
Advances in Infectious Disease Vaccine Adjuvants

 ,
, 
 and
and 
Abstract
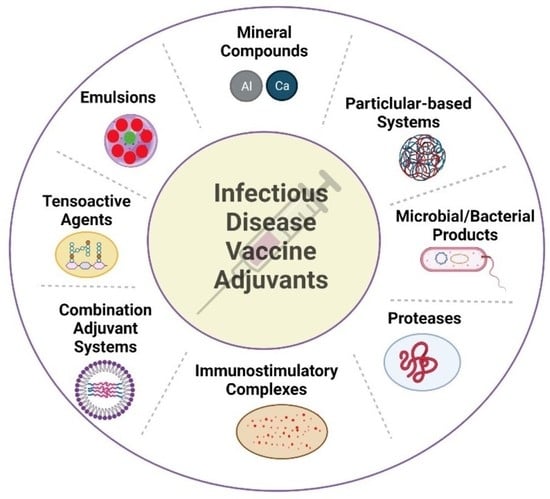
1. Introduction
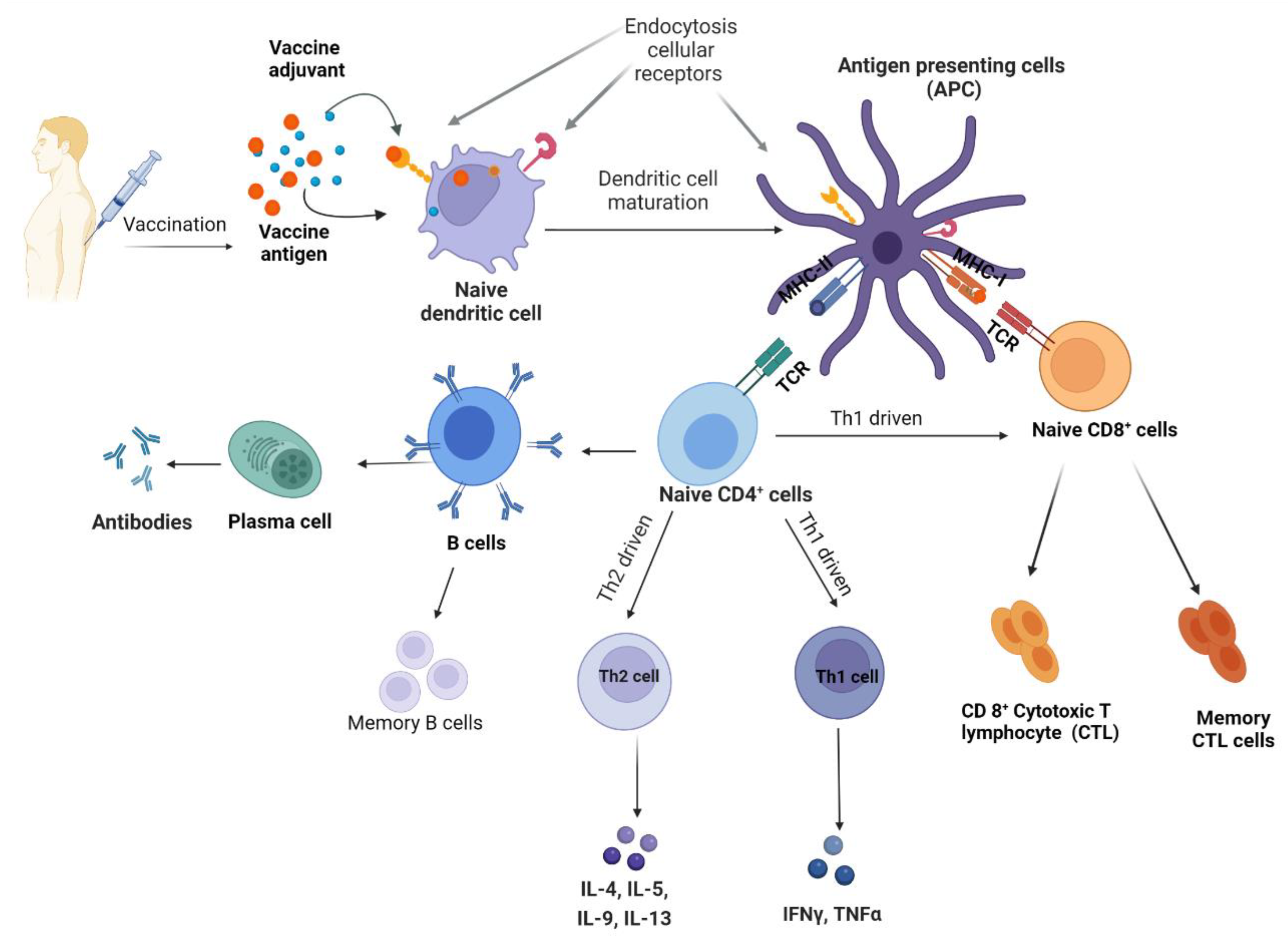
2. Vaccine Adjuvants
2.1. T Helper Interplay in Adjuvant Function
2.2. Importance of Toll-like Receptors in Adjuvant Function
3. Mineral-Based Adjuvants
3.1. Pre-Clinical and Clinical Development of Mineral-Based Adjuvants
3.1.1. Aluminum Adjuvants
3.1.2. Calcium Phosphate Adjuvants
3.1.3. Other Mineral Adjuvants
4. Microbial/Bacterial Adjuvants
4.1. Flagellin Adjuvants
Pre-Clinical and Clinical Development of Flagellin Adjuvants
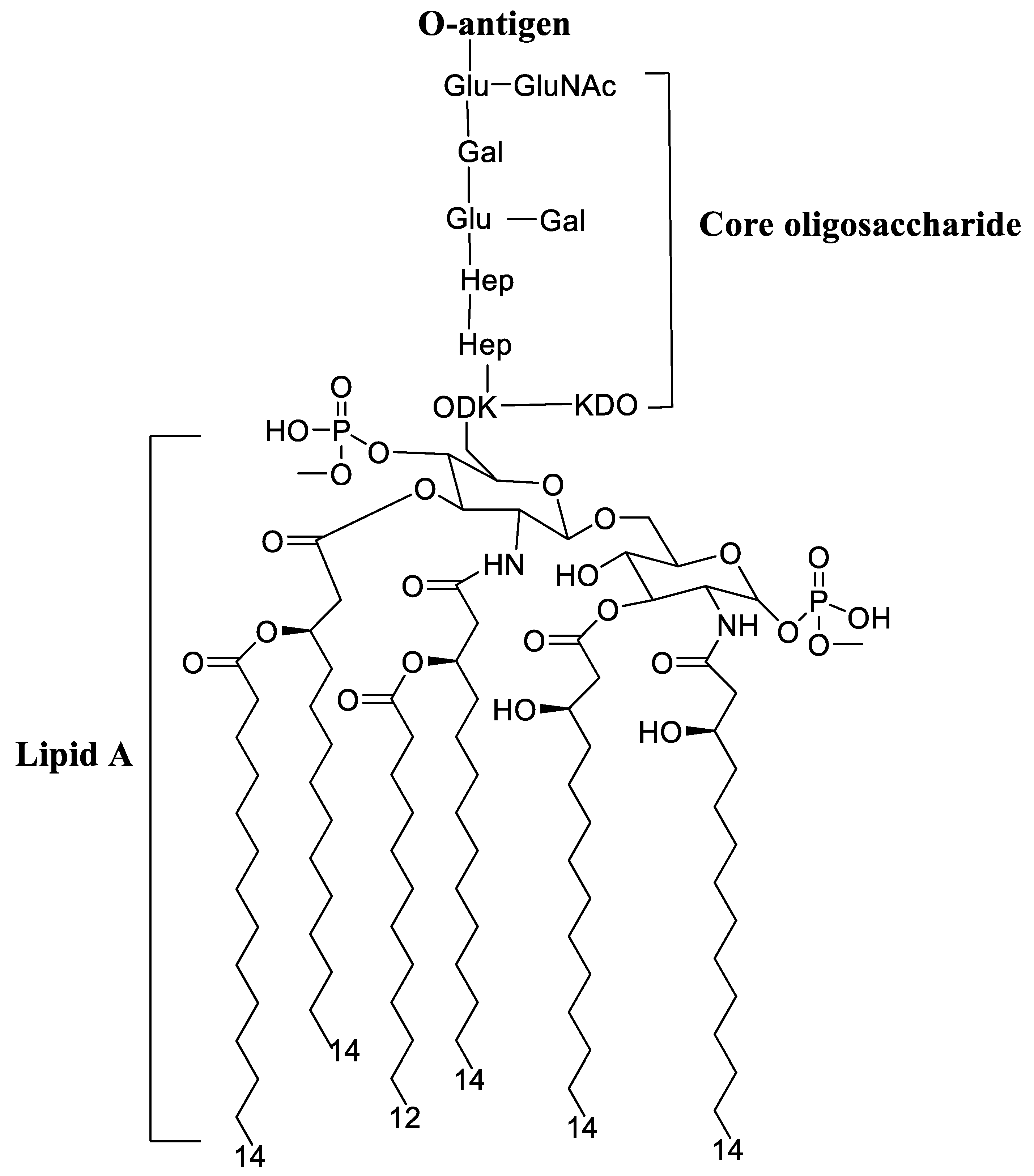
4.2. Lipopolysaccharide Adjuvants
Pre-Clinical and Clinical Development of Lipopolysaccharide Adjuvants
4.3. Cholera Toxin Adjuvant
Pre-Clinical Development of Cholera Toxin Adjuvants
4.4. Bacillus Calmette–Guérin
Pre-Clinical and Clinical Development of Bacillus Calmette–Guérin
5. Emulsions Adjuvants
5.1. Complete and Incomplete Freund’s Adjuvants
Pre-Clinical and Clinical Development of Freund’s Adjuvants
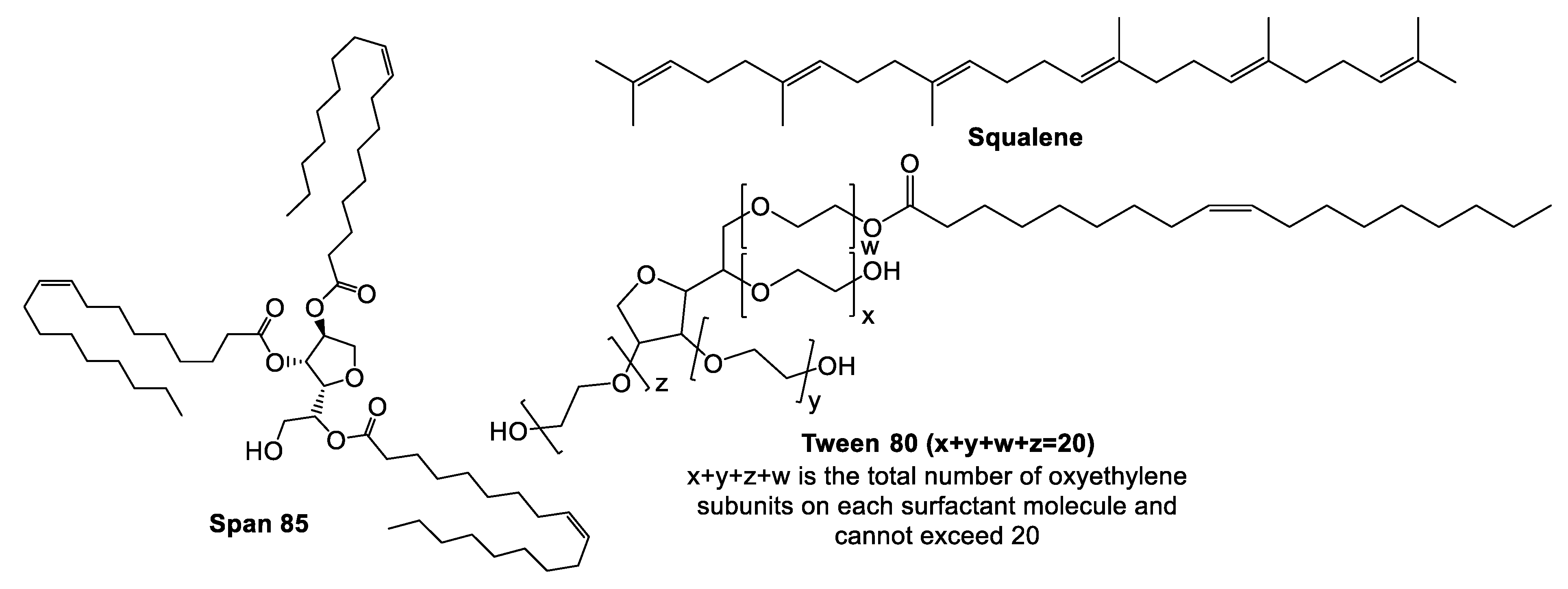
5.2. Montanides
Pre-Clinical and Clinical Development of Montanide Adjuvants
5.3. MF59®
Pre-Clinical and Clinical Development of MF59®
5.4. GLA-SE Adjuvant
Pre-Clinical and Clinical Development of GLA-SE
5.5. TiterMAX Adjuvants
Pre-Clinical Development of TiterMAX Adjuvants
5.6. RIBI Adjuvant
Pre-Clinical Development of RIBI Adjuvant System
6. Immunostimulatory Complexes
6.1. Cytokines
Pre-Clinical and Clinical Development of Cytokines
6.2. Chemokines
Pre-Clinical Development of Chemokines
7. Particulate Adjuvants
7.1. Imidazoquinolines
Pre-Clinical Development of Imidazoquinolines
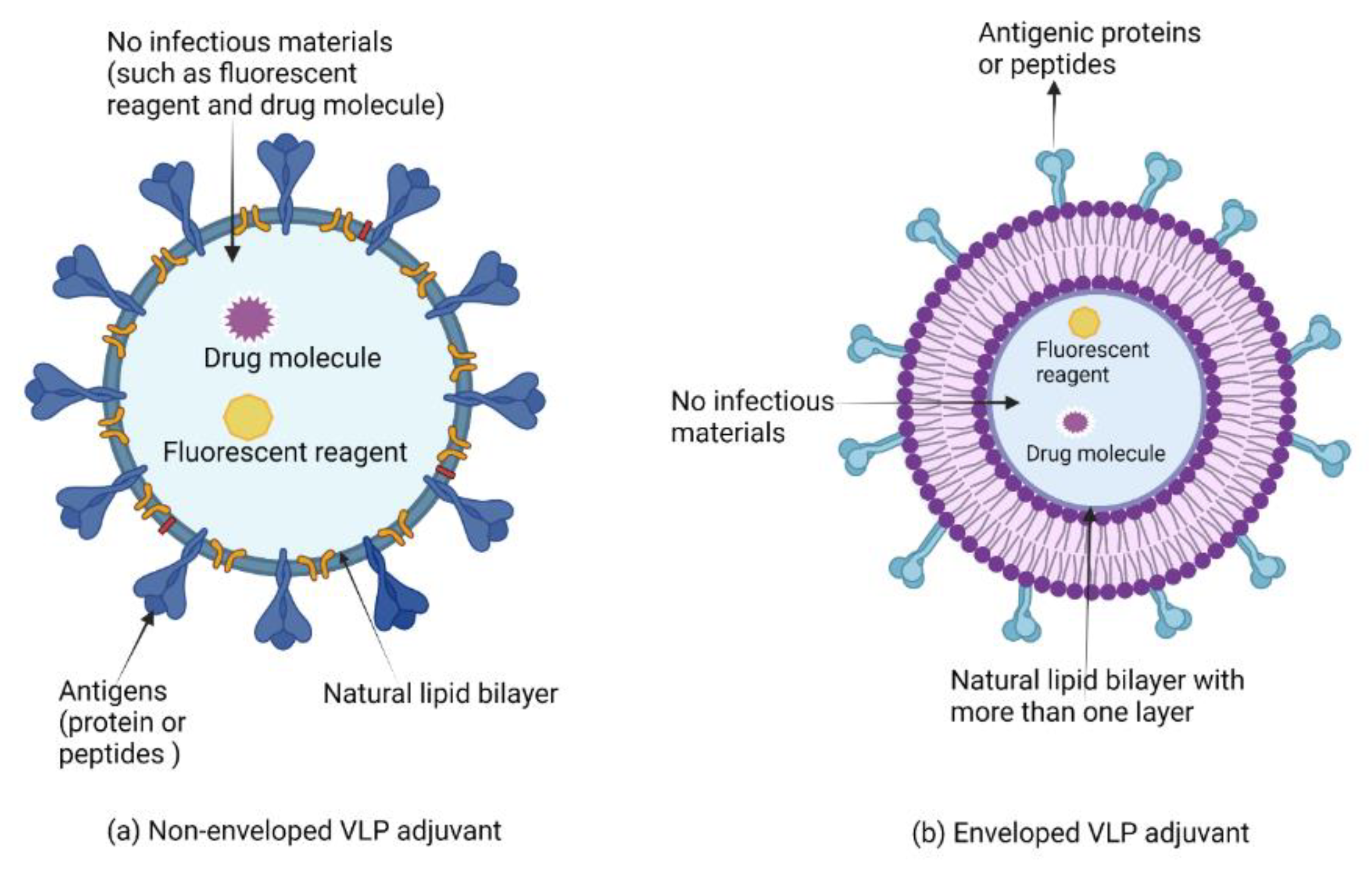
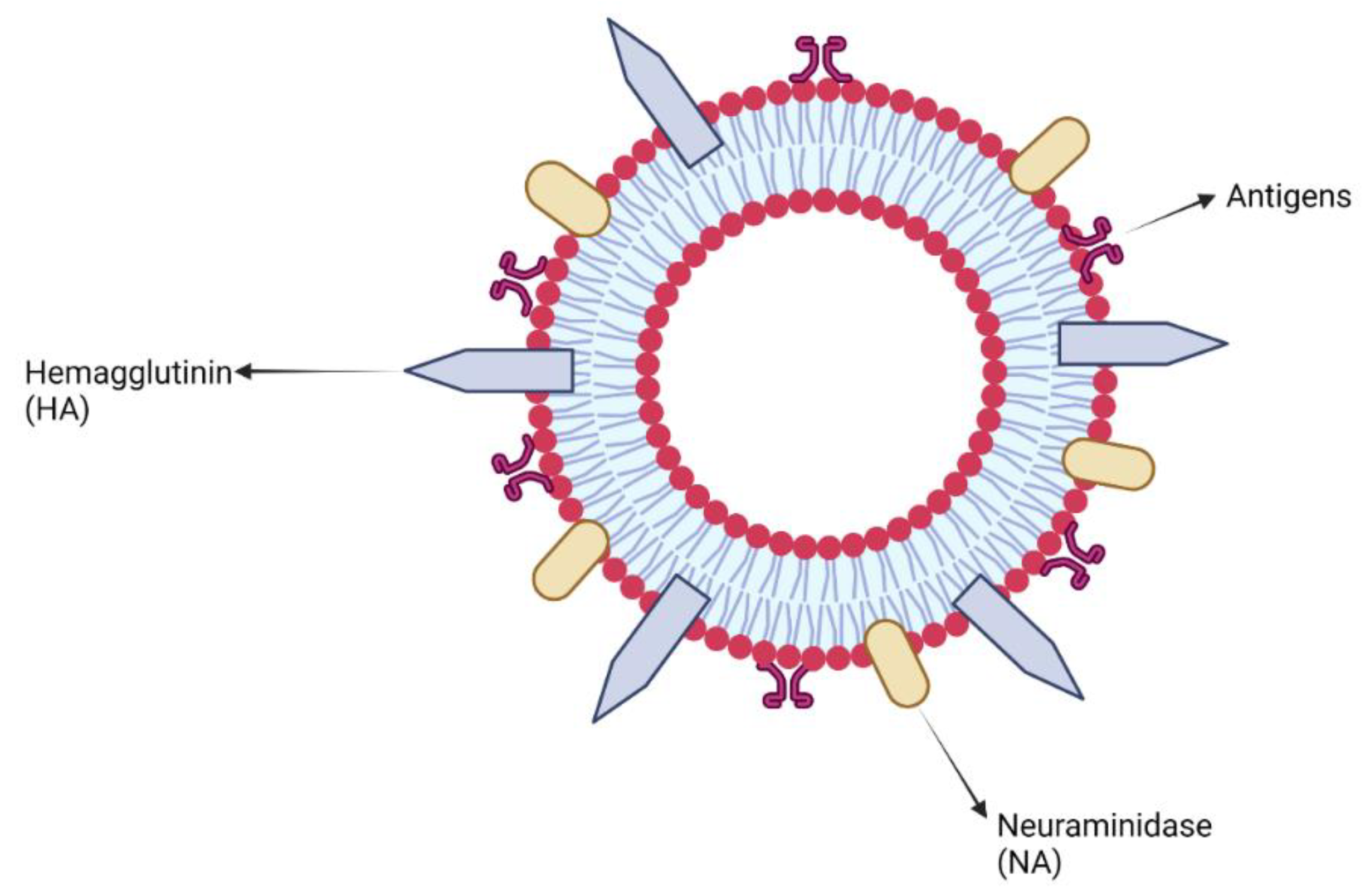
7.2. Virus-like Particles and Virosomes Adjuvants
Clinical Development of Virus-like Particles and Virosomes Adjuvants
7.3. Synthetic Polynucleotides Adjuvants
Pre-Clinical and Clinical Development of Polynucleotide Adjuvants
7.4. Liposomes as Adjuvants (Mucoadhesive Antigen Delivery Systems)
Pre-Clinical and Clinical Development of Liposome Adjuvants (and/or Delivery Systems)
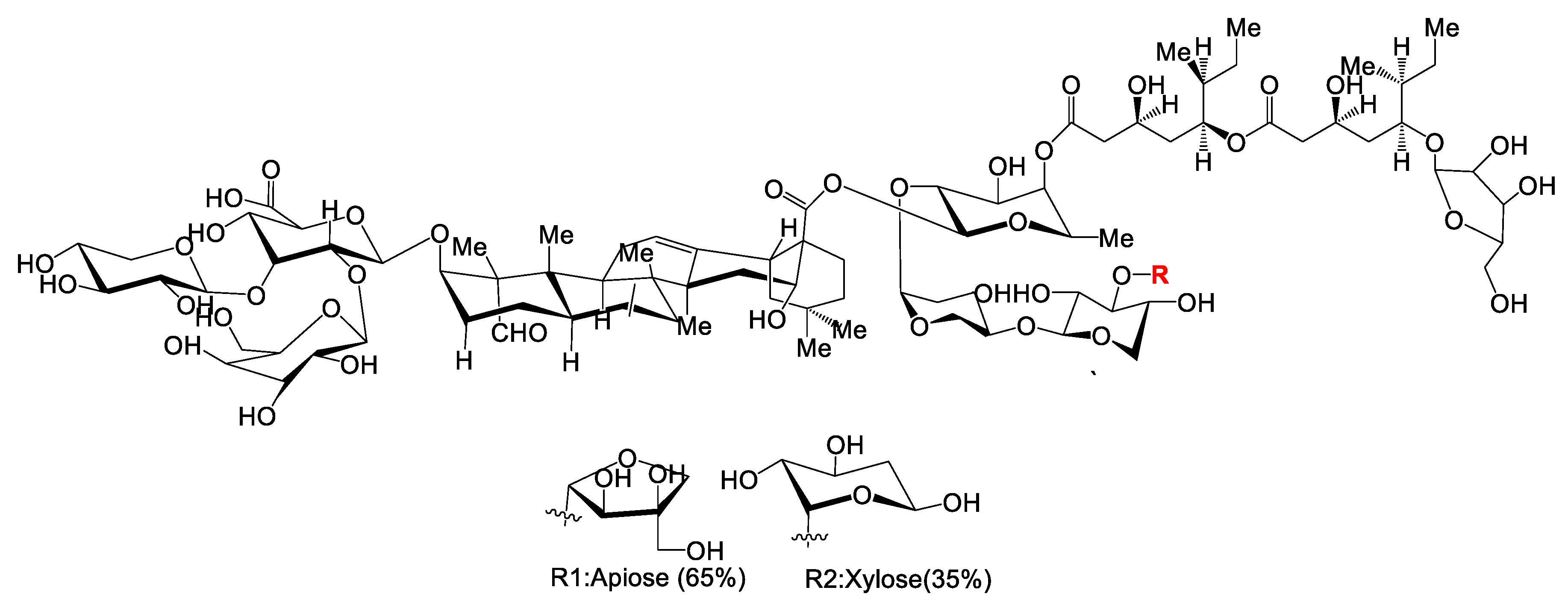
7.5. Polysaccharides as Adjuvants
Pre-Clinical and Clinical Development of Polysaccharide Adjuvants
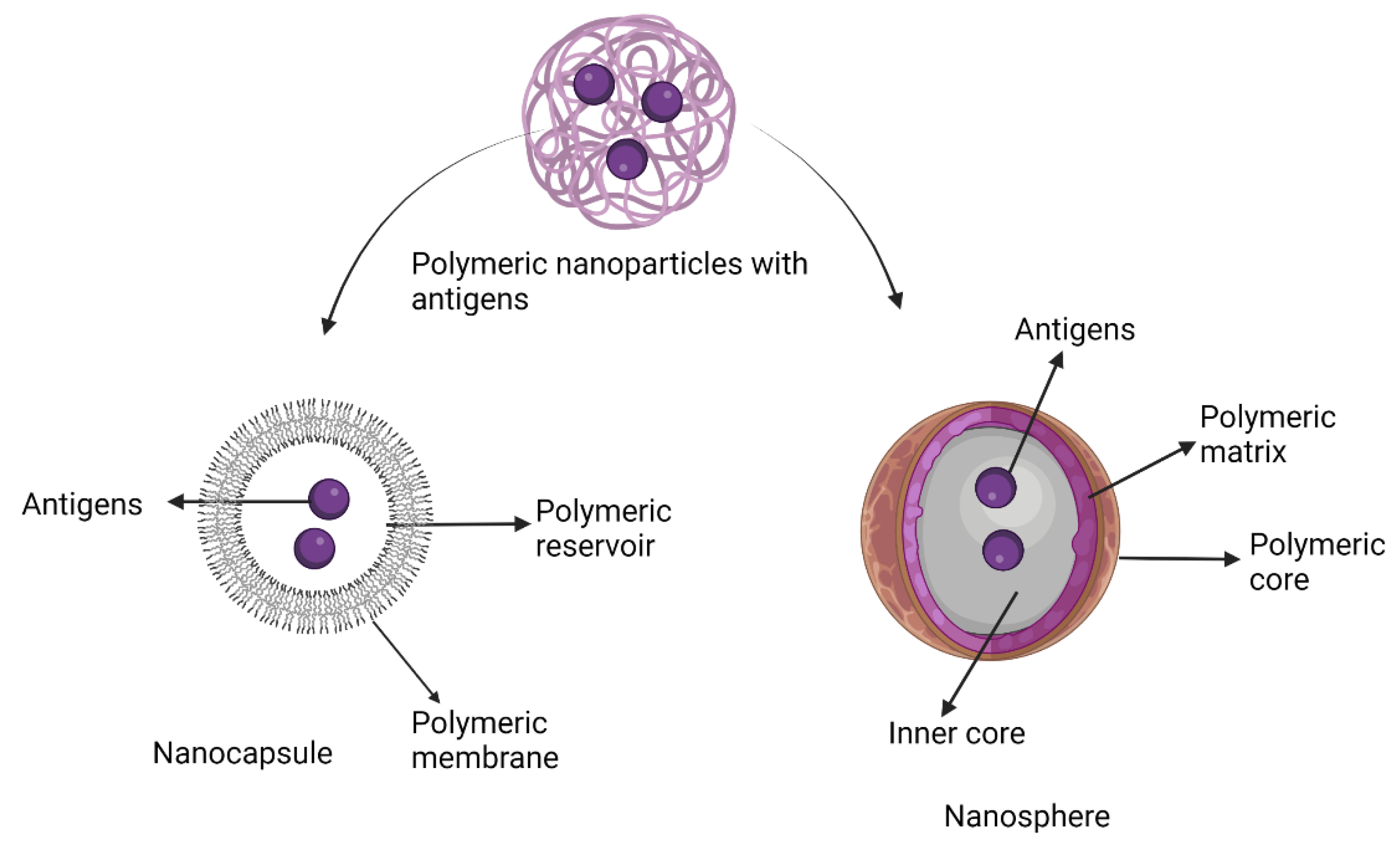
7.6. Polymeric Nanoparticle Adjuvants
Pre-Clinical and Clinical Development of Polymeric Nanoparticle Adjuvants
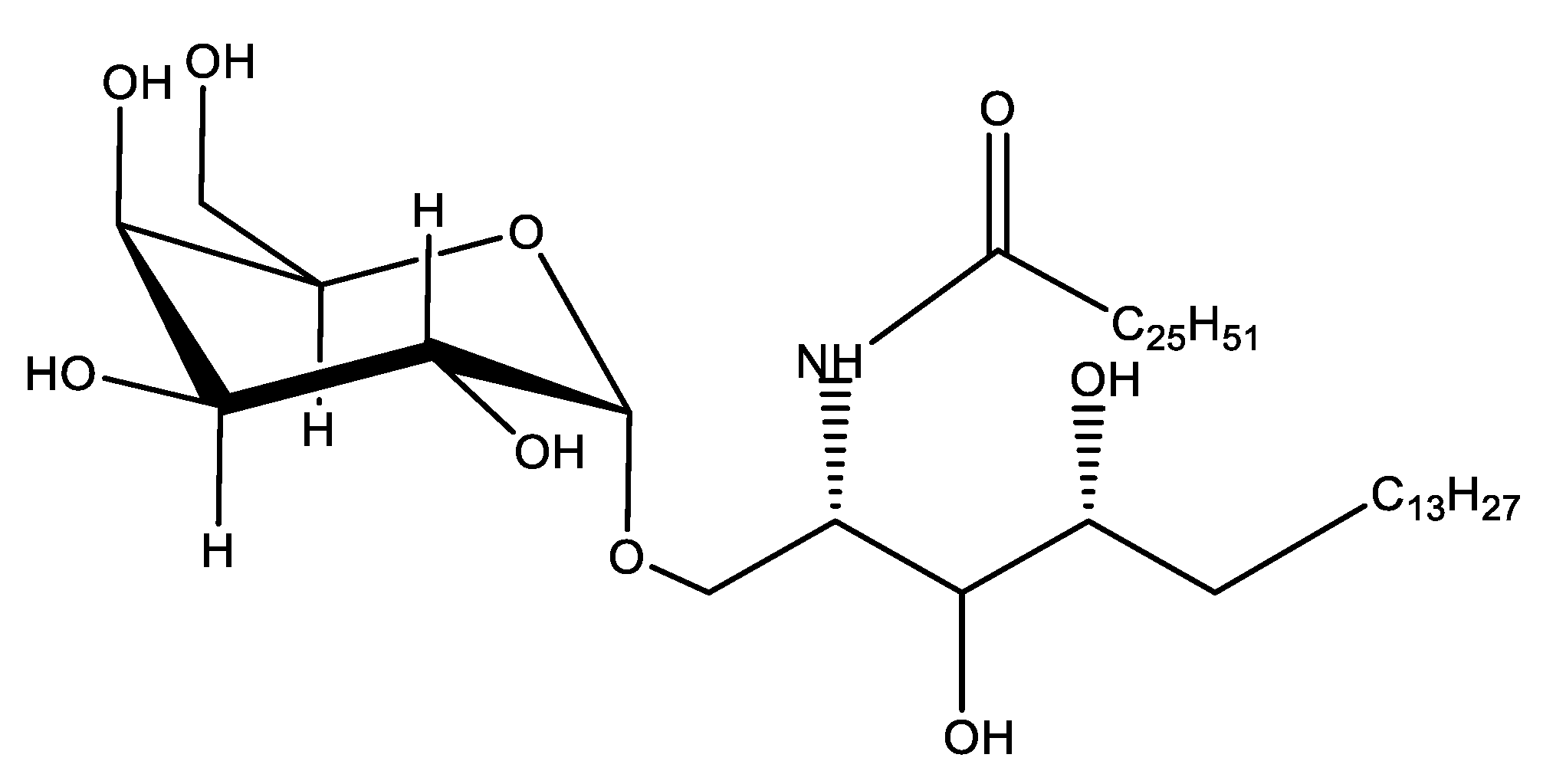
7.7. Glycosphingolipids as Adjuvants
Pre-Clinical and Clinical Development of Glycosphingolipids Adjuvants
8. Tensoactive Adjuvants
Pre-Clinical and Clinical Development of Saponin and Its Derivatives
9. Protease Adjuvants
Pre-Clinical Development of Protease Adjuvants
10. Combination Adjuvant Systems
Pre-Clinical and Clinical Development of Combination Adjuvant Systems
11. Perspective and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.F.; et al. Infectious disease in an era of global change. Nat. Rev. Microbiol. 2022, 20, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Talisuna, A.O.; Bloland, P.; D’Alessandro, U. History, dynamics, and public health importance of malaria parasite resistance. Clin. Microbiol. Rev. 2004, 17, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Andreano, E.; D’Oro, U.; Rappuoli, R.; Finco, O. Vaccine Evolution and Its Application to Fight Modern Threats. Front. Immunol. 2019, 10, 1722. [Google Scholar] [CrossRef] [PubMed]
- Dworetzky, M.; Cohen, S.; Mullin, D. Prometheus in Gloucestershire: Edward Jenner, 1749-1823. J. Allergy Clin. Immunol. 2003, 112, 810–814. [Google Scholar] [CrossRef]
- Azuar, A.; Jin, W.; Mukaida, S.; Hussein, W.M.; Toth, I.; Skwarczynski, M. Recent advances in the development of peptide vaccines and their delivery systems against Group A Streptococcus. Vaccines 2019, 7, 58. [Google Scholar] [CrossRef]
- Greenwood, B. The contribution of vaccination to global health: Past, present and future. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130433. [Google Scholar] [CrossRef]
- Skwarczynskim, M.; Zaman, M.; Toth, I. Lipo-peptides/saccharides in peptide vaccine delivery. In Handbook of Biologically Active Peptides; Kastin, A.J., Ed.; Academic Press/Elsevier: Amsterdam, The Netherlands, 2013; pp. 571–579. [Google Scholar]
- Plotkin, S.A. Vaccines: Past, present and future. Nat. Med. 2005, 11, S5–S11. [Google Scholar] [CrossRef]
- Kaufmann, S.H. The contribution of immunology to the rational design of novel antibacterial vaccines. Nat. Rev. Microbiol. 2007, 5, 491–504. [Google Scholar] [CrossRef]
- Moyle, P.M.; Toth, I. Modern subunit vaccines: Development, components, and research opportunities. ChemMedChem 2013, 8, 360–376. [Google Scholar] [CrossRef]
- Karch, C.P.; Burkhard, P. Vaccine technologies: From whole organisms to rationally designed protein assemblies. Biochem. Pharmacol. 2016, 120, 1–14. [Google Scholar] [CrossRef]
- Stephenson, R.; You, H.; McManus, D.P.; Toth, I. Schistosome Vaccine Adjuvants in Preclinical and Clinical Research. Vaccines 2014, 2, 654–685. [Google Scholar] [CrossRef] [PubMed]
- Apostolico Jde, S.; Lunardelli, V.A.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, Modus Operandi, and Licensing. J. Immunol. Res. 2016, 2016, 1459394. [Google Scholar] [CrossRef]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef]
- Lim, Y.T. Vaccine adjuvant materials for cancer immunotherapy and control of infectious disease. Clin. Exp. Vaccine Res. 2015, 4, 54–58. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.; Paust, S. Dynamic Natural Killer Cell and T Cell Responses to Influenza Infection. Front. Cell. Infect. Microbiol. 2020, 10, 425. [Google Scholar] [CrossRef]
- Vazquez, M.I.; Catalan-Dibene, J.; Zlotnik, A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine 2015, 74, 318–326. [Google Scholar] [CrossRef]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Ben Mkaddem, S.; Benhamou, M.; Monteiro, R.C. Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front. Immunol. 2019, 10, 811. [Google Scholar] [CrossRef]
- Koenderman, L. Inside-Out Control of Fc-Receptors. Front. Immunol. 2019, 10, 544. [Google Scholar] [CrossRef]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef]
- Gnjatic, S.; Sawhney, N.B.; Bhardwaj, N. Toll-like receptor agonists: Are they good adjuvants? Cancer J. 2010, 16, 382–391, reprinted in Cancer J. 2011, 16, 382–391. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.K.; Mansell, A. Toll-like receptors: The swiss army knife of immunity and vaccine development. Clin. Transl. Immunology 2016, 5, e85. [Google Scholar] [CrossRef] [PubMed]
- Matijevic, T.; Pavelic, J. Toll-like receptors: Cost or benefit for cancer? Curr. Pharm Des. 2010, 16, 1081–1090. [Google Scholar] [CrossRef]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A. Targeting Toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef]
- Jeong, S.K.; Heo, Y.K.; Jeong, J.H.; Ham, S.J.; Yum, J.S.; Ahn, B.C.; Song, C.S.; Chun, E.Y. COVID-19 Subunit Vaccine with a Combination of TLR1/2 and TLR3 Agonists Induces Robust and Protective Immunity. Vaccines 2021, 9, 957. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Abdelnoor, A.M. The potential use of Toll-like receptor (TLR) agonists and antagonists as prophylactic and/or therapeutic agents. Immunopharmacol. Immunotoxicol. 2009, 31, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.C.; Coulter, A.R. Adjuvants—A classification and review of their modes of action. Vaccine 1997, 15, 248–256. [Google Scholar] [CrossRef]
- HogenEsch, H.; O’Hagan, D.T.; Fox, C.B. Optimizing the utilization of aluminum adjuvants in vaccines: You might just get what you want. NPJ. Vaccines 2018, 3, 51. [Google Scholar] [CrossRef]
- Powell, B.S.; Andrianov, A.K.; Fusco, P.C. Polyionic vaccine adjuvants: Another look at aluminum salts and polyelectrolytes. Clin. Exp. Vaccine Res. 2015, 4, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Xia, T. Nanomaterial-Based Vaccine Adjuvants. J. Mater. Chem. B 2016, 4, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhu, H.; Xia, X.; Liang, Z.; Ma, X.; Sun, B. Vaccine adjuvants: Understanding the structure and mechanism of adjuvanticity. Vaccine 2019, 37, 3167–3178. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B.; Kramer, R.M.; Barnes, V.L.; Dowling, Q.M.; Vedvick, T.S. Working together: Interactions between vaccine antigens and adjuvants. Ther. Adv. Vaccines 2013, 1, 7–20. [Google Scholar] [CrossRef]
- Awate, S.; Babiuk, L.A.; Mutwiri, G. Mechanisms of action of adjuvants. Front. Immunol. 2013, 4, 114. [Google Scholar] [CrossRef]
