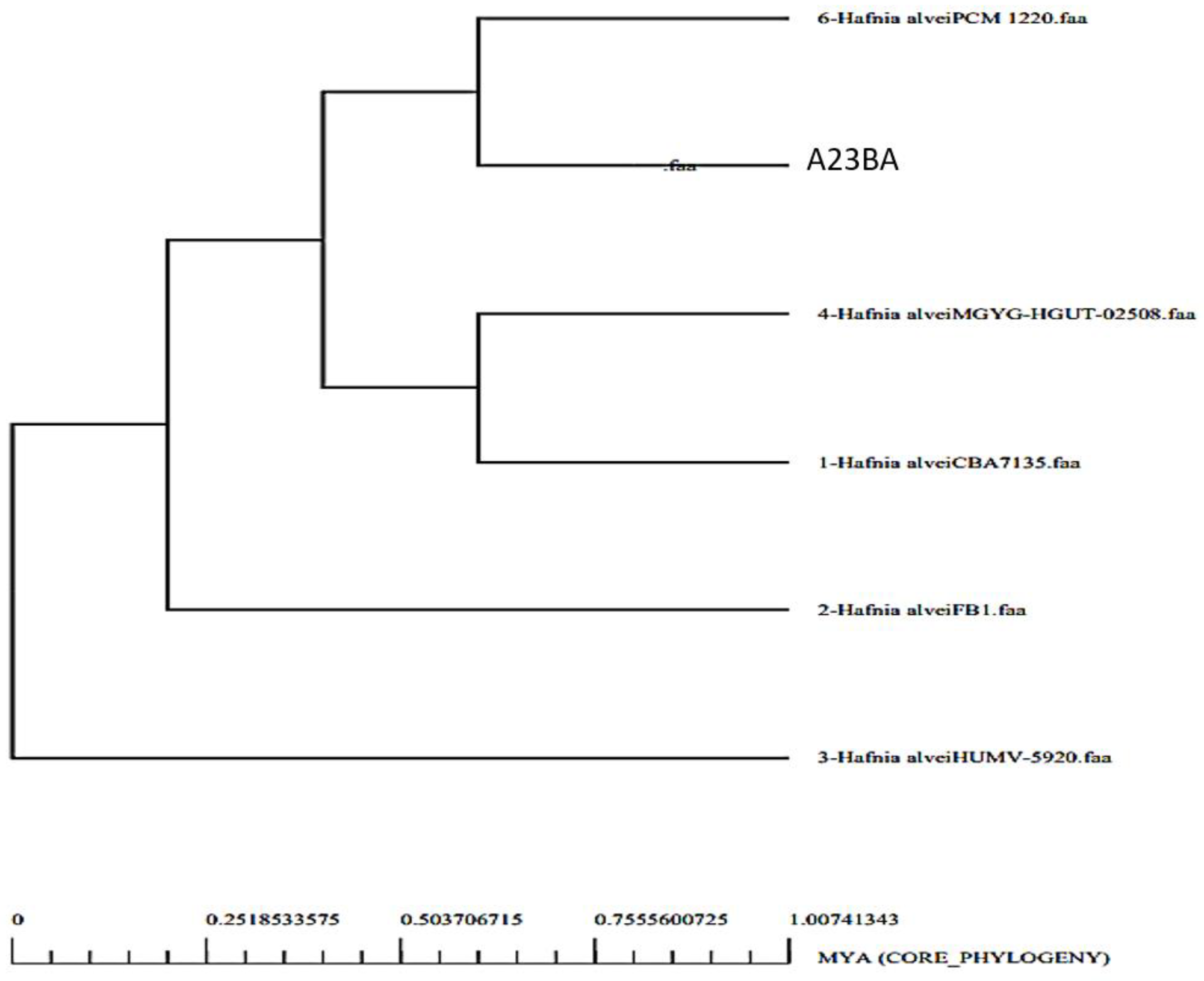
3.1. Genomes Retrieval of H. alvei
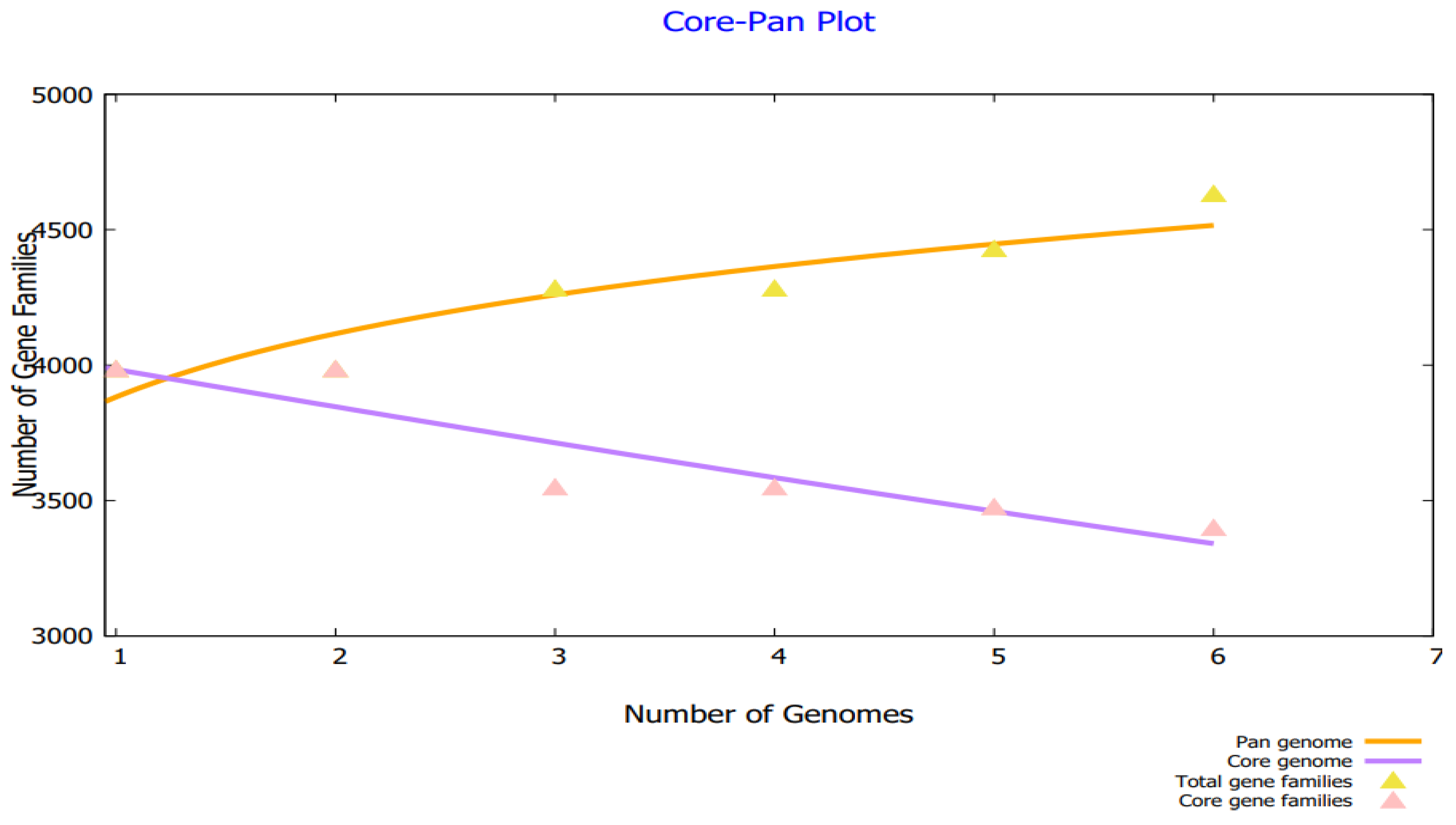
3.2. Bacterial Pan-Genome Analysis
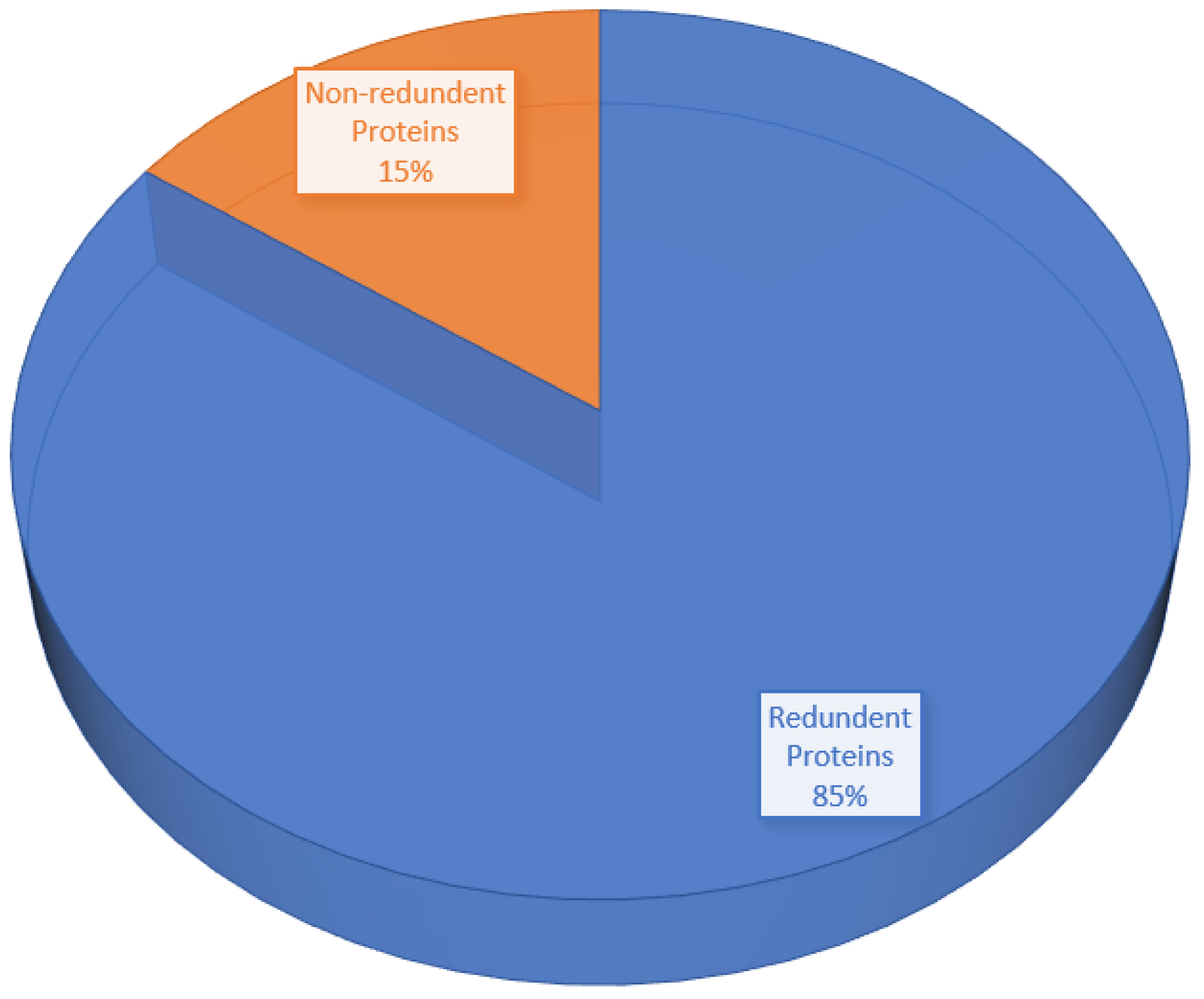
3.3. CD-HIT Analysis
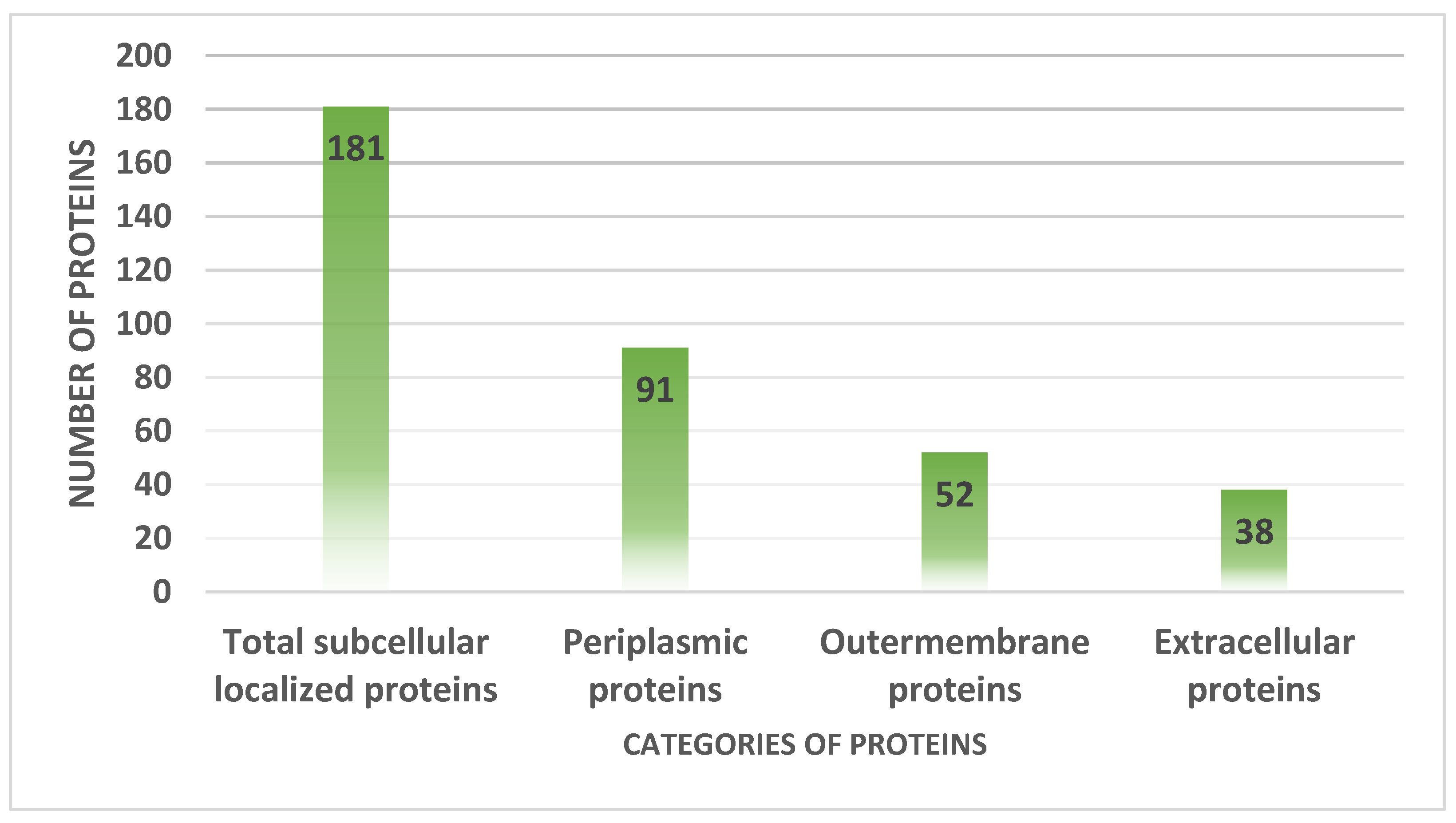
3.4. Proteins Subcellular Localization
3.5. VFDB Analysis
3.6. Transmembrane Helices and Physiochemical Analysis
3.7. Similarity with Human Genome and Prediction of Antigenicity and Allergenicity
3.8. Homology Check of Normal Flora
3.9. B-Cell Epitopes Prediction
3.10. MHC-I and MHC-II Epitopes Prediction
3.11. Epitope Prioritization Phase
3.12. MHCPred Analysis
3.13. Allergenicity and Antigenicity
3.14. Analysis of Solubility and Toxicity
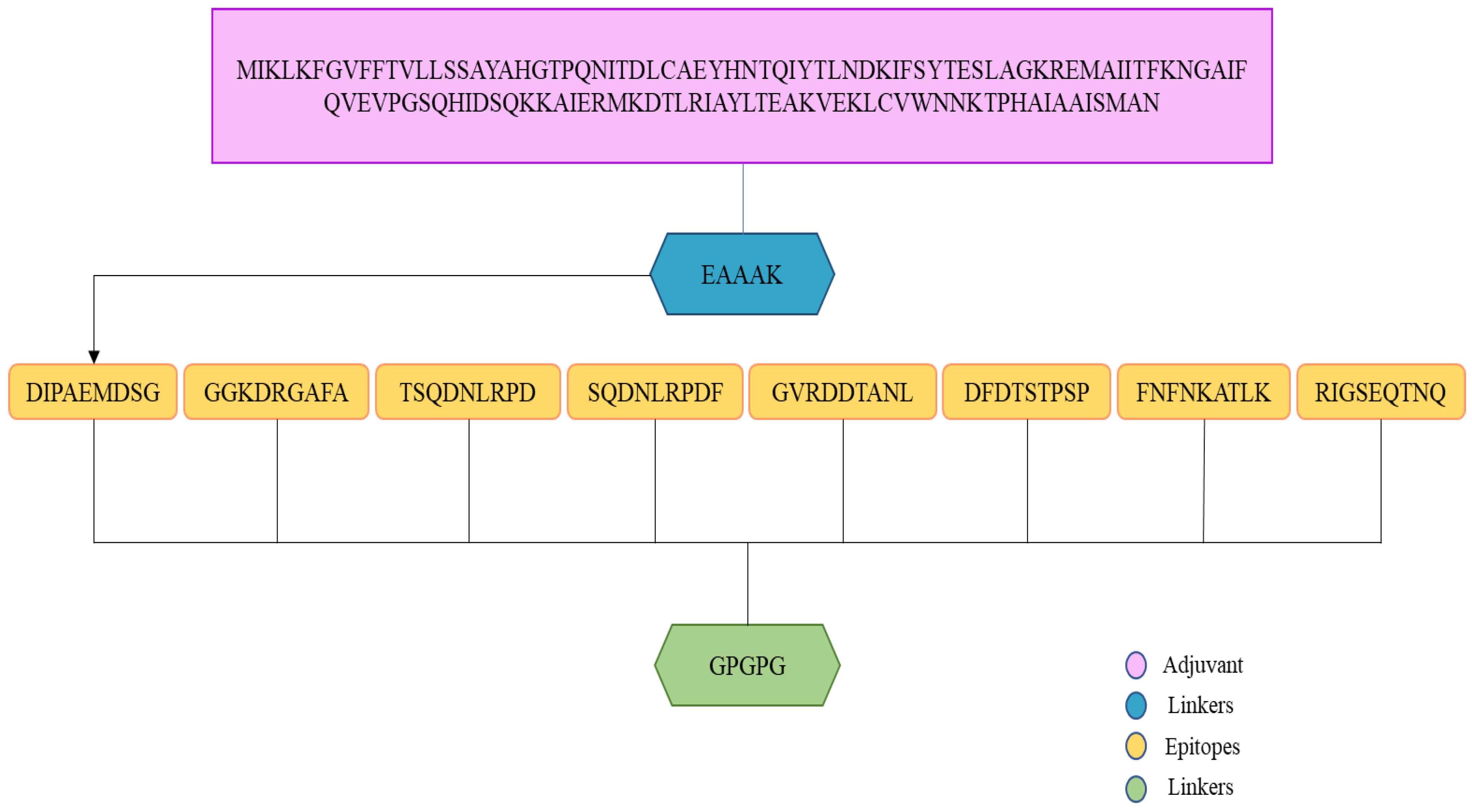
3.15. Multi-Epitopes Vaccine Designing
3.16. Vaccine Structure Modeling
3.17. Loop Modeling and Refinement
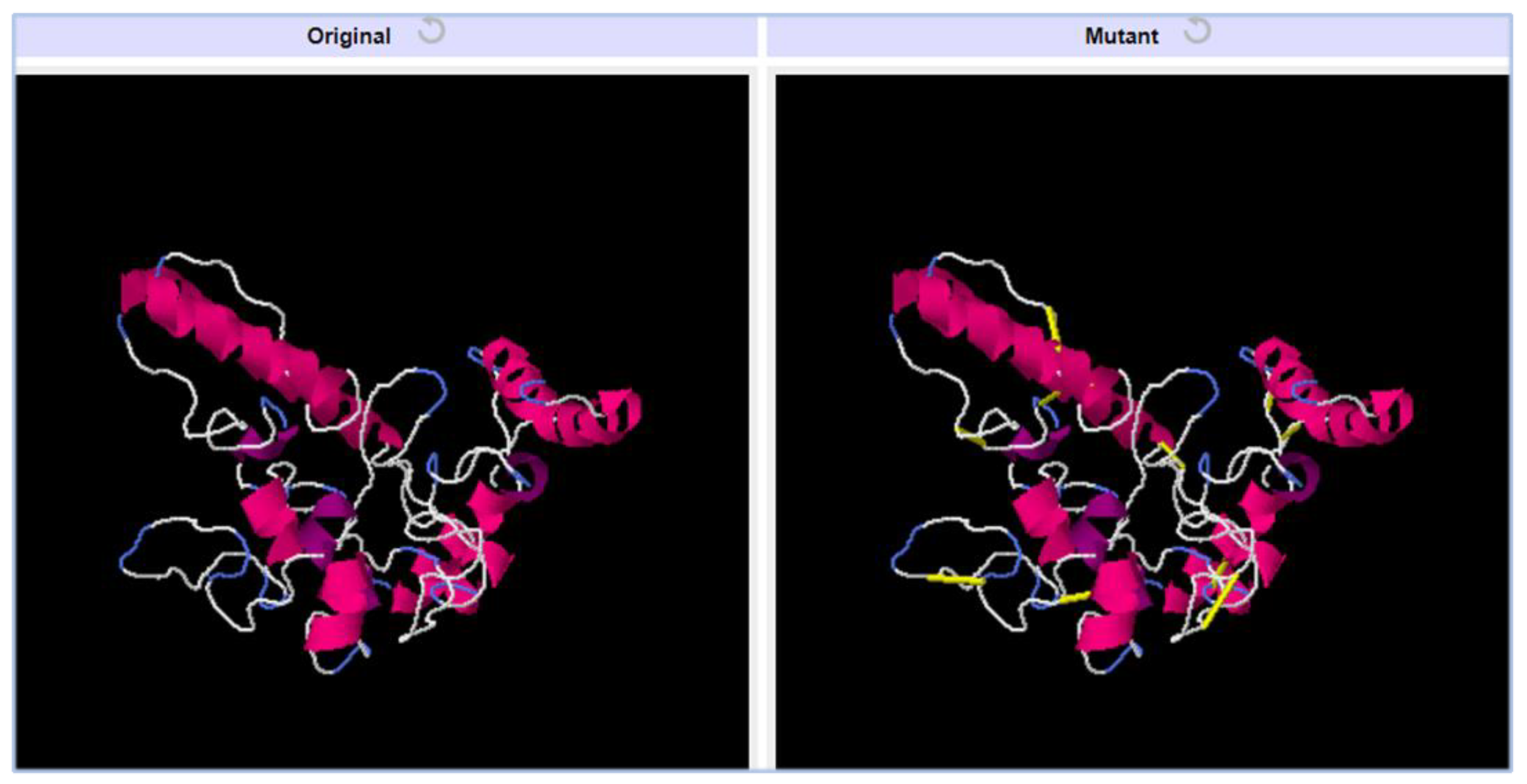
3.18. Disulfide Engineering
3.19. Codon Optimization
3.20. Analysis of Molecular Docking
3.21. Docked Complexes Refinement
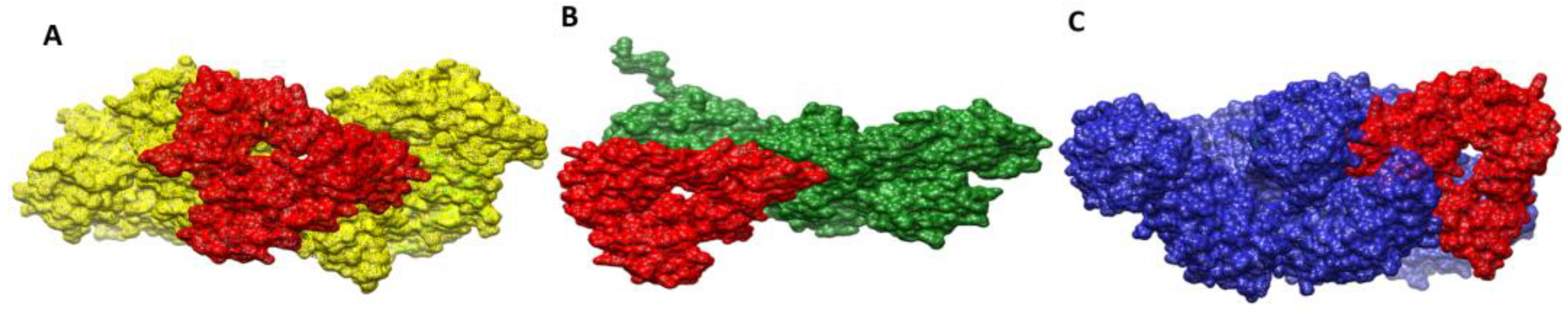
3.22. Docked Confirmation of Vaccine with Immune Receptors
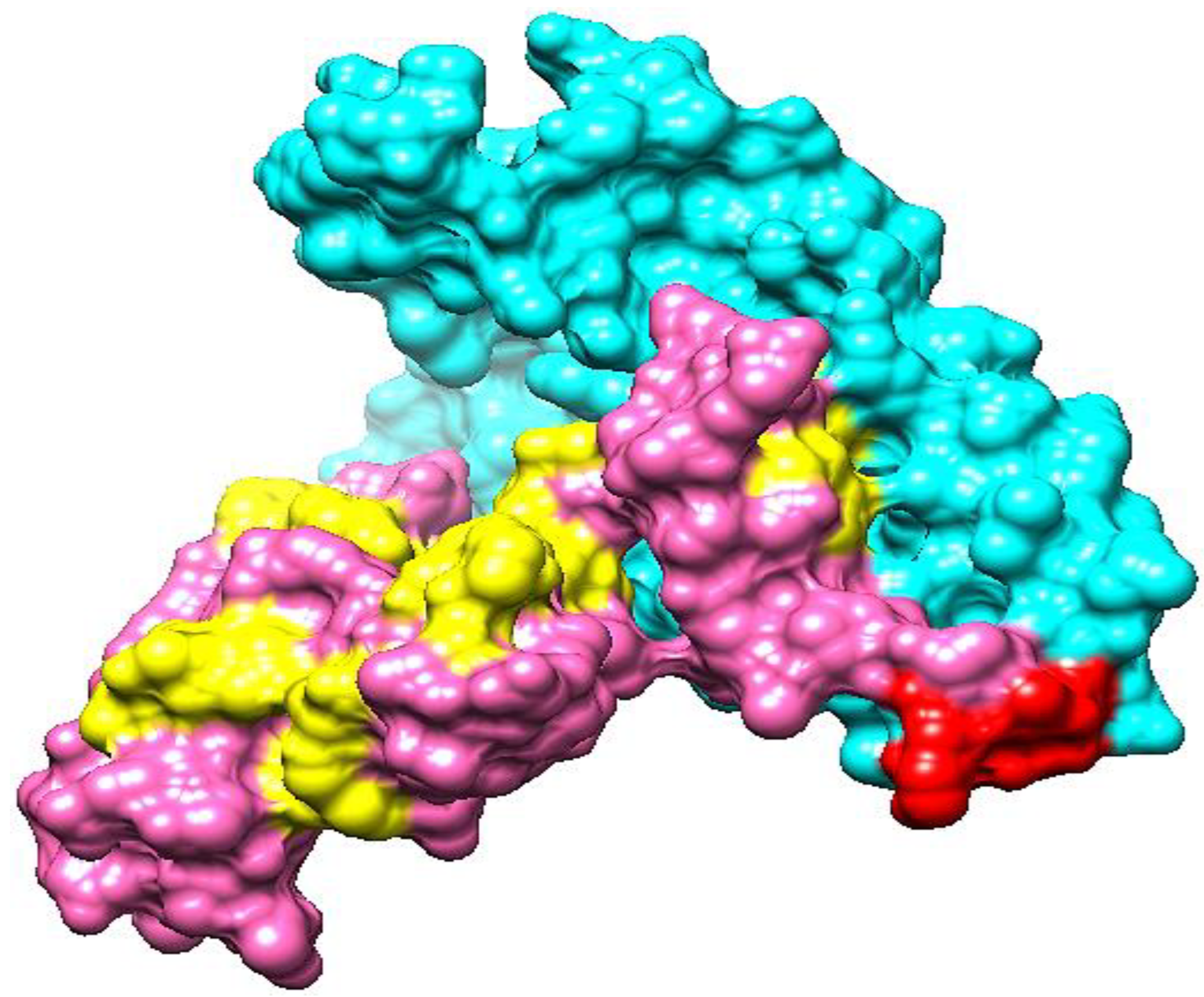
3.23. Interactions of Vaccine to Immune Receptors
3.24. Molecular Dynamic Simulation
3.25. Calculation of Binding Free Energies
4. Discussion
5. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shallcross, L.J.; Davies, D.S.C. Antibiotic overuse: A key driver of antimicrobial resistance. Br. J. Gen. Pract. 