- Hogenesch, H. Mechanism of immunopotentiation and safety of aluminum adjuvants. Front. Immunol. 2013, 3. [Google Scholar] [CrossRef]
- Gao, Q.; Bao, L.; Mao, H.; Wang, L.; Xu, K.; Yang, M.; Li, Y.; Zhu, L.; Wang, N.; Lv, Z.; et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science 2020, 369, 77–81. [Google Scholar] [CrossRef]
- Langley, J.M.; Aggarwal, N.; Toma, A.; Halperin, S.A.; McNeil, S.A.; Fissette, L.; Dewe, W.; Leyssen, M.; Toussaint, J.F.; Dieussaert, I. A Randomized, Controlled, Observer-Blinded Phase 1 Study of the Safety and Immunogenicity of a Respiratory Syncytial Virus Vaccine with or Without Alum Adjuvant. J. Infect. Dis. 2017, 215, 24–33. [Google Scholar] [CrossRef]
- Shang, Z.; Tan, S.; Ma, D. Respiratory syncytial virus: From pathogenesis to potential therapeutic strategies. Int. J. Biol. Sci. 2021, 17, 4073–4091. [Google Scholar] [CrossRef]
- Muse, D.; Christensen, S.; Bhuyan, P.; Absalon, J.; Eiden, J.J.; Jones, T.R.; York, L.J.; Jansen, K.U.; O’Neill, R.E.; Harris, S.L.; et al. A Phase 2, Randomized, Active-controlled, Observer-blinded Study to Assess the Immunogenicity, Tolerability and Safety of Bivalent rLP2086, a Meningococcal Serogroup B Vaccine, Coadministered With Tetanus, Diphtheria and Acellular Pertussis Vaccine and Serogroup A, C, Y and W-135 Meningococcal Conjugate Vaccine in Healthy US Adolescents. Pediatr. Infect. Dis. J. 2016, 35, 673–682. [Google Scholar]
- Glenn, G.M.; Fries, L.F.; Thomas, D.N.; Smith, G.; Kpamegan, E.; Lu, H.; Flyer, D.; Jani, D.; Hickman, S.P.; Piedra, P.A. A Randomized, Blinded, Controlled, Dose-Ranging Study of a Respiratory Syncytial Virus Recombinant Fusion (F) Nanoparticle Vaccine in Healthy Women of Childbearing Age. J. Infect. Dis. 2016, 213, 411–422. [Google Scholar] [CrossRef]
- Pitisuttithum, P.; Gilbert, P.; Gurwith, M.; Heyward, W.; Martin, M.; van Griensven, F.; Hu, D.; Tappero, J.W.; Choopanya, K.; Bangkok Vaccine Evaluation, G. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J. Infect. Dis. 2006, 194, 1661–1671. [Google Scholar] [CrossRef]
- Richmond, P.; Hatchuel, L.; Dong, M.; Ma, B.; Hu, B.; Smolenov, I.; Li, P.; Liang, P.; Han, H.H.; Liang, J.; et al. Safety and immunogenicity of S-Trimer (SCB-2019), a protein subunit vaccine candidate for COVID-19 in healthy adults: A phase 1, randomised, double-blind, placebo-controlled trial. Lancet 2021, 397, 682–694. [Google Scholar] [CrossRef]
- He, Q.; Mitchell, A.R.; Johnson, S.L.; Wagner-Bartak, C.; Morcol, T.; Bell, S.J. Calcium phosphate nanoparticle adjuvant. Clin. Diagn. Lab. Immunol. 2000, 7, 899–903. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, X.; Huang, X.; Zhang, J.; Xia, N.; Zhao, Q. Calcium phosphate nanoparticles as a new generation vaccine adjuvant. Expert Rev. Vaccines 2017, 16, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.I.; Omar, A.R.; Hussein, M.Z.; Elkhidir, I.M.; Sekawi, Z. Systemic antibody response to nano-size calcium phospate biocompatible adjuvant adsorbed HEV-71 killed vaccine. Clin. Exp. Vaccine Res. 2015, 4, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Koppad, S.; Raj, G.D.; Gopinath, V.P.; Kirubaharan, J.J.; Thangavelu, A.; Thiagarajan, V. Calcium phosphate coupled Newcastle disease vaccine elicits humoral and cell mediated immune responses in chickens. Res. Vet. Sci. 2011, 91, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Joyappa, D.H.; Kumar, C.A.; Banumathi, N.; Reddy, G.R.; Suryanarayana, V.V. Calcium phosphate nanoparticle prepared with foot and mouth disease virus P1-3CD gene construct protects mice and guinea pigs against the challenge virus. Vet. Microbiol. 2009, 139, 58–66. [Google Scholar] [CrossRef]
- Turley, C.B.; Rupp, R.E.; Johnson, C.; Taylor, D.N.; Wolfson, J.; Tussey, L.; Kavita, U.; Stanberry, L.; Shaw, A. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults. Vaccine 2011, 29, 5145–5152. [Google Scholar] [CrossRef]
- Treanor, J.J.; Taylor, D.N.; Tussey, L.; Hay, C.; Nolan, C.; Fitzgerald, T.; Liu, G.; Kavita, U.; Song, L.; Dark, I.; et al. Safety and immunogenicity of a recombinant hemagglutinin influenza-flagellin fusion vaccine (VAX125) in healthy young adults. Vaccine 2010, 28, 8268–8274. [Google Scholar] [CrossRef]
- Taylor, D.N.; Treanor, J.J.; Strout, C.; Johnson, C.; Fitzgerald, T.; Kavita, U.; Ozer, K.; Tussey, L.; Shaw, A. Induction of a potent immune response in the elderly using the TLR-5 agonist, flagellin, with a recombinant hemagglutinin influenza-flagellin fusion vaccine (VAX125, STF2.HA1 SI). Vaccine 2011, 29, 4897–4902. [Google Scholar] [CrossRef] [PubMed]
- Tussey, L.; Strout, C.; Davis, M.; Johnson, C.; Lucksinger, G.; Umlauf, S.; Song, L.; Liu, G.; Abraham, K.; White, C.J. Phase 1 Safety and Immunogenicity Study of a Quadrivalent Seasonal Flu Vaccine Comprising Recombinant Hemagglutinin-Flagellin Fusion Proteins. Open Forum Infect. Dis. 2016, 3, ofw015. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Liu, X.; Fang, Y.; Zhou, P.; Zhang, Y.; Wang, Y. Flagellin as a vaccine adjuvant. Expert Rev. Vaccines 2018, 17, 335–349. [Google Scholar] [CrossRef]
- Carter, D.; van Hoeven, N.; Baldwin, S.; Levin, Y.; Kochba, E.; Magill, A.; Charland, N.; Landry, N.; Nu, K.; Frevol, A.; et al. The adjuvant GLA-AF enhances human intradermal vaccine responses. Sci. Adv. 2018, 4, eaas9930. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yoo, J.K.; Sohn, H.J.; Kang, H.K.; Kim, D.; Shin, H.J.; Kim, J.H. Protective immunity against Naegleria fowleri infection on mice immunized with the rNfa1 protein using mucosal adjuvants. Parasitol. Res. 2015, 114, 1377–1385. [Google Scholar] [CrossRef]
- Miyata, T.; Harakuni, T.; Tsuboi, T.; Sattabongkot, J.; Kohama, H.; Tachibana, M.; Matsuzaki, G.; Torii, M.; Arakawa, T. Plasmodium vivax ookinete surface protein Pvs25 linked to cholera toxin B subunit induces potent transmission-blocking immunity by intranasal as well as subcutaneous immunization. Infect. Immun. 2010, 78, 3773–3782. [Google Scholar] [CrossRef]
- Maeto, C.; Rodriguez, A.M.; Holgado, M.P.; Falivene, J.; Gherardi, M.M. Novel mucosal DNA-MVA HIV vaccination in which DNA-IL-12 plus cholera toxin B subunit (CTB) cooperates to enhance cellular systemic and mucosal genital tract immunity. PLoS ONE 2014, 9, e107524. [Google Scholar] [CrossRef]
- Darrah, P.A.; Zeppa, J.J.; Maiello, P.; Hackney, J.A.; Wadsworth, M.H., 2nd; Hughes, T.K.; Pokkali, S.; Swanson, P.A., 2nd; Grant, N.L.; Rodgers, M.A.; et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 2020, 577, 95–102. [Google Scholar] [CrossRef]
- Nemes, E.; Geldenhuys, H.; Rozot, V.; Rutkowski, K.T.; Ratangee, F.; Bilek, N.; Mabwe, S.; Makhethe, L.; Erasmus, M.; Toefy, A.; et al. Prevention of M. tuberculosis Infection with H4:IC31 Vaccine or BCG Revaccination. N. Engl. J. Med. 2018, 379, 138–149. [Google Scholar] [CrossRef]
- Bekker, L.G.; Dintwe, O.; Fiore-Gartland, A.; Middelkoop, K.; Hutter, J.; Williams, A.; Randhawa, A.K.; Ruhwald, M.; Kromann, I.; Andersen, P.L.; et al. A phase 1b randomized study of the safety and immunological responses to vaccination with H4:IC31, H56:IC31, and BCG revaccination in Mycobacterium tuberculosis-uninfected adolescents in Cape Town, South Africa. EClinicalMedicine 2020, 21, 100313. [Google Scholar] [CrossRef]
- Lobo, N.; Brooks, N.A.; Zlotta, A.R.; Cirillo, J.D.; Boorjian, S.; Black, P.C.; Meeks, J.J.; Bivalacqua, T.J.; Gontero, P.; Steinberg, G.D.; et al. 100 years of Bacillus Calmette-Guerin immunotherapy: From cattle to COVID-19. Nat. Rev. Urol. 2021, 18, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Escobar, L.E.; Molina-Cruz, A.; Barillas-Mury, C. BCG vaccine protection from severe coronavirus disease 2019 (COVID-19). Proc. Natl. Acad. Sci. USA 2020, 117, 17720–17726. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.M.; Groshen, S.; Allen, J.; Munson, K.M.; Carlo, D.J.; Daigle, A.E.; Ferre, F.; Jensen, F.C.; Richieri, S.P.; Trauger, R.J.; et al. Initial studies on active immunization of HIV-infected subjects using a gp120-depleted HIV-1 Immunogen: Long-term follow-up. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 11, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.E.; Oliveira, F.; Coutinho-Abreu, I.V.; Herbert, S.; Meneses, C.; Kamhawi, S.; Baus, H.A.; Han, A.; Czajkowski, L.; Rosas, L.A.; et al. Safety and immunogenicity of a mosquito saliva peptide-based vaccine: A randomised, placebo-controlled, double-blind, phase 1 trial. Lancet 2020, 395, 1998–2007. [Google Scholar] [CrossRef]
- Lindert, K.; Leav, B.; Heijnen, E.; Barrett, J.; Nicolay, U. Cumulative clinical experience with MF59-adjuvanted trivalent seasonal influenza vaccine in young children and adults 65 years of age and older. Int. J. Infect. Dis. 2019, 85, S10–S17. [Google Scholar] [CrossRef]
- Chappell, K.J.; Mordant, F.L.; Li, Z.; Wijesundara, D.K.; Ellenberg, P.; Lackenby, J.A.; Cheung, S.T.M.; Modhiran, N.; Avumegah, M.S.; Henderson, C.L.; et al. Safety and immunogenicity of an MF59-adjuvanted spike glycoprotein-clamp vaccine for SARS-CoV-2: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Infect. Dis. 2021, 21, 1383–1394. [Google Scholar] [CrossRef]
- Tendler, M.; Almeida, M.; Simpson, A. Development of the Brazilian Anti Schistosomiasis Vaccine Based on the Recombinant Fatty Acid Binding Protein Sm14 Plus GLA-SE Adjuvant. Front. Immunol. 2015, 6, 218. [Google Scholar] [CrossRef]
- Coler, R.N.; Day, T.A.; Ellis, R.; Piazza, F.M.; Beckmann, A.M.; Vergara, J.; Rolf, T.; Lu, L.; Alter, G.; Hokey, D.; et al. The TLR-4 agonist adjuvant, GLA-SE, improves magnitude and quality of immune responses elicited by the ID93 tuberculosis vaccine: First-in-human trial. NPJ. Vaccines 2018, 3, 34. [Google Scholar] [CrossRef]
- Cargnelutti, D.E.; Sanchez, M.A.; Alvarez, P.; Boado, L.; Mattion, N.; Scodeller, E.A. Enhancement of Th1 immune responses to recombinant influenza nucleoprotein by Ribi adjuvant. New Microbiol. 2013, 36, 145–151. [Google Scholar]
- Mullerad, J.; Michal, I.; Fishman, Y.; Hovav, A.H.; Barletta, R.G.; Bercovier, H. The immunogenicity of Mycobacterium paratuberculosis 85B antigen. Med. Microbiol. Immunol. 2002, 190, 179–187. [Google Scholar] [CrossRef]
- Ma, H.; Lim, T.H.; Leerapun, A.; Weltman, M.; Jia, J.; Lim, Y.S.; Tangkijvanich, P.; Sukeepaisarnjaroen, W.; Ji, Y.; Le Bert, N.; et al. Therapeutic vaccine BRII-179 restores HBV-specific immune responses in patients with chronic HBV in a phase Ib/IIa study. JHEP Rep. 2021, 3, 100361. [Google Scholar] [CrossRef] [PubMed]
- Brekke, K.; Sommerfelt, M.; Okvist, M.; Dyrhol-Riise, A.M.; Kvale, D. The therapeutic HIV Env C5/gp41 vaccine candidate Vacc-C5 induces specific T cell regulation in a phase I/II clinical study. BMC Infect. Dis. 2017, 17, 228. [Google Scholar] [CrossRef] [PubMed]
- Petrina, M.; Martin, J.; Basta, S. Granulocyte macrophage colony-stimulating factor has come of age: From a vaccine adjuvant to antiviral immunotherapy. Cytokine Growth Factor Rev. 2021, 59, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Launay, O.; Grabar, S.; Bloch, F.; Desaint, C.; Jegou, D.; Lallemand, C.; Erickson, R.; Lebon, P.; Tovey, M.G. Effect of sublingual administration of interferon-alpha on the immune response to influenza vaccination in institutionalized elderly individuals. Vaccine 2008, 26, 4073–4079. [Google Scholar] [CrossRef]
- Mohan, T.; Zhu, W.; Wang, Y.; Wang, B.Z. Applications of chemokines as adjuvants for vaccine immunotherapy. Immunobiology 2018, 223, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, B.C.; Kim, J.R.; Blevins, L.K.; Jorgensen, M.J.; Kock, N.D.; D’Agostino, R.B., Jr.; Aycock, S.T.; Hadimani, M.B.; King, S.B.; Parks, G.D.; et al. A Novel R848-Conjugated Inactivated Influenza Virus Vaccine Is Efficacious and Safe in a Neonate Nonhuman Primate Model. J. Immunol. 2016, 197, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, L.; Van Hoecke, L.; Deswarte, K.; Schepens, B.; Li, Y.; Lambrecht, B.N.; De Koker, S.; David, S.A.; Saelens, X.; De Geest, B.G. Potent anti-viral vaccine adjuvant based on pH-degradable nanogels with covalently linked small molecule imidazoquinoline TLR7/8 agonist. Biomaterials 2018, 178, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Meijer, C.J.L.M.; Kieninger, D.; Schuyleman, A.; Thomas, S.; Luxembourg, A.; Baudin, M. A phase III clinical study to compare the immunogenicity and safety of the 9-valent and quadrivalent HPV vaccines in men. Vaccine 2016, 34, 4205–4212. [Google Scholar] [CrossRef]
- Chen, G.L.; Coates, E.E.; Plummer, S.H.; Carter, C.A.; Berkowitz, N.; Conan-Cibotti, M.; Cox, J.H.; Beck, A.; O’Callahan, M.; Andrews, C.; et al. Effect of a Chikungunya Virus-Like Particle Vaccine on Safety and Tolerability Outcomes: A Randomized Clinical Trial. JAMA 2020, 323, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Shinde, V.; Fries, L.; Wu, Y.; Agrawal, S.; Cho, I.; Thomas, D.N.; Spindler, M.; Lindner, E.; Hahn, T.; Plested, J.; et al. Improved Titers against Influenza Drift Variants with a Nanoparticle Vaccine. N. Engl. J. Med. 2018, 378, 2346–2348. [Google Scholar] [CrossRef]
- Asadi, K.; Gholami, A. Virosome-based nanovaccines; a promising bioinspiration and biomimetic approach for preventing viral diseases: A review. Int. J. Biol. Macromol. 2021, 182, 648–658. [Google Scholar] [CrossRef]
- Thompson, F.M.; Porter, D.W.; Okitsu, S.L.; Westerfeld, N.; Vogel, D.; Todryk, S.; Poulton, I.; Correa, S.; Hutchings, C.; Berthoud, T.; et al. Evidence of blood stage efficacy with a virosomal malaria vaccine in a phase IIa clinical trial. PLoS ONE 2008, 3, e1493. [Google Scholar] [CrossRef] [PubMed]
- Overton, E.T.; Goepfert, P.A.; Cunningham, P.; Carter, W.A.; Horvath, J.; Young, D.; Strayer, D.R. Intranasal seasonal influenza vaccine and a TLR-3 agonist, rintatolimod, induced cross-reactive IgA antibody formation against avian H5N1 and H7N9 influenza HA in humans. Vaccine 2014, 32, 5490–5495. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Sabado, R.L.; La Mar, M.; Mohri, H.; Salazar, A.M.; Dong, H.; Correa Da Rosa, J.; Markowitz, M.; Bhardwaj, N.; Miller, E. Poly-ICLC, a TLR3 Agonist, Induces Transient Innate Immune Responses in Patients with Treated HIV-Infection: A Randomized Double-Blinded Placebo Controlled Trial. Front. Immunol. 2019, 10, 725. [Google Scholar] [CrossRef]
- Scheiermann, J.; Klinman, D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef] [PubMed]
- Mullen, G.E.; Ellis, R.D.; Miura, K.; Malkin, E.; Nolan, C.; Hay, M.; Fay, M.P.; Saul, A.; Zhu, D.; Rausch, K.; et al. Phase 1 trial of AMA1-C1/Alhydrogel plus CPG 7909: An asexual blood-stage vaccine for Plasmodium falciparum malaria. PLoS ONE 2008, 3, e2940. [Google Scholar] [CrossRef]
- Norrby, M.; Vesikari, T.; Lindqvist, L.; Maeurer, M.; Ahmed, R.; Mahdavifar, S.; Bennett, S.; McClain, J.B.; Shepherd, B.M.; Li, D.; et al. Safety and immunogenicity of the novel H4:IC31 tuberculosis vaccine candidate in BCG-vaccinated adults: Two phase I dose escalation trials. Vaccine 2017, 35, 1652–1661. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef]
- Van Dissel, J.T.; Joosten, S.A.; Hoff, S.T.; Soonawala, D.; Prins, C.; Hokey, D.A.; O’Dee, D.M.; Graves, A.; Thierry-Carstensen, B.; Andreasen, L.V.; et al. A novel liposomal adjuvant system, CAF01, promotes long-lived Mycobacterium tuberculosis-specific T-cell responses in human. Vaccine 2014, 32, 7098–7107. [Google Scholar] [CrossRef]
- Abraham, S.; Juel, H.B.; Bang, P.; Cheeseman, H.M.; Dohn, R.B.; Cole, T.; Kristiansen, M.P.; Korsholm, K.S.; Lewis, D.; Olsen, A.W.; et al. Safety and immunogenicity of the chlamydia vaccine candidate CTH522 adjuvanted with CAF01 liposomes or aluminium hydroxide: A first-in-human, randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Infect. Dis. 2019, 19, 1091–1100. [Google Scholar] [CrossRef]
- Atmar, R.L.; Bernstein, D.I.; Harro, C.D.; Al-Ibrahim, M.S.; Chen, W.H.; Ferreira, J.; Estes, M.K.; Graham, D.Y.; Opekun, A.R.; Richardson, C.; et al. Norovirus vaccine against experimental human Norwalk Virus illness. N. Engl. J. Med. 2011, 365, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Kelley, P.; Heinzel, S.; Cooper, P.; Petrovsky, N. Immunogenicity and safety of Advax, a novel polysaccharide adjuvant based on delta inulin, when formulated with hepatitis B surface antigen: A randomized controlled Phase 1 study. Vaccine 2014, 32, 6469–6477. [Google Scholar] [CrossRef] [PubMed]
- Vasan, S.; Schlesinger, S.J.; Huang, Y.; Hurley, A.; Lombardo, A.; Chen, Z.; Than, S.; Adesanya, P.; Bunce, C.; Boaz, M.; et al. Phase 1 safety and immunogenicity evaluation of ADVAX, a multigenic, DNA-based clade C/B’ HIV-1 candidate vaccine. PLoS ONE 2010, 5, e8617. [Google Scholar] [CrossRef] [PubMed]
- Aline, F.; Brand, D.; Pierre, J.; Roingeard, P.; Severine, M.; Verrier, B.; Dimier-Poisson, I. Dendritic cells loaded with HIV-1 p24 proteins adsorbed on surfactant-free anionic PLA nanoparticles induce enhanced cellular immune responses against HIV-1 after vaccination. Vaccine 2009, 27, 5284–5291. [Google Scholar] [CrossRef] [PubMed]