2014, 64, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Llor, C.; Bjerrum, L. Antimicrobial resistance: Risk associated with antibiotic overuse and initiatives to reduce the problem. Ther. Adv. Drug Saf. 2014, 5, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Albekairi, T.H.; Alshammari, A.; Alharbi, M.; Alshammary, A.F.; Tahir ul Qamar, M.; Ullah, A.; Irfan, M.; Ahmad, S. Designing of a Novel Multi-Antigenic Epitope-Based Vaccine against E. hormaechei: An Intergraded Reverse Vaccinology and Immunoinformatics Approach. Vaccines 2022, 10, 665. [Google Scholar]
- Albekairi, T.H.; Alshammari, A.; Alharbi, M.; Alshammary, A.F.; Tahir ul Qamar, M.; Anwar, T.; Ismail, S.; Shaker, B.; Ahmad, S. Design of a Multi-Epitope Vaccine against Tropheryma whipplei Using Immunoinformatics and Molecular Dynamics Simulation Techniques. Vaccines 2022, 10, 691. [Google Scholar] [CrossRef]
- Lucien, M.A.B.; Canarie, M.F.; Kilgore, P.E.; Jean-Denis, G.; Fénélon, N.; Pierre, M.; Cerpa, M.; Joseph, G.A.; Maki, G.; Zervos, M.J. Antibiotics and antimicrobial resistance in the COVID-19 era: Perspective from resource-limited settings. Int. J. Infect. Dis. 2021, 104, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Ud-Din, M.; Albutti, A.; Ullah, A.; Ismail, S.; Ahmad, S.; Naz, A.; Khurram, M.; Haq, M.; Afsheen, Z.; Bakri, Y. El Vaccinomics to Design a Multi-Epitopes Vaccine for Acinetobacter baumannii. Int. J. Environ. Res. Public Health 2022, 19, 5568. [Google Scholar]
- Suleman, M.; ul Qamar, M.T.; Rasool, S.; Rasool, A.; Albutti, A.; Alsowayeh, N.; Alwashmi, A.S.S.; Aljasir, M.A.; Ahmad, S.; Hussain, Z. Immunoinformatics and Immunogenetics-Based Design of Immunogenic Peptides Vaccine against the Emerging Tick-Borne Encephalitis Virus (TBEV) and Its Validation through In Silico Cloning and Immune Simulation. Vaccines 2021, 9, 1210. [Google Scholar] [CrossRef]
- Clem, A.S. Fundamentals of vaccine immunology. J. Glob. Infect. Dis. 2011, 3, 73–78. [Google Scholar] [CrossRef]
- Jalal, K.; Khan, K.; Ahmad, D.; Hayat, A.; Basharat, Z.; Abbas, M.N.; Alghamdi, S.; Almehmadi, M.; Sahibzada, M.U.K. Pan-genome reverse vaccinology approach for the design of multi-epitope vaccine construct against escherichia albertii. Int. J. Mol. Sci. 2021, 22, 2814. [Google Scholar] [CrossRef]
- Alharbi, M.; Alshammari, A.; Alasmari, A.F.; Alharbi, S.; Tahir ul Qamar, M.; Abbasi, S.W.; Shaker, B.; Ahmad, S. Whole Proteome-Based Therapeutic Targets Annotation and Designing of Multi-Epitope-Based Vaccines against the Gram-Negative XDR-Alcaligenes faecalis Bacterium. Vaccines 2022, 10, 462. [Google Scholar] [CrossRef]
- Alharbi, M.; Alshammari, A.; Alasmari, A.F.; Alharbi, S.M.; Tahir ul Qamar, M.; Ullah, A.; Ahmad, S.; Irfan, M.; Khalil, A.A.K. Designing of a Recombinant Multi-Epitopes Based Vaccine against Enterococcus mundtii Using Bioinformatics and Immunoinformatics Approaches. Int. J. Environ. Res. Public Health 2022, 19, 3729. [Google Scholar] [PubMed]
- Naz, S.; Ahmad, S.; Abbasi, S.W.; Ismail, S.; Waseem, S.; Ul Qamar, M.T.; Almatroudi, A.; Ali, Z. Identification of immunodominant epitopes in allelic variants VK210 and VK247 of Plasmodium Vivax Circumsporozoite immunogen. Infect. Genet. Evol. 2021, 96, 105120. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Stanic, M.; Meusburger, E.; Hartmann, G.; Lhotta, K. Hafnia alvei urosepsis in a kidney transplant patient. Case Rep. Transplant. 2015, 2015, 863131. [Google Scholar] [PubMed]