- Woltman, A.M.; Ter Borg, M.J.; Binda, R.S.; Sprengers, D.; von Blomberg, B.M.; Scheper, R.J.; Hayashi, K.; Nishi, N.; Boonstra, A.; van der Molen, R.; et al. Alpha-galactosylceramide in chronic hepatitis B infection: Results from a randomized placebo-controlled Phase I/II trial. Antivir. Ther. 2009, 14, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Guebre-Xabier, M.; Patel, N.; Tian, J.H.; Zhou, B.; Maciejewski, S.; Lam, K.; Portnoff, A.D.; Massare, M.J.; Frieman, M.B.; Piedra, P.A.; et al. NVX-CoV2373 vaccine protects cynomolgus macaque upper and lower airways against SARS-CoV-2 challenge. Vaccine 2020, 38, 7892–7896. [Google Scholar] [CrossRef]
- Heath, P.T.; Galiza, E.P.; Baxter, D.N.; Boffito, M.; Browne, D.; Burns, F.; Chadwick, D.R.; Clark, R.; Cosgrove, C.; Galloway, J.; et al. Safety and Efficacy of NVX-CoV2373 COVID-19 Vaccine. N. Engl. J. Med. 2021, 385, 1172–1183. [Google Scholar] [CrossRef]
- Chung, K.Y.; Coyle, E.M.; Jani, D.; King, L.R.; Bhardwaj, R.; Fries, L.; Smith, G.; Glenn, G.; Golding, H.; Khurana, S. ISCOMATRIX adjuvant promotes epitope spreading and antibody affinity maturation of influenza A H7N9 virus like particle vaccine that correlate with virus neutralization in humans. Vaccine 2015, 33, 3953–3962. [Google Scholar] [CrossRef]
- El Ridi, R.; Tallima, H. Vaccine-induced protection against murine schistosomiasis mansoni with larval excretory-secretory antigens and papain or type-2 cytokines. J. Parasitol. 2013, 99, 194–202. [Google Scholar] [CrossRef]
- Rts, S.C.T.P.; Agnandji, S.T.; Lell, B.; Soulanoudjingar, S.S.; Fernandes, J.F.; Abossolo, B.P.; Conzelmann, C.; Methogo, B.G.; Doucka, Y.; Flamen, A.; et al. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N. Engl. J. Med. 2011, 365, 1863–1875. [Google Scholar] [CrossRef]
- Graves, S.F.; Kouriba, B.; Diarra, I.; Daou, M.; Niangaly, A.; Coulibaly, D.; Keita, Y.; Laurens, M.B.; Berry, A.A.; Vekemans, J.; et al. Strain-specific Plasmodium falciparum multifunctional CD4+ T cell cytokine expression in Malian children immunized with the FMP2.1/AS02A vaccine candidate. Vaccine 2016, 34, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Francica, J.R.; Flynn, B.J.; Foulds, K.E.; Noe, A.T.; Werner, A.P.; Moore, I.N.; Gagne, M.; Johnston, T.S.; Tucker, C.; Davis, R.L.; et al. Vaccination with SARS-CoV-2 Spike Protein and AS03 Adjuvant Induces Rapid Anamnestic Antibodies in the Lung and Protects Against Virus Challenge in Nonhuman Primates. bioRxiv 2021. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; Walls, A.C.; Golden, N.; Atyeo, C.; Fischinger, S.; Li, C.; Aye, P.; Navarro, M.J.; Lai, L.; Edara, V.V.; et al. Adjuvanting a subunit COVID-19 vaccine to induce protective immunity. Nature 2021, 594, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.I.; Muscatello, D.J.; Soh, A.C.Y.; Dwyer, D.E.; Turner, R.M. An indirect comparison meta-analysis of AS03 and MF59 adjuvants in pandemic influenza A(H1N1)pdm09 vaccines. Vaccine 2019, 37, 4246–4255. [Google Scholar] [CrossRef] [PubMed]
- Rumke, H.C.; Bayas, J.M.; de Juanes, J.R.; Caso, C.; Richardus, J.H.; Campins, M.; Rombo, L.; Duval, X.; Romanenko, V.; Schwarz, T.F.; et al. Safety and reactogenicity profile of an adjuvanted H5N1 pandemic candidate vaccine in adults within a phase III safety trial. Vaccine 2008, 26, 2378–2388. [Google Scholar] [CrossRef] [PubMed]
- Tota, J.E.; Struyf, F.; Hildesheim, A.; Gonzalez, P.; Ryser, M.; Herrero, R.; Schussler, J.; Karkada, N.; Rodriguez, A.C.; Folschweiller, N.; et al. Efficacy of AS04-Adjuvanted Vaccine Against Human Papillomavirus (HPV) Types 16 and 18 in Clearing Incident HPV Infections: Pooled Analysis of Data From the Costa Rica Vaccine Trial and the PATRICIA Study. J. Infect. Dis. 2021, 223, 1576–1581. [Google Scholar] [CrossRef]
- Gupta, R.K. Aluminum compounds as vaccine adjuvants. Adv. Drug Deliv. Rev. 1998, 32, 155–172. [Google Scholar] [CrossRef]
- Cao, P.; Han, F.Y.; Grondahl, L.; Xu, Z.P.; Li, L. Enhanced Oral Vaccine Efficacy of Polysaccharide-Coated Calcium Phosphate Nanoparticles. ACS Omega 2020, 5, 18185–18197. [Google Scholar] [CrossRef]
- Lee, S.; Nguyen, M.T. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 2015, 15, 51–57. [Google Scholar] [CrossRef]
- Khoomrung, S.; Nookaew, I.; Sen, P.; Olafsdottir, T.A.; Persson, J.; Moritz, T.; Andersen, P.; Harandi, A.M.; Nielsen, J. Metabolic Profiling and Compound-Class Identification Reveal Alterations in Serum Triglyceride Levels in Mice Immunized with Human Vaccine Adjuvant Alum. J. Proteome Res. 2020, 19, 269–278. [Google Scholar] [CrossRef]
- Oleszycka, E.; McCluskey, S.; Sharp, F.A.; Muñoz-Wolf, N.; Hams, E.; Gorman, A.L.; Fallon, P.G.; Lavelle, E.C. The vaccine adjuvant alum promotes IL-10 production that suppresses Th1 responses. Eur. J. Immunol. 2018, 48, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Petrovsky, N. Comparative safety of vaccine adjuvants: A summary of current evidence and future needs. Drug Saf. 2015, 38, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Torrents de la Pena, A.; Sanders, R.W. Stabilizing HIV-1 envelope glycoprotein trimers to induce neutralizing antibodies. Retrovirology 2018, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, N.; Hickey, J.M.; Kaur, K.; Xiong, J.; Sawant, N.; Cupo, A.; Lee, W.; Ozorowski, G.; Medina-Ramírez, M.; Ward, A.B.; et al. Developability assessment of physicochemical properties and stability profiles of HIV-1 BG505 SOSIP.664 and BG505 SOSIP.v4.1-GT1.1 gp140 envelope glycoprotein trimers as candidate vaccine antigens. J. Pharm. Sci. 2019, 108, 2264–2277. [Google Scholar] [CrossRef]
- Bontempo, A.; Garcia, M.M.; Rivera, N.; Cayabyab, M.J. A Systematic Approach to HIV-1 Vaccine Immunogen Selection. AIDS Res. Hum. Retrovir. 2020, 36, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Pena, A.T.; Korzun, J.; et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef]
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): A global pandemic and treatment strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef]
- Pulendran, B.; Arunachalam, P.S.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- Marrack, P.; McKee, A.S.; Munks, M.W. Towards an understanding of the adjuvant action of aluminium. Nat. Rev. Immunol. 2009, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tscharke, D.C. Herpes Simplex Virus Latency Is Noisier the Closer We Look. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Pusic, K.; Aguilar, Z.; McLoughlin, J.; Kobuch, S.; Xu, H.; Tsang, M.; Wang, A.; Hui, G. Iron oxide nanoparticles as a clinically acceptable delivery platform for a recombinant blood-stage human malaria vaccine. FASEB J. 2013, 27, 1153–1166. [Google Scholar] [CrossRef]
- Hajam, I.A.; Dar, P.A.; Shahnawaz, I.; Jaume, J.C.; Lee, J.H. Bacterial flagellin-a potent immunomodulatory agent. Exp. Mol. Med. 2017, 49, e373. [Google Scholar] [CrossRef] [PubMed]
- Auvray, F.; Thomas, J.; Fraser, G.M.; Hughes, C. Flagellin polymerisation control by a cytosolic export chaperone. J. Mol. Biol. 2001, 308, 221–229. [Google Scholar] [CrossRef]
- Aspinall, R.; Del Giudice, G.; Effros, R.B.; Grubeck-Loebenstein, B.; Sambhara, S. Challenges for vaccination in the elderly. Immun. Ageing 2007, 4, 9. [Google Scholar] [CrossRef]
- Mizel, S.B.; Graff, A.H.; Sriranganathan, N.; Ervin, S.; Lees, C.J.; Lively, M.O.; Hantgan, R.R.; Thomas, M.J.; Wood, J.; Bell, B. Flagellin-F1-V fusion protein is an effective plague vaccine in mice and two species of nonhuman primates. Clin. Vaccine Immunol. 2009, 16, 21–28. [Google Scholar] [CrossRef]
- Chilton, P.M.; Hadel, D.M.; To, T.T.; Mitchell, T.C.; Darveau, R.P. Adjuvant activity of naturally occurring monophosphoryl lipopolysaccharide preparations from mucosa-associated bacteria. Infect. Immun. 2013, 81, 3317–3325. [Google Scholar] [CrossRef]
- Zariri, A.; van der Ley, P. Biosynthetically engineered lipopolysaccharide as vaccine adjuvant. Expert Rev. Vaccines 2015, 14, 861–876. [Google Scholar] [CrossRef]
- Cai, S.; Zemans, R.L.; Young, S.K.; Worthen, G.S.; Jeyaseelan, S. Myeloid differentiation protein-2-dependent and -independent neutrophil accumulation during Escherichia coli pneumonia. Am. J. Respir. Cell. Mol. Biol. 2009, 40, 701–709. [Google Scholar] [CrossRef]
- Mata-Haro, V.; Cekic, C.; Martin, M.; Chilton, P.M.; Casella, C.R.; Mitchell, T.C. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 2007, 316, 1628–1632. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Bazin-Lee, H.; Evans, J.T.; Casella, C.R.; Mitchell, T.C. MPL Adjuvant Contains Competitive Antagonists of Human TLR4. Front. Immunol. 2020, 11, 577823. [Google Scholar] [CrossRef] [PubMed]
- Casella, C.R.; Mitchell, T.C. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. 2008, 65, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Clegg, C.H.; Rininger, J.A.; Baldwin, S.L. Clinical vaccine development for H5N1 influenza. Expert Rev. Vaccines 2013, 12, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Coler, R.N.; Bertholet, S.; Moutaftsi, M.; Guderian, J.A.; Windish, H.P.; Baldwin, S.L.; Laughlin, E.M.; Duthie, M.S.; Fox, C.B.; Carter, D.; et al. Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS ONE 2011, 6, e16333. [Google Scholar] [CrossRef]