- Begbey, A.; Guppy, J.H.; Mohan, C.; Webster, S. Hafnia alvei pneumonia: A rare cause of infection in the multimorbid or immunocompromised. BMJ Case Rep. CP 2020, 13, e237061. [Google Scholar] [CrossRef]
- Günthard, H.; Pennekamp, A. Clinical significance of extraintestinal Hafnia alvei isolates from 61 patients and review of the literature. Clin. Infect. Dis. 1996, 22, 1040–1045. [Google Scholar] [CrossRef]
- Mourgues, R.; Vassal, L.; Auclair, J.; Mocquot, G.; Vandeweghe, J. Origine et développement des bactéries coliformes dans les fromages à pâte molle. Lait 1977, 57, 131–149. [Google Scholar] [CrossRef][Green Version]
- Gul, S.; Ahmad, S.; Ullah, A.; Ismail, S.; Khurram, M.; Tahir ul Qamar, M.; Hakami, A.R.; Alkhathami, A.G.; Alrumaihi, F.; Allemailem, K.S. Designing a Recombinant Vaccine against Providencia rettgeri Using Immunoinformatics Approach. Vaccines 2022, 10, 189. [Google Scholar] [CrossRef]
- Ullah, A.; Ahmad, S.; Ismail, S.; Afsheen, Z.; Khurram, M.; Tahir ul Qamar, M.; AlSuhaymi, N.; Alsugoor, M.H.; Allemailem, K.S. Towards A Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella morganii. Int. J. Environ. Res. Public Health 2021, 18, 10961. [Google Scholar] [CrossRef]
- Fatima, I.; Ahmad, S.; Abbasi, S.W.; Ashfaq, U.A.; Shahid, F.; ul Qamar, M.T.; Rehman, A.; Allemailem, K.S. Designing of a multi-epitopes-based peptide vaccine against rift valley fever virus and its validation through integrated computational approaches. Comput. Biol. Med. 2021, 14, 105151. [Google Scholar] [CrossRef]
- Alamri, M.A.; Mirza, M.U.; Adeel, M.M.; Ashfaq, U.A.; Tahir ul Qamar, M.; Shahid, F.; Ahmad, S.; Alatawi, E.A.; Albalawi, G.M.; Allemailem, K.S. Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors. Pharmaceuticals 2022, 15, 659. [Google Scholar] [CrossRef] [PubMed]
- Fatima, I.; Ahmad, S.; Alamri, M.A.; Mirza, M.U.; Tahir ul Qamar, M.; Rehman, A.; Shahid, F.; Alatawi, E.A.; Alkhayl, F.F.; Al-Megrin, W.A. Discovery of Rift Valley fever virus natural pan-inhibitors by targeting its multiple key proteins through computational approaches. Sci. Rep. 2022, 12, 9260. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Hosen, M.A.; Ahmad, S.; ul Qamar, M.T.; Dey, S.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, antimicrobial, anticancer activities, PASS prediction, molecular docking, molecular dynamics and pharmacokinetic studies of designed methyl α-D-glucopyranoside esters. J. Mol. Struct. 2022, 1260, 132761. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017, 45, D12. [Google Scholar]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA—An ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Combating tigecycline resistant Acinetobacter baumannii: A leap forward towards multi-epitope based vaccine discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef]
- Tusnady, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- ExPASy-ProtParam Tool 2017. Available online: https://web.expasy.org/protparam/ (accessed on 5 April 2022).
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2018, 47, D339–D343. [Google Scholar] [CrossRef]
- Guan, P.; Doytchinova, I.A.; Zygouri, C.; Flower, D.R. MHCPred: A server for quantitative prediction of peptide–MHC binding. Nucleic Acids Res. 2003, 31, 3621–3624. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P.S. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Stratmann, T. Cholera toxin subunit B as adjuvant––An accelerator in protective immunity and a break in autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Lee, J.; Hitzenberger, M.; Rieger, M.; Kern, N.R.; Zacharias, M.; Im, W. CHARMM-GUI supports the Amber force fields. J. Chem. Phys. 2020, 153, 35103. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [PubMed]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein disulfide engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Adu-Bobie, J.; Capecchi, B.; Serruto, D.; Rappuoli, R.; Pizza, M. Two years into reverse vaccinology. Vaccine 2003, 21, 605–610. [Google Scholar] [CrossRef]
- Seib, K.L.; Zhao, X.; Rappuoli, R. Developing vaccines in the era of genomics: A decade of reverse vaccinology. Clin. Microbiol. Infect. 2012, 18, 109–116. [Google Scholar] [CrossRef]
- Aslam, S.; Ahmad, S.; Noor, F.; Ashfaq, U.A.; Shahid, F.; Rehman, A.; Tahir ul Qamar, M.; Alatawi, E.A.; Alshabrmi, F.M.; Allemailem, K.S. Designing a Multi-Epitope Vaccine against Chlamydia trachomatis by Employing Integrated Core Proteomics, Immuno-Informatics and In Silico Approaches. Biology 2021, 10, 997. [Google Scholar] [CrossRef]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; ul Qamar, M.T. Pan-Vaccinomics Approach towards a Universal Vaccine Candidate against WHO Priority Pathogens to Address Growing Global Antibiotic Resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Organism Name | Strain | Size (Mb) | GC% |
|---|---|---|---|---|
| 1. | H. alvei | A23BA | 4.77 | 48.77 |
| 2. | H. alvei | PCM_1220 | 4.55 | 48.90 |
| 3. | H. alvei | HUMV-5920 | 4.63 | 48.70 |
| 4. | H. alvei | CBA7135 | 4.50 | 48.90 |
| 5. | H. alvei | MGYG-HGUT-02508 | 4.50 | 48.90 |
| 6. | H. alvei | FB1 | 4.71 | 49.00 |
| Extracellular Proteins | Bit Score | Sequence Identity |
|---|---|---|
| core/462/1/Org1_Gene2554 | 591 bits | 57% |
| core/770/1/Org1_Gene3263 | 168 bits | 37% |
| core/845/1/Org1_Gene1577 | 250 bits | 30% |
| core/847/1/Org1_Gene3025 | 194 bits | 30% |
| core/935/1/Org1_Gene2294 | 268 bits | 36% |
| core/979/1/Org1_Gene3259 | 345 bits | 50% |
| core/1075/1/Org1_Gene1236 | 464 bits | 56% |
| core/2716/1/Org1_Gene1237 | 279 bits | 61% |
| core/3338/1/Org1_Gene755 | 111 bits | 39% |
| core/3797/1/Org1_Gene1966 | 135 bits | 49% |
| Outer membrane Proteins | ||
| core/1/1/Org1_Gene3744 | 2148 bits | 39% |
| core/120/1/Org1_Gene3603 | 407 bits | 32% |
| core/149/1/Org1_Gene2889 | 786 bits | 48% |
| core/243/1/Org1_Gene2268 | 894 bits | 63% |
| core/303/1/Org1_Gene3723 | 293 bits | 31% |
| core/969/1/Org1_Gene2163 | 246 bits | 36% |
| core/1517/1/Org1_Gene912 | 466 bits | 70% |
| core/2828/1/Org1_Gene1578 | 204 bits | 48% |
| core/168/3/Org3_Gene3997 | 438 bits | 33% |
| Periplasmic proteins | ||
| core/406/1/Org1_Gene3346 | 464 bits | 46% |
| core/738/1/Org1_Gene745 | 243 bits | 35% |
| core/1381/1/Org1_Gene1348 | 520 bits | 78% |
| core/2485/1/Org1_Gene1235 | 357 bits | 70% |
| core/2509/1/Org1_Gene738 | 158 bits | 34% |
| core/2667/1/Org1_Gene3604 | 147 bits | 39% |
| core/2786/1/Org1_Gene1969 | 207 bits | 50% |
| core/2972/1/Org1_Gene3596 | 182 bits | 46% |
| core/3741/1/Org1_Gene572 | 149 bits | 53% |
| core/3781/1/Org1_Gene1239 | 230 bits | 81% |
| core/4130/1/Org1_Gene147 | 129 bits | 62% |
| Vaccine Target | Physiochemical Properties | |||||||
|---|---|---|---|---|---|---|---|---|
| Extracellular Proteins | HMMTOP | TMHMM | Amino Acid | M.W | T. PI | I. I | A. I | GRAVY |
| core/770/1/Org1_Gene3263 | 0 | 0 | 469 | 48.76661 | 6.09 | 39.75 | 84.48 | −0.39 |
| core/845/1/Org1_Gene1577 | 0 | 0 | 456 | 48.51884 | 4.4 | 29.03 | 95.39 | −0.345 |
| core/935/1/Org1_Gene2294 | 0 | 0 | 440 | 47.62321 | 4.78 | 19.79 | 78.32 | −0.512 |
| core/3338/1/Org1_Gene755 | 0 | 0 | 175 | 18.74466 | 5.9 | 20.37 | 105.26 | 0.129 |
| core/3797/1/Org1_Gene1966 | 1 | 0 | 136 | 14.82368 | 5.15 | 28.26 | 83.82 | −0.254 |
| Outer membrane proteins | ||||||||
| core/120/1/Org1_Gene3603 | 1 | 0 | 865 | 95.03412 | 6.51 | 33.61 | 69.69 | −0.501 |
| core/149/1/Org1_Gene2889 | 1 | 0 | 824 | 91.28249 | 5.55 | 36.3 | 70.91 | −0.437 |
| core/243/1/Org1_Gene2268 | 1 | 0 | 705 | 76.71293 | 5.17 | 25.66 | 63.12 | −0.517 |
| core/1517/1/Org1_Gene912 | 1 | 0 | 351 | 38.01163 | 5.73 | 26.46 | 78.15 | −0.341 |
| Periplasmic proteins | ||||||||
| core/2485/1/Org1_Gene1235 | 0 | 0 | 251 | 26.78204 | 4.74 | 29.41 | 85.22 | −0.294 |
| core/2667/1/Org1_Gene3604 | 0 | 0 | 237 | 26.36229 | 9.32 | 36.11 | 82.24 | −0.201 |
| core/4130/1/Org1_Gene147 | 0 | 0 | 104 | 11.07165 | 5.14 | 32.46 | 98.56 | 0.018 |
| Extracellular Proteins | Antigenicity | Allergenicity | Human (taxid:9606) |
|---|---|---|---|
| core/770/1/Org1_Gene3263 | |||
| core/845/1/Org1_Gene1577 | |||
| core/935/1/Org1_Gene2294 | Antigen | Non-Allergen | No Similarity |
| core/3338/1/Org1_Gene755 | |||
| core/3797/1/Org1_Gene1966 | |||
| Outer membrane Proteins | |||
| core/120/1/Org1_Gene3603 | |||
| core/149/1/Org1_Gene2889 | Antigen | Non-Allergen | No Similarity |
| core/243/1/Org1_Gene2268 | |||
| core/1517/1/Org1_Gene912 | |||
| Periplasmic Proteins | |||
| core/2485/1/Org1_Gene1235 | |||
| core/2667/1/Org1_Gene3604 | Antigen | Non-Allergen | No Similarity |
| core/4130/1/Org1_Gene147 |
| Extracellular Proteins | Lactobacillus casei (taxid:1582) | Lactobacillus jhonsoni (taxid:33959) | Lactobacillus rhamnosus (taxid:47715) |
|---|---|---|---|
| core/770/1/Org1_Gene3263 | |||
| core/845/1/Org1_Gene1577 | |||
| core/935/1/Org1_Gene2294 | |||
| core/3338/1/Org1_Gene755 | |||
| core/3797/1/Org1_Gene1966 | |||
| Outer membrane Proteins | |||
| core/120/1/Org1_Gene3603 | No Similarity | ||
| core/149/1/Org1_Gene2889 | |||
| core/243/1/Org1_Gene2268 | |||
| core/1517/1/Org1_Gene912 | |||
| Periplasmic Proteins | |||
| core/2485/1/Org1_Gene1235 | |||
| core/2667/1/Org1_Gene3604 | |||
| core/4130/1/Org1_Gene147 |
| B-Cell Epitopes | Peptides |
|---|---|
| core/770/1/Org1_Gene3263 Flagellar hook length control protein FliK |
GSEAQPQWNGSQQNASDRQATSGGFSVD
GNNDDRIVTASVSAKSVRVGGV |
| core/845/1/Org1_Gene1577 Flagellar hook associated protein FlgK | SKANYYDSGQKYIG |
| QKIAELEATGGNTNVLRDQRDELVKQMS | |
|
QPDGSQLLTLKYAGSEYSINPATGGQLGA
LNDYEQG | |
| QNQRDNLSAVDQDEE | |
| core/935/1/Org1_Gene2294 Flagellar filament capping protein FliD | QAALKKQQTSLTGQQD |
| LNKTNNGLLTNKVTTSLD | |
| VAFDDMTDDAVNNATG | |
| NKTNDGSEKSPA | |
| SGDIPAEMDSGKKTTISEAK | |
| KLTASGSGGKDRGAFAGDAG | |
| core/3338/1/Org1_Gene755 Type 1 fimbrial protein | TLPVSELARTGQGPEK |
| CELTSQDNLRPDFKSVHL | |
| FDGVRDDTANLLSIHGEAS | |
| core/3797/1/Org1_Gene1966 Curli production assembly/transport protein CsgF | AQNSYKDPNGYDFDTSTPSPLDN |
| core/120/1/Org1_Gene3603 Fimbria/pilus outer membrane usher protein | FLGGGFAKGLKRFNTDNNTAT |
| LTRSPRDAVPVESWDAG | |
| QSRYRTGSGGTSQY | |
| GERANKKQGSNNVFKSDTLNQRN | |
| YQQNRENQAGSTKNWG | |
| core/149/1/Org1_Gene2889 Fimbria/pilus outer membrane usher protein | DDLVEFNTDVLDASDRTHV |
| LALKEEARLKVEQVSENCFVLQ | |
| EYQTSYYNSTHQFDF | |
| VPAGPFNIQDLSSSVR | |
| core/243/1/Org1_Gene2268 TonB-dependent hemoglobin /transferrin/lactoferrin family receptor | RDRGNLRMSNGFDSPNDET |
| INASPTGSSYEKRKQTTNG | |
| KLENRSRLFADSFAS | |
| KQKQTPGGATTGFPQAD | |
| KGSSDGYDDVNADKW | |
| VTMDMGFVNGRFGCIDCS | |
| KDQKTGEWLDNINP | |
| FADRNNQVNAGTAPQA | |
| EYYTSQGVIQDGI | |
| core/1517/1/Org1_Gene912 Porin OmpA | SHYYDNSINHFGSTNVRPDQLGG |
| DWLGRMEYRGNNNGAFKS | |
| DGSANSETRGRYIDSHDTGVSP | |
| FNFNKATLKPQGQQA | |
| RIGSEQYNQKLSEQ | |
| core/2485/1/Org1_Gene1235 Flagellar basal body rod protein FlgF | QLTSQGNLVIGDNGPIAIPDRAEVT |
| core/2667/1/Org1_Gene3604 Molecular chaperone | ENKVTQLGNKMV |
| core/4130/1/Org1_Gene147 Flagellar hook-basal body complex protein FliE | LQQLQATAVSAANRSQNSEAPQGA |
| AQNFEMGVPGVAL |
| MHC-I | Percentile Score | MHC-II | Percentile Score |
|---|---|---|---|
| RIVTASVSAK | 0.1 | DRIVTASVSAKSVRVG | 0.73 |
| SVSAKSVRV | 0.13 | ||
| QATSGGFSV | 0.56 | DRQATSGGFSVDGN | 17 |
| SEAQPQWNG | 0.64 | GSEAQPQWNGSQQNAS | 46 |
| QWNGSQQNA | 8.1 | ||
| AYYDSGQKY | 0.04 | SKANYYDSGQKYI | 7 |
| EATGGNTNV | 0.1 | QKIAELEATGGNTNVL | 24 |
| KIAELEATG | 15 | ||
| VLRDQRDEL | 0.31 | VLRDQRDELVKQMS | 3.5 |
| QRDELVKQM | 1.7 | ||
| SEYSINPAT | 0.47 | AGSEYSINPATGGQL | 4 |
| SINPATGGQL | 2.1 | ||
| GSQLLTLKY | 0.06 | QPDGSQLLTLKYAG | 13 |
| GQLGALNDY | 0.11 | PATGGQLGALNDYEQG | 17 |
| ATGGQLGAL | 3.3 | ||
| RDNLSAVDQ | 26 | RDNLSAVDQDE | 0.88 |
| ALKKQQTSL | 0.02 | AALKKQQTSLTG | 1.3 |
| LTNKVTTSL | 0.19 | GLLTNKVTTSL | 0.7 |
| KTNNGLLTNK | 0.02 | LNKTNNGLLTNKVTTSL | 5.3 |
| LTNKVTTSL | 0.15 | ||
| FDDMTDDAV | 5.5 | VAFDDMTDDAVNN | 21 |
| KTNDGSEKS | 5.4 | NKTNDGSEKSPA | 59 |
| DIPAEMDSGK | 1.7 | SGDIPAEMDSGK | 6.5 |
| EMDSGKKTTI | 1.8 | EMDSGKKTTISEAK | 34 |
| KKTTISEAK | 3.8 | ||
| KLTASGSGGK | 0.23 | KLTASGSGGKDRGAFA | 21 |
| GSGGKDRGAF | 5 | ||
| GGKDRGAFA | 2.8 | GSGGKDRGAFAGDAG | 31 |
| KDRGAFAGDA | 20 | ||
| TLPVSELAR | 1.2 | TLPVSELARTGQGP | 31 |
| SELARTGQGP | 2.5 | ||
| NLRPDFKSV | 0.1 | DNLRPDFKSVH | 1.8 |
| TSQDNLRPDF | 2.5 | ELTSQDNLRPDFKSVH | 8.1 |
| GVRDDTANL | 0.53 | DGVRDDTANLL | 1.2 |
| DTANLLSIH | 0.24 | DGVRDDTANLLSIHG | 3.6 |
| DGVRDDTANL | 1.2 | ||
| SYKDPNGYDF | 0.05 | AQNSYKDPNGYDF | 14 |
| DFDTSTPSPL | 1.3 | NGYDFDTSTPSPLDN | 6.1 |
| GFAKGLKRF | 0.29 | GFAKGLKRFNTDNNTAT | 1.1 |
| RFNTDNNTA | 3.5 | ||
| RDAVPVESW | 0.13 | RSPRDAVPVESWDA | 11 |
| RYRTGSGGT | 2.7 | RYRTGSGGTSQ | 4.7 |
| VFKSDTLNQR | 0.16 | VFKSDTLNQRN | 1.2 |
| KKQGSNNVFK | 1.4 | RANKKQGSNNVFKSDTLNQR | 3.7 |
| NQAGSTKNW | 0.25 | QQNRENQAGSTKNW | 37 |
| DLVEFNTDV | 1.1 | DDLVEFNTDVLDAS | 3.9 |
| VEFNTDVLDA | 3.5 | ||
| NTDVLDASDR | 1.4 | LVEFNTDVLDASDRT | 4.3 |
| LVEFNTDVL | 2.7 | ||
| ALKEEARLK | 0.11 | LALKEEARLKVE | 3.1 |
| VEQVSENCF | 0.29 | ARLKVEQVSENCFVLQ | 20 |
| RLKVEQVSE | 2.6 | ||
| TSYYNSTHQF | 0.01 | QTSYYNSTHQFDF | 6.2 |
| NIQDLSSSVR | 0.62 | GPFNIQDLSSSVR | 6.5 |
| RGNLRMSNGF | 6.3 | RGNLRMSNGFDSP | 5.5 |
| NASPTGSSY | 0.01 | INASPTGSSYEKRKQTTNG | 13 |
| SYEKRKQTT | 2.4 | ||
| RSRLFADSF | 0.21 | NRSRLFADSFA | 3.3 |
| QTPGGATTGF | 0.38 | QKQTPGGATTGFP | 5.7 |
| SSDGYDDVNA | 4.5 | GSSDGYDDVNA | 6.2 |
| FVNGRFGCI | 1.2 | DMGFVNGRFGCI | 3.3 |
| KTGEWLDNI | 0.82 | DQKTGEWLDNIN | 5.3 |
| QVNAGTAPQA | 2.4 | FADRNNQVNAGTAPQA | 8.1 |
| FADRNNQVNA | 3.3 | ||
| EYYTSQGVI | 0.78 | EYYTSQGVIQD | 7 |
| SINHFGSTNV | 1.1 | NSINHFGSTNVR | 1.3 |
| EYRGNNNGAF | 0.92 | WLGRMEYRGNNNGAFKS | 1.1 |
| WLGRMEYRG | 48 | ||
| SANSETRGRY | 0.18 | SANSETRGRYID | 7.6 |
| YIDSHDTGV | 0.11 | RGRYIDSHDTGVSP | 29 |
| RGRYIDSHDT | 17 | ||
| FNFNKATLK | 1 | FNFNKATLKPQ | 0.85 |
| RIGSEQYNQK | 0.2 | RIGSEQYNQKLSE | 46 |
| DNGPIAIPDR | 1.2 | NLVIGDNGPIAIPDRAE | 2.9 |
| VIGDNGPIAI | 2.4 | ||
| LTSQGNLVI | 1.1 | QLTSQGNLVIGDNGPIAI | 1.7 |
| IGDNGPIAI | 2.1 | ||
| KVTQLGNKM | 0.7 | NKVTQLGNKMV | 2.7 |
| ATAVSAANR | 0.15 | LQQLQATAVSAANRSQNSEAPQGA | 0.33 |
| LQQLQATAV | 1.1 | ||
| FEMGVPGVAL | 0.05 | AQNFEMGVPGVAL | 0.15 |
| MHC-Pred | MHC-Pred IC50 Value (nM) | Antigenicity | Allergenicity | Solubility | Toxin Pred |
|---|---|---|---|---|---|
| DIPAEMDSG | 84.72 | ||||
| GGKDRGAFA | 95.28 | ||||
| TSQDNLRPD | 9.48 | ||||
| SQDNLRPDF | 16.48 | Antigen | Antigen | Good water solubility | Non-Toxin |
| GVRDDTANL | 18.49 | ||||
| DFDTSTPSP | 20.75 | ||||
| FNFNKATLK | 4.44 | ||||
| RIGSEQYNQ | 24.72 |
| Vaccine Complex | Interactive Residues |
|---|---|
| MHC-I | Ala15, Asp 39, Arg181, Asp76, Glu89, Gln98, Glu44, Gly237, His13, Leu130, Lys3, Pro5, Phe56, Ser52, Trp60, Thr73, Val85. |
| MHC-II | Asp17, Asn78, Arg100, Asp181, Ala140, Glu4, Gly125, Gln311, His143, Leu147, Lys 122, Phe137, Pro127, Ser221, Thr80, Val97. |
| TLR-4 | Arg69, Asn213, Asp50, Cys29, Glu135, Gly110, Gln41, Ile66, Ile285, Leu109, Met40, Met414, Pro53, Ser368, Ser98, Tyr191. |
| Energy Parameter | TLR-4-Vaccine Complex | MHC-I-Vaccine Complex | MHC-II-Vaccine Complex |
|---|---|---|---|
| MM-GBSA | |||
| VDWAALS | −115.36 | −87.30 | −110.00 |
| EEL | −50.00 | −53.60 | −61.96 |
| EGB | 53.54 | 50.00 | 55.33 |
| ESURF | −11.49 | −12.22 | −10.00 |
| Delta G gas | −165.36 | −140.9 | −171.96 |
| Delta G solv | 42.05 | 37.78 | 45.33 |
| Delta Total | −123.31 | −178.68 | −126.63 |
| MM-PBSA | |||
| VDWAALS | −115.36 | −87.30 | −110.00 |
| EEL | −50.00 | −53.60 | −61.96 |
| EPB | 46.31 | 48.00 | 43.10 |
| ENPOLAR | −15.00 | −10.67 | −8.06 |
| Delta G gas | −165.36 | −140.9 | −171.96 |
| Delta G solv | 31.31 | 37.33 | 35.04 |
| Delta Total | −134.05 | −103.57 | −136.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshabrmi, F.M.; Alrumaihi, F.; Alrasheedi, S.F.; Al-Megrin, W.A.I.; Almatroudi, A.; Allemailem, K.S. An In-Silico Investigation to Design a Multi-Epitopes Vaccine against Multi-Drug Resistant Hafnia alvei. Vaccines 2022, 10, 1127. https://doi.org/10.3390/vaccines10071127
Alshabrmi FM, Alrumaihi F, Alrasheedi SF, Al-Megrin WAI, Almatroudi A, Allemailem KS. An In-Silico Investigation to Design a Multi-Epitopes Vaccine against Multi-Drug Resistant Hafnia alvei. Vaccines. 2022; 10(7):1127. https://doi.org/10.3390/vaccines10071127
Chicago/Turabian StyleAlshabrmi, Fahad M., Faris Alrumaihi, Sahar Falah Alrasheedi, Wafa Abdullah I. Al-Megrin, Ahmad Almatroudi, and Khaled S. Allemailem. 2022. "An In-Silico Investigation to Design a Multi-Epitopes Vaccine against Multi-Drug Resistant Hafnia alvei" Vaccines 10, no. 7: 1127. https://doi.org/10.3390/vaccines10071127
APA StyleAlshabrmi, F. M., Alrumaihi, F., Alrasheedi, S. F., Al-Megrin, W. A. I., Almatroudi, A., & Allemailem, K. S. (2022). An In-Silico Investigation to Design a Multi-Epitopes Vaccine against Multi-Drug Resistant Hafnia alvei. Vaccines, 10(7), 1127. https://doi.org/10.3390/vaccines10071127