- Du, G.; Hathout, R.M.; Nasr, M.; Nejadnik, M.R.; Tu, J.; Koning, R.I.; Koster, A.J.; Slutter, B.; Kros, A.; Jiskoot, W.; et al. Intradermal vaccination with hollow microneedles: A comparative study of various protein antigen and adjuvant encapsulated nanoparticles. J. Control. Release 2017, 266, 109–118. [Google Scholar] [CrossRef]
- Levin, Y.; Kochba, E.; Hung, I.; Kenney, R. Intradermal vaccination using the novel microneedle device MicronJet600: Past, present, and future. Hum. Vaccines Immunother. 2015, 11, 991–997. [Google Scholar] [CrossRef]
- Gongal, G.; Wright, A.E. Human Rabies in the WHO Southeast Asia Region: Forward Steps for Elimination. Adv. PRev. Med. 2011, 2011, 383870. [Google Scholar] [CrossRef]
- Coler, R.N.; Baldwin, S.L.; Shaverdian, N.; Bertholet, S.; Reed, S.J.; Raman, V.S.; Lu, X.; DeVos, J.; Hancock, K.; Katz, J.M.; et al. A synthetic adjuvant to enhance and expand immune responses to influenza vaccines. PLoS ONE 2010, 5, e13677. [Google Scholar] [CrossRef]
- Goji, N.A.; Nolan, C.; Hill, H.; Wolff, M.; Noah, D.L.; Williams, T.B.; Rowe, T.; Treanor, J.J. Immune responses of healthy subjects to a single dose of intramuscular inactivated influenza A/Vietnam/1203/2004 (H5N1) vaccine after priming with an antigenic variant. J. Infect. Dis. 2008, 198, 635–641. [Google Scholar] [CrossRef]
- Rivera-Chavez, F.; Mekalanos, J.J. Cholera toxin promotes pathogen acquisition of host-derived nutrients. Nature 2019, 572, 244–248. [Google Scholar] [CrossRef]
- Gagliardi, M.C.; De Magistris, M.T. Maturation of human dendritic cells induced by the adjuvant cholera toxin: Role of cAMP on chemokine receptor expression. Vaccine 2003, 21, 856–861. [Google Scholar] [CrossRef]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant—An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Snider, D.P.; Marshall, J.S.; Perdue, M.H.; Liang, H. Production of IgE antibody and allergic sensitization of intestinal and peripheral tissues after oral immunization with protein Ag and cholera toxin. J. Immunol. 1994, 153, 647–657. [Google Scholar]
- Tamura, S.; Miyata, K.; Matsuo, K.; Asanuma, H.; Takahashi, H.; Nakajima, K.; Suzuki, Y.; Aizawa, C.; Kurata, T. Acceleration of influenza virus clearance by Th1 cells in the nasal site of mice immunized intranasally with adjuvant-combined recombinant nucleoprotein. J. Immunol. 1996, 156, 3892–3900. [Google Scholar] [PubMed]
- Datta, S.K.; Sabet, M.; Nguyen, K.P.; Valdez, P.A.; Gonzalez-Navajas, J.M.; Islam, S.; Mihajlov, I.; Fierer, J.; Insel, P.A.; Webster, N.J.; et al. Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc. Natl. Acad. Sci. USA 2010, 107, 10638–10643. [Google Scholar] [CrossRef]
- Yoder, J.S.; Eddy, B.A.; Isvesvara, G.S.V.; Capewell, L.; Beach, A.M.J. The epidemiology of primary amoebic meningoencephalitis in the USA, 1962–2008. Epidemiol. Infect. 2009, 138, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, I.; Garg, R.; van Drunen Littel-van den Hurk, S. Selection of adjuvants for vaccines targeting specific pathogens. Expert Rev. Vaccines 2019, 18, 505–521. [Google Scholar] [CrossRef]
- Herr, H.W.; Morales, A. History of bacillus Calmette-Guerin and bladder cancer: An immunotherapy success story. J. Urol. 2008, 179, 53–56. [Google Scholar] [CrossRef]
- Nishida, S.; Tsuboi, A.; Tanemura, A.; Ito, T.; Nakajima, H.; Shirakata, T.; Morimoto, S.; Fujiki, F.; Hosen, N.; Oji, Y.; et al. Immune adjuvant therapy using Bacillus Calmette-Guerin cell wall skeleton (BCG-CWS) in advanced malignancies: A phase 1 study of safety and immunogenicity assessments. Medicine 2019, 98, e16771. [Google Scholar] [CrossRef]
- Nachega, J.B.; Maeurer, M.; Sam-Agudu, N.A.; Chakaya, J.; Katoto, P.D.M.; Zumla, A. Bacille Calmette-Guerin (BCG) vaccine and potential cross-protection against SARS-CoV-2 infection-Assumptions, knowns, unknowns and need for developing an accurate scientific evidence base. Int. J. Infect. Dis. 2021, 113 (Suppl. S1), S78–S81. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.; Tili, E.; Suster, D.; Matys, E.; Hupp, L.; Magro, C. Strong homology between SARS-CoV-2 envelope protein and a Mycobacterium sp. antigen allows rapid diagnosis of Mycobacterial infections and may provide specific anti-SARS-CoV-2 immunity via the BCG vaccine. Ann. Diagn. Pathol. 2020, 48, 151600. [Google Scholar] [CrossRef] [PubMed]
- Behzad, H.; Huckriede, A.L.; Haynes, L.; Gentleman, B.; Coyle, K.; Wilschut, J.C.; Kollmann, T.R.; Reed, S.G.; McElhaney, J.E. GLA-SE, a synthetic toll-like receptor 4 agonist, enhances T-cell responses to influenza vaccine in older adults. J. Infect. Dis. 2012, 205, 466–473. [Google Scholar] [CrossRef]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef]
- Jensen, F.C.; Savary, J.R.; Diveley, J.P.; Chang, J.C. Adjuvant activity of incomplete Freund’s adjuvant. Adv. Drug Deliv. Rev. 1998, 32, 173–186. [Google Scholar] [CrossRef]
- Wilson-Welder, J.H.; Torres, M.P.; Kipper, M.J.; Mallapragada, S.K.; Wannemuehler, M.J.; Narasimhan, B. Vaccine adjuvants: Current challenges and future approaches. J. Pharm. Sci. 2009, 98, 1278–1316. [Google Scholar] [CrossRef]
- Stills, H.F., Jr. Adjuvants and antibody production: Dispelling the myths associated with Freund’s complete and other adjuvants. ILAR J. 2005, 46, 280–293. [Google Scholar] [CrossRef]
- Van Doorn, E.; Liu, H.; Huckriede, A.; Hak, E. Safety and tolerability evaluation of the use of Montanide ISA™51 as vaccine adjuvant: A systematic review. Hum. Vaccines Immunother. 2016, 12, 159–169. [Google Scholar] [CrossRef]
- Aucouturier, J.; Dupuis, L.; Deville, S.; Ascarateil, S.; Ganne, V. Montanide ISA 720 and 51: A new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev. Vaccines 2002, 1, 111–118. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; Ott, G.S.; Nest, G.V.; Rappuoli, R.; Giudice, G.D. The history of MF59((R)) adjuvant: A phoenix that arose from the ashes. Expert Rev. Vaccines 2013, 12, 13–30. [Google Scholar] [CrossRef]
- O’Hagan, D.T.; Ott, G.S.; De Gregorio, E.; Seubert, A. The mechanism of action of MF59–An innately attractive adjuvant formulation. Vaccine 2011, 30, 4341–4348. [Google Scholar] [CrossRef]
- Zhang, J.; Miao, J.; Han, X.; Lu, Y.; Deng, B.; Lv, F.; Zhao, Y.; Ding, C.; Hou, J. Development of a novel oil-in-water emulsion and evaluation of its potential adjuvant function in a swine influenza vaccine in mice. BMC Vet. Res. 2018, 14, 415. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B.; Haensler, J. An update on safety and immunogenicity of vaccines containing emulsion-based adjuvants. Expert Rev. Vaccines 2013, 12, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.-J.; Kang, S.-M. Immunology and efficacy of MF59-adjuvanted vaccines. Hum. Vaccin Immunother. 2018, 14, 3041–3045. [Google Scholar] [CrossRef]
- Fox, C.B.; Van Hoeven, N.; Granger, B.; Lin, S.; Guderian, J.A.; Hartwig, A.; Marlenee, N.; Bowen, R.A.; Soultanov, V.; Carter, D. Vaccine adjuvant activity of emulsified oils from species of the Pinaceae family. Phytomedicine 2019, 64, 152927. [Google Scholar] [CrossRef] [PubMed]
- Popa, O.; Băbeanu, N.E.; Popa, I.; Niță, S.; Dinu-Pârvu, C.E. Methods for obtaining and determination of squalene from natural sources. BioMed. Res. Int. 2015, 2015, 367202–367216. [Google Scholar] [CrossRef]
- Dubois Cauwelaert, N.; Desbien, A.L.; Hudson, T.E.; Pine, S.O.; Reed, S.G.; Coler, R.N.; Orr, M.T. The TLR4 Agonist Vaccine Adjuvant, GLA-SE, Requires Canonical and Atypical Mechanisms of Action for TH1 Induction. PLoS ONE 2016, 11, e0146372. [Google Scholar] [CrossRef]
- Reed, S.G.; Carter, D.; Casper, C.; Duthie, M.S.; Fox, C.B. Correlates of GLA family adjuvants’ activities. Semin. Immunol. 2018, 39, 22–29. [Google Scholar] [CrossRef]
- Patton, K.; Aslam, S.; Shambaugh, C.; Lin, R.; Heeke, D.; Frantz, C.; Zuo, F.; Esser, M.T.; Paliard, X.; Lambert, S.L. Enhanced immunogenicity of a respiratory syncytial virus (RSV) F subunit vaccine formulated with the adjuvant GLA-SE in cynomolgus macaques. Vaccine 2015, 33, 4472–4478. [Google Scholar] [CrossRef]
- Henker, L.C.; Schwertz, C.I.; Lucca, N.J.; Piva, M.M.; Prior, K.C.; Baska, P.; Norbury, L.; Januszkiewicz, K.; Dezen, D.; Duarte, M.; et al. Immune protection conferred by recombinant MRLC (myosin regulatory light chain) antigen in TiterMax Gold(R) adjuvant against experimental fasciolosis in rats. Vaccine 2017, 35, 663–671. [Google Scholar] [CrossRef]
- Leenaars, P.P.; Koedam, M.A.; Wester, P.W.; Baumans, V.; Claassen, E.; Hendriksen, C.F. Assessment of side effects induced by injection of different adjuvant/antigen combinations in rabbits and mice. Lab. Anim. 1998, 32, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Azuma, I.; Ribi, E.E.; Meyer, T.J.; Zbar, B. Biologically active components from mycobacterial cell walls. I. Isolation and composition of cell wall skeleton and component P3. J. Natl. Cancer Inst. 1974, 52, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Jennings, V.M. Review of Selected Adjuvants Used in Antibody Production. ILAR J. 1995, 37, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.G. Molecular adjuvants and immunomodulators: New approaches to immunization. Clin. Microbiol. Rev. 1994, 7, 277–289. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deeb, B.J.; DiGiacomo, R.F.; Kunz, L.L.; Stewart, J.L. Comparison of Freund’s and Ribi adjuvants for inducing antibodies to the synthetic antigen (TG)-AL in rabbits. J. Immunol. Methods 1992, 152, 105–113. [Google Scholar] [CrossRef]
- Lipman, N.S.; Trudel, L.J.; Murphy, J.C.; Sahali, Y. Comparison of immune response potentiation and in vivo inflammatory effects of Freund’s and RIBI adjuvants in mice. Lab. Anim. Sci. 1992, 42, 193–197. [Google Scholar] [PubMed]
- Tovey, M.G.; Lallemand, C. Adjuvant activity of cytokines. Methods Mol. Biol. 2010, 626, 287–309. [Google Scholar] [CrossRef]
- Boyaka, P.N.; McGhee, J.R. Cytokines as adjuvants for the induction of mucosal immunity. Adv. Drug Deliv Rev. 2001, 51, 71–79. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef]
- Kayamuro, H.; Yoshioka, Y.; Abe, Y.; Arita, S.; Katayama, K.; Nomura, T.; Yoshikawa, T.; Kubota-Koketsu, R.; Ikuta, K.; Okamoto, S.; et al. Interleukin-1 family cytokines as mucosal vaccine adjuvants for induction of protective immunity against influenza virus. J. Virol. 2010, 84, 12703–12712. [Google Scholar] [CrossRef]
- Sabbaghi, A.; Ghaemi, A. Molecular Adjuvants for DNA Vaccines: Application, Design, Preparation, and Formulation. Methods Mol. Biol. 2021, 2197, 87–112. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Petrovsky, N. Molecular Adjuvants for DNA Vaccines. Curr. Issues Mol. Biol. 2017, 22, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Suschak, J.J.; Williams, J.A.; Schmaljohn, C.S. Advancements in DNA vaccine vectors, non-mechanical delivery methods, and molecular adjuvants to increase immunogenicity. Hum. Vaccin Immunother. 2017, 13, 2837–2848. [Google Scholar] [CrossRef]
- Blyszczuk, P.; Behnke, S.; Luscher, T.F.; Eriksson, U.; Kania, G. GM-CSF promotes inflammatory dendritic cell formation but does not contribute to disease progression in experimental autoimmune myocarditis. Biochim. Biophys. Acta 2013, 1833, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N. The Role of Interferons in Inflammation and Inflammasome Activation. Front. Immunol. 2017, 8, 873. [Google Scholar] [CrossRef] [PubMed]
- Tovey, M.G.; Lallemand, C.; Thyphronitis, G. Adjuvant activity of type I interferons. Biol. Chem. 2008, 389, 541–545. [Google Scholar] [CrossRef]
- Zanetti, B.F.; Ferreira, C.P.; Vasconcelos, J.R.C.; Han, S.W. Adjuvant properties of IFN-gamma and GM-CSF in the scFv6.C4 DNA vaccine against CEA-expressing tumors. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Mohan, T.; Verma, P.; Rao, D.N. Novel adjuvants & delivery vehicles for vaccines development: A road ahead. Indian J. Med. Res. 2013, 138, 779–795. [Google Scholar]
- Perera, P.Y.; Lichy, J.H.; Waldmann, T.A.; Perera, L.P. The role of interleukin-15 in inflammation and immune responses to infection: Implications for its therapeutic use. Microbes Infect. 2012, 14, 247–261. [Google Scholar] [CrossRef]
- Kasahara, S.; Jhingran, A.; Dhingra, S.; Salem, A.; Cramer, R.A.; Hohl, T.M. Role of Granulocyte-Macrophage Colony-Stimulating Factor Signaling in Regulating Neutrophil Antifungal Activity and the Oxidative Burst During Respiratory Fungal Challenge. J. Infect. Dis. 2016, 213, 1289–1298. [Google Scholar] [CrossRef]
- Yoon, H.A.; Aleyas, A.G.; George, J.A.; Park, S.O.; Han, Y.W.; Lee, J.H.; Cho, J.G.; Eo, S.K. Cytokine GM-CSF genetic adjuvant facilitates prophylactic DNA vaccine against pseudorabies virus through enhanced immune responses. Microbiol. Immunol. 2006, 50, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.L.; Montefiori, D.C.; Villinger, F.; Robinson, J.E.; Sharma, S.; Wyatt, L.S.; Earl, P.L.; McClure, H.M.; Moss, B.; Amara, R.R. Studies on GM-CSF DNA as an adjuvant for neutralizing Ab elicited by a DNA/MVA immunodeficiency virus vaccine. Virology 2006, 352, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Fabrizi, F.; Ganeshan, S.V.; Dixit, V.; Martin, P. Meta-analysis: The adjuvant role of granulocyte macrophage-colony stimulating factor on immunological response to hepatitis B virus vaccine in end-stage renal disease. Aliment. Pharmacol. Ther. 2006, 24, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Guilhaudis, L.; Jacobs, A.; Caffrey, M. Solution structure of the HIV gp120 C5 domain. Eur. J. BioChem. 2002, 269, 4860–4867. [Google Scholar] [CrossRef]
- Bracci, L.; Canini, I.; Puzelli, S.; Sestili, P.; Venditti, M.; Spada, M.; Donatelli, I.; Belardelli, F.; Proietti, E. Type I IFN is a powerful mucosal adjuvant for a selective intranasal vaccination against influenza virus in mice and affects antigen capture at mucosal level. Vaccine 2005, 23, 2994–3004. [Google Scholar] [CrossRef]
- Ye, L.; Ohnemus, A.; Ong, L.C.; Gad, H.H.; Hartmann, R.; Lycke, N.; Staeheli, P. Type I and Type III Interferons Differ in Their Adjuvant Activities for Influenza Vaccines. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Couch, R.B.; Atmar, R.L.; Cate, T.R.; Quarles, J.M.; Keitel, W.A.; Arden, N.H.; Wells, J.; Nino, D.; Wyde, P.R. Contrasting effects of type I interferon as a mucosal adjuvant for influenza vaccine in mice and humans. Vaccine 2009, 27, 5344–5348. [Google Scholar] [CrossRef][Green Version]
- Sturchler, D.; Berger, R.; Etlinger, H.; Fernex, M.; Matile, H.; Pink, R.; Schlumbom, V.; Just, M. Effects of interferons on immune response to a synthetic peptide malaria sporozoite vaccine in non-immune adults. Vaccine 1989, 7, 457–461. [Google Scholar] [CrossRef]
- Rizza, P.; Capone, I.; Moretti, F.; Proietti, E.; Belardelli, F. IFN-alpha as a vaccine adjuvant: Recent insights into the mechanisms and perspectives for its clinical use. Expert Rev. Vaccines 2011, 10, 487–498. [Google Scholar] [CrossRef]
- Henke, A.; Rohland, N.; Zell, R.; Wutzler, P. Co-expression of interleukin-2 by a bicistronic plasmid increases the efficacy of DNA immunization to prevent influenza virus infections. Intervirology 2006, 49, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tao, L.; Wang, Y.; Chen, L.; Yang, J.; Wang, H. Enhancing immune responses against SARS-CoV nucleocapsid DNA vaccine by co-inoculating interleukin-2 expressing vector in mice. Biotechnol. Lett. 2009, 31, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Wolf, N.; Lavelle, E.C. A Guide to IL-1 family cytokines in adjuvanticity. FEBS J. 2018, 285, 2377–2401. [Google Scholar] [CrossRef] [PubMed]
- Arulanandam, B.P.; O’Toole, M.; Metzger, D.W. Intranasal interleukin-12 is a powerful adjuvant for protective mucosal immunity. J. Infect. Dis. 1999, 180, 940–949. [Google Scholar] [CrossRef]
- Kumar, D.; Kirimanjeswara, G.; Metzger, D.W. Intranasal administration of an inactivated Yersinia pestis vaccine with interleukin-12 generates protective immunity against pneumonic plague. Clin. Vaccine Immunol. 2011, 18, 1925–1935. [Google Scholar] [CrossRef]
- Kalams, S.A.; Parker, S.; Jin, X.; Elizaga, M.; Metch, B.; Wang, M.; Hural, J.; Lubeck, M.; Eldridge, J.; Cardinali, M.; et al. Safety and immunogenicity of an HIV-1 gag DNA vaccine with or without IL-12 and/or IL-15 plasmid cytokine adjuvant in healthy, HIV-1 uninfected adults. PLoS ONE 2012, 7, e29231. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Fantuzzi, G. Interleukin-18 and host defense against infection. J. Infect. Dis. 2003, 187 (Suppl. S2), S370–S384. [Google Scholar] [CrossRef]
- Zhu, M.; Xu, X.; Liu, H.; Liu, X.; Wang, S.; Dong, F.; Yang, B.; Song, G. Enhancement of DNA vaccine potency against herpes simplex virus 1 by co-administration of an interleukin-18 expression plasmid as a genetic adjuvant. J. Med. Microbiol. 2003, 52, 223–228. [Google Scholar] [CrossRef]
- Oh, Y.K.; Park, J.S.; Yoon, H.; Kim, C.K. Enhanced mucosal and systemic immune responses to a vaginal vaccine coadministered with RANTES-expressing plasmid DNA using in situ-gelling mucoadhesive delivery system. Vaccine 2003, 21, 1980–1988. [Google Scholar] [CrossRef]
- AEgan, M.; RIsrael, Z. The use of cytokines and chemokines as genetic adjuvants for plasmid DNA vaccines. Clinical. Appl. Immunol. Rev. 2002, 2, 255–287. [Google Scholar] [CrossRef]
- Eo, S.K.; Lee, S.; Chun, S.; Rouse, B.T. Modulation of immunity against herpes simplex virus infection via mucosal genetic transfer of plasmid DNA encoding chemokines. J. Virol. 2001, 75, 569–578. [Google Scholar] [CrossRef]
- Tsuji, T.; Fukushima, J.; Hamajima, K.; Ishii, N.; Aoki, I.; Bukawa, H.; Ishigatsubo, Y.; Tani, K.; Okubo, T.; Dorf, M.E.; et al. HIV-1-specific cell-mediated immunity is enhanced by co-inoculation of TCA3 expression plasmid with DNA vaccine. Immunology 1997, 90, 1–6. [Google Scholar] [CrossRef]
- Smith, K.J.; Hamza, S.; Skelton, H. The imidazoquinolines and their place in the therapy of cutaneous disease. Expert Opin Pharmacother. 2003, 4, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Van Hoeven, N.; Fox, C.B.; Granger, B.; Evers, T.; Joshi, S.W.; Nana, G.I.; Evans, S.C.; Lin, S.; Liang, H.; Liang, L.; et al. A Formulated TLR7/8 Agonist is a Flexible, Highly Potent and Effective Adjuvant for Pandemic Influenza Vaccines. Sci. Rep. 2017, 7, 46426. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Nanoparticles and microparticles for drug and vaccine delivery. J. Anat. 1996, 189 Pt 3, 503–505. [Google Scholar] [PubMed]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: Preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 2021, 19, 59. [Google Scholar] [CrossRef]
- Cimica, V.; Galarza, J.M. Adjuvant formulations for virus-like particle (VLP) based vaccines. Clin. Immunol. 2017, 183, 99–108. [Google Scholar] [CrossRef]
- Qian, C.; Liu, X.; Xu, Q.; Wang, Z.; Chen, J.; Li, T.; Zheng, Q.; Yu, H.; Gu, Y.; Li, S.; et al. Recent Progress on the Versatility of Virus-Like Particles. Vaccines 2020, 8, 139. [Google Scholar] [CrossRef]
- Gao, Y.; Wijewardhana, C.; Mann, J.F.S. Virus-Like Particle, Liposome, and Polymeric Particle-Based Vaccines against HIV-1. Front. Immunol. 2018, 9, 345. [Google Scholar] [CrossRef]
- Felnerova, D.; Viret, J.-F.; Glück, R.; Moser, C. Liposomes and virosomes as delivery systems for antigens, nucleic acids and drugs. Curr. Opin. Biotechnol. 2004, 15, 518–529. [Google Scholar] [CrossRef]
- Liu, H.; Tu, Z.; Feng, F.; Shi, H.; Chen, K.; Xu, X. Virosome, a hybrid vehicle for efficient and safe drug delivery and its emerging application in cancer treatment. Acta Pharm. 2015, 65, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.J.; Dowd, K.A.; Mendoza, F.H.; Saunders, J.G.; Sitar, S.; Plummer, S.H.; Yamshchikov, G.; Sarwar, U.N.; Hu, Z.; Enama, M.E.; et al. Safety and tolerability of chikungunya virus-like particle vaccine in healthy adults: A phase 1 dose-escalation trial. Lancet 2014, 384, 2046–2052. [Google Scholar] [CrossRef]
- Luxembourg, A.; Moeller, E. 9-Valent human papillomavirus vaccine: A review of the clinical development program. Expert Rev. Vaccines 2017, 16, 1119–1139. [Google Scholar] [CrossRef]
- Toh, Z.Q.; Kosasih, J.; Russell, F.M.; Garland, S.M.; Mulholland, E.K.; Licciardi, P.V. Recombinant human papillomavirus nonavalent vaccine in the prevention of cancers caused by human papillomavirus. Infect. Drug Resist. 2019, 12, 1951–1967. [Google Scholar] [CrossRef]
- Yilmaz, I.C.; Ipekoglu, E.M.; Bulbul, A.; Turay, N.; Yildirim, M.; Evcili, I.; Yilmaz, N.S.; Guvencli, N.; Aydin, Y.; Gungor, B.; et al. Development and preclinical evaluation of virus-like particle vaccine against COVID-19 infection. Allergy 2022, 77, 258–270. [Google Scholar] [CrossRef]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, G.; Rappuoli, R.; Didierlaurent, A.M. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin Immunol. 2018, 39, 14–21. [Google Scholar] [CrossRef]
- Fujimoto, C.; Nakagawa, Y.; Ohara, K.; Takahashi, H. Polyriboinosinic polyribocytidylic acid [poly(I:C)]/TLR3 signaling allows class I processing of exogenous protein and induction of HIV-specific CD8+ cytotoxic T lymphocytes. Int. Immunol. 2004, 16, 55–63. [Google Scholar] [CrossRef]
- Ichinohe, T.; Watanabe, I.; Ito, S.; Fujii, H.; Moriyama, M.; Tamura, S.; Takahashi, H.; Sawa, H.; Chiba, J.; Kurata, T.; et al. Synthetic double-stranded RNA poly(I:C) combined with mucosal vaccine protects against influenza virus infection. J. Virol. 2005, 79, 2910–2919. [Google Scholar] [CrossRef]
- De Waele, J.; Verhezen, T.; van der Heijden, S.; Berneman, Z.N.; Peeters, M.; Lardon, F.; Wouters, A.; Smits, E. A systematic review on poly(I:C) and poly-ICLC in glioblastoma: Adjuvants coordinating the unlocking of immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 213. [Google Scholar] [CrossRef]
- Van der Velden, M.V.; Geisberger, A.; Dvorak, T.; Portsmouth, D.; Fritz, R.; Crowe, B.A.; Herr, W.; Distler, E.; Wagner, E.M.; Zeitlinger, M.; et al. Safety and immunogenicity of a vero cell culture-derived whole-virus H5N1 influenza vaccine in chronically ill and immunocompromised patients. Clin. Vaccine Immunol. 2014, 21, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.F.; Wang, C.C.; Wu, T.C.; Wu, K.G.; Lee, C.C.; Peng, H.J. Neonatal sublingual vaccination with Salmonella proteins and adjuvant cholera toxin or CpG oligodeoxynucleotides induces mucosal and systemic immunity in mice. J. Pediatr. Gastroenterol. Nutr. 2008, 46, 262–271. [Google Scholar] [CrossRef]
- Klinman, D.M.; Currie, D.; Lee, G.; Grippe, V.; Merkel, T. Systemic but not mucosal immunity induced by AVA prevents inhalational anthrax. Microbes Infect. 2007, 9, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Lentino, J.; Kopp, J.; Murray, L.; Ellison, W.; Rhee, M.; Shockey, G.; Akella, L.; Erby, K.; Heyward, W.L.; et al. Immunogenicity of a two-dose investigational hepatitis B vaccine, HBsAg-1018, using a toll-like receptor 9 agonist adjuvant compared with a licensed hepatitis B vaccine in adults. Vaccine 2018, 36, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.Y.; Lin, M.Y.; Coffman, R.L.; Campbell, J.D.; Traquina, P.; Lin, Y.J.; Liu, L.T.; Cheng, J.; Wu, Y.C.; Wu, C.C.; et al. Development of CpG-adjuvanted stable prefusion SARS-CoV-2 spike antigen as a subunit vaccine against COVID-19. Sci. Rep. 2020, 10, 20085. [Google Scholar] [CrossRef]
- Hopkins, R.J.; Daczkowski, N.F.; Kaptur, P.E.; Muse, D.; Sheldon, E.; LaForce, C.; Sari, S.; Rudge, T.L.; Bernton, E. Randomized, double-blind, placebo-controlled, safety and immunogenicity study of 4 formulations of Anthrax Vaccine Adsorbed plus CPG 7909 (AV7909) in healthy adult volunteers. Vaccine 2013, 31, 3051–3058. [Google Scholar] [CrossRef]
- Cooper, C.L.; Davis, H.L.; Morris, M.L.; Efler, S.M.; Krieg, A.M.; Li, Y.; Laframboise, C.; Al Adhami, M.J.; Khaliq, Y.; Seguin, I.; et al. Safety and immunogenicity of CPG 7909 injection as an adjuvant to Fluarix influenza vaccine. Vaccine 2004, 22, 3136–3143. [Google Scholar] [CrossRef]
- Olafsdottir, T.A.; Lingnau, K.; Nagy, E.; Jonsdottir, I. IC31, a two-component novel adjuvant mixed with a conjugate vaccine enhances protective immunity against pneumococcal disease in neonatal mice. Scand. J. Immunol. 2009, 69, 194–202. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett 2013, 8, 102. [Google Scholar] [CrossRef]
- Giddam, A.K.; Zaman, M.; Skwarczynski, M.; Toth, I. Liposome-based delivery system for vaccine candidates: Constructing an effective formulation. Nanomedicine 2012, 7, 1877–1893. [Google Scholar] [CrossRef]
- Schwendener, R.A. Liposomes as vaccine delivery systems: A review of the recent advances. Ther. Adv. Vaccines 2014, 2, 159–182. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Szoka, F.C., Jr. Lipid-based nanoparticles for nucleic acid delivery. Pharm. Res. 2007, 24, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ma, Y.; Zhang, J.; Kuo, J.C.; Zhang, Z.; Xie, H.; Zhu, J.; Liu, T. Modification of Lipid-Based Nanoparticles: An Efficient Delivery System for Nucleic Acid-Based Immunotherapy. Molecules 2022, 27, 1943. [Google Scholar] [CrossRef] [PubMed]
- Agger, E.M.; Rosenkrands, I.; Hansen, J.; Brahimi, K.; Vandahl, B.S.; Aagaard, C.; Werninghaus, K.; Kirschning, C.; Lang, R.; Christensen, D.; et al. Cationic liposomes formulated with synthetic mycobacterial cordfactor (CAF01): A versatile adjuvant for vaccines with different immunological requirements. PLoS ONE 2008, 3, e3116. [Google Scholar] [CrossRef]
- Pedersen, G.K.; Andersen, P.; Christensen, D. Immunocorrelates of CAF family adjuvants. Semin. Immunol. 2018, 39, 4–13. [Google Scholar] [CrossRef]
- Christensen, D.; Foged, C.; Rosenkrands, I.; Lundberg, C.V.; Andersen, P.; Agger, E.M.; Nielsen, H.M. CAF01 liposomes as a mucosal vaccine adjuvant: In vitro and in vivo investigations. Int. J. Pharm. 2010, 390, 19–24. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Ghaffar, K.A.; Marasini, N.; Giddam, A.K.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. Liposome-based intranasal delivery of lipopeptide vaccine candidates against group A streptococcus. Acta Biomater. 2016, 41, 161–168. [Google Scholar] [CrossRef]
- Dai, C.C.; Huang, W.; Yang, J.; Hussein, W.M.; Wang, J.; Khalil, Z.G.; Capon, R.J.; Toth, I.; Stephenson, R.J. Polyethylenimine quantity and molecular weight influence its adjuvanting properties in liposomal peptide vaccines. Bioorg. Med. Chem. Lett. 2021, 40, 127920. [Google Scholar] [CrossRef]
- Huang, W.C.; Deng, B.; Lin, C.; Carter, K.A.; Geng, J.; Razi, A.; He, X.; Chitgupi, U.; Federizon, J.; Sun, B.; et al. A malaria vaccine adjuvant based on recombinant antigen binding to liposomes. Nat. Nanotechnol. 2018, 13, 1174–1181. [Google Scholar] [CrossRef]
- Abhyankar, M.M.; Mann, B.J.; Sturek, J.M.; Brovero, S.; Moreau, G.B.; Sengar, A.; Richardson, C.M.; Agah, S.; Pomes, A.; Kasson, P.M.; et al. Development of COVID-19 vaccine using a dual Toll-like receptor ligand liposome adjuvant. NPJ Vaccines 2021, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Wloch, M.K.; Ye, M.; Reyes, L.R.; Boutsaboualoy, S.; Dunne, C.E.; Chaplin, J.A.; Rusalov, D.; Rolland, A.P.; Fisher, C.L.; et al. Phase 1 clinical trials of the safety and immunogenicity of adjuvanted plasmid DNA vaccines encoding influenza A virus H5 hemagglutinin. Vaccine 2010, 28, 2565–2572. [Google Scholar] [CrossRef] [PubMed]
- Schautteet, K.; De Clercq, E.; Vanrompay, D. Chlamydia trachomatis vaccine research through the years. Infect. Dis. Obstet Gynecol. 2011, 2011, 963513. [Google Scholar] [CrossRef]
- Soni, D.; Bobbala, S.; Li, S.; Scott, E.A.; Dowling, D.J. The sixth revolution in pediatric vaccinology: Immunoengineering and delivery systems. Pediatr. Res. 2021, 89, 1364–1372. [Google Scholar] [CrossRef]
- Sun, B.; Yu, S.; Zhao, D.; Guo, S.; Wang, X.; Zhao, K. Polysaccharides as vaccine adjuvants. Vaccine 2018, 36, 5226–5234. [Google Scholar] [CrossRef]
- Li, P.; Wang, F. Polysaccharides: Candidates of promising vaccine adjuvants. Drug Discov. Ther. 2015, 9, 88–93. [Google Scholar] [CrossRef]
- McNeela, E.A.; O’Connor, D.; Jabbal-Gill, I.; Illum, L.; Davis, S.S.; Pizza, M.; Peppoloni, S.; Rappuoli, R.; Mills, K.H. A mucosal vaccine against diphtheria: Formulation of cross reacting material (CRM(197)) of diphtheria toxin with chitosan enhances local and systemic antibody and Th2 responses following nasal delivery. Vaccine 2000, 19, 1188–1198. [Google Scholar] [CrossRef]
- Mann, A.J.; Noulin, N.; Catchpole, A.; Stittelaar, K.J.; de Waal, L.; Veldhuis Kroeze, E.J.; Hinchcliffe, M.; Smith, A.; Montomoli, E.; Piccirella, S.; et al. Intranasal H5N1 vaccines, adjuvanted with chitosan derivatives, protect ferrets against highly pathogenic influenza intranasal and intratracheal challenge. PLoS ONE 2014, 9, e93761. [Google Scholar] [CrossRef]
- Sahni, J.K.; Chopra, S.; Ahmad, F.J.; Khar, R.K. Potential prospects of chitosan derivative trimethyl chitosan chloride (TMC) as a polymeric absorption enhancer: Synthesis, characterization and applications. J. Pharm. Pharmacol. 2008, 60, 1111–1119. [Google Scholar] [CrossRef]
- Zielinska, A.; Carreiro, F.; Oliveira, A.M.; Neves, A.; Pires, B.; Venkatesh, D.N.; Durazzo, A.; Lucarini, M.; Eder, P.; Silva, A.M.; et al. Polymeric Nanoparticles: Production, Characterization, Toxicology and Ecotoxicology. Molecules 2020, 25, 3731. [Google Scholar] [CrossRef] [PubMed]
- Pavot, V.; Berthet, M.; Resseguier, J.; Legaz, S.; Handke, N.; Gilbert, S.C.; Paul, S.; Verrier, B. Poly(lactic acid) and poly(lactic-co-glycolic acid) particles as versatile carrier platforms for vaccine delivery. Nanomedicine 2014, 9, 2703–2718. [Google Scholar] [CrossRef]
- Jin, Z.; Gao, S.; Cui, X.; Sun, D.; Zhao, K. Adjuvants and delivery systems based on polymeric nanoparticles for mucosal vaccines. Int. J. Pharm. 2019, 572, 118731. [Google Scholar] [CrossRef] [PubMed]
- Roopngam, P. Poly (Lactic-Co-Glycolic) Acid (PLGA) Adjuvant for Immunotherapy. Immunol. Disord. Immunother. 2017, 2, 1–2. [Google Scholar] [CrossRef]
- Lori, F.; Calarota, S.A.; Lisziewicz, J. Nanochemistry-based immunotherapy for HIV-1. Curr. Med. Chem. 2007, 14, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Lori, F. DermaVir: A plasmid DNA-based nanomedicine therapeutic vaccine for the treatment of HIV/AIDS. Expert Rev. Vaccines 2011, 10, 1371–1384. [Google Scholar] [CrossRef]
- Okuda, T.; Shimizu, K.; Hasaba, S.; Date, M. Induction of specific adaptive immune responses by immunization with newly designed artificial glycosphingolipids. Sci. Rep. 2019, 9, 18803. [Google Scholar] [CrossRef]
- Subrahmanyam, P.; Webb, T.J. Boosting the Immune Response: The Use of iNKT cell ligands as vaccine adjuvants. Front. Biol. 2012, 7, 436–444. [Google Scholar] [CrossRef]
- Van Kaer, L.; Parekh, V.V.; Wu, L. Invariant natural killer T cells: Bridging innate and adaptive immunity. Cell. Tissue Res. 2011, 343, 43–55. [Google Scholar] [CrossRef]
- Fujii, S.; Shimizu, K.; Hemmi, H.; Fukui, M.; Bonito, A.J.; Chen, G.; Franck, R.W.; Tsuji, M.; Steinman, R.M. Glycolipid alpha-C-galactosylceramide is a distinct inducer of dendritic cell function during innate and adaptive immune responses of mice. Proc. Natl. Acad. Sci. USA 2006, 103, 11252–11257. [Google Scholar] [CrossRef]
- Fotouhi, F.; Shaffifar, M.; Farahmand, B.; Shirian, S.; Saeidi, M.; Tabarraei, A.; Gorji, A.; Ghaemi, A. Adjuvant use of the NKT cell agonist alpha-galactosylceramide leads to enhancement of M2-based DNA vaccine immunogenicity and protective immunity against influenza A virus. Arch. Virol. 2017, 162, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Rajput, Z.I.; Hu, S.H.; Xiao, C.W.; Arijo, A.G. Adjuvant effects of saponins on animal immune responses. J. Zhejiang Univ. Sci. B 2007, 8, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Tuo, W. QS-21: A Potent Vaccine Adjuvant. Nat. Prod. Chem. Res. 2016, 3, e113. [Google Scholar] [CrossRef] [PubMed]
- Moses, T.; Papadopoulou, K.K.; Osbourn, A. Metabolic and functional diversity of saponins, biosynthetic intermediates and semi-synthetic derivatives. Crit Rev. BioChem. Mol. Biol. 2014, 49, 439–462. [Google Scholar] [CrossRef] [PubMed]
- Lacaille-Dubois, M.A. Updated insights into the mechanism of action and clinical profile of the immunoadjuvant QS-21: A review. Phytomedicine 2019, 60, 152905. [Google Scholar] [CrossRef]
- Katayama, S.; Oda, K.; Ohgitani, T.; Hirahara, T.; Shimizu, Y. Influence of antigenic forms and adjuvants on the IgG subclass antibody response to Aujeszky’s disease virus in mice. Vaccine 1999, 17, 2733–2739. [Google Scholar] [CrossRef]
- Magnusson, S.E.; Altenburg, A.F.; Bengtsson, K.L.; Bosman, F.; de Vries, R.D.; Rimmelzwaan, G.F.; Stertman, L. Matrix-M adjuvant enhances immunogenicity of both protein- and modified vaccinia virus Ankara-based influenza vaccines in mice. Immunol. Res. 2018, 66, 224–233. [Google Scholar] [CrossRef]
- Wang, P. Natural and Synthetic Saponins as Vaccine Adjuvants. Vaccines 2021, 9, 222. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Oliveira, C.L.; Hubschmann, H.B.; Arleth, L.; Manniche, S.; Kirkby, N.; Nielsen, H.M. Structure of immune stimulating complex matrices and immune stimulating complexes in suspension determined by small-angle X-ray scattering. Biophys. J. 2012, 102, 2372–2380. [Google Scholar] [CrossRef][Green Version]
- Myschik, J.; Lendemans, D.G.; McBurney, W.T.; Demana, P.H.; Hook, S.; Rades, T. On the preparation, microscopic investigation and application of ISCOMs. Micron 2006, 37, 724–734. [Google Scholar] [CrossRef]
- Fernandez-Tejada, A.; Tan, D.S.; Gin, D.Y. Development of Improved Vaccine Adjuvants Based on the Saponin Natural Product QS-21 through Chemical Synthesis. Acc. Chem. Res. 2016, 49, 1741–1756. [Google Scholar] [CrossRef] [PubMed]
- Suartha, I.N.; Suartini, G.A.A.; Wirata, I.W.; Dewi, N.M.A.R.K.; Putra, G.N.N.; Kencana, G.A.Y.; Mahardika, G.N. Intranasal administration of inactivated avian influenza virus of H5N1 subtype vaccine-induced systemic immune response in chicken and mice. Vet. World 2018, 11, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Baz Morelli, A.; Becher, D.; Koernig, S.; Silva, A.; Drane, D.; Maraskovsky, E. ISCOMATRIX: A novel adjuvant for use in prophylactic and therapeutic vaccines against infectious diseases. J. Med. Microbiol. 2012, 61, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, M.; Wang, Q.; Cheng, L.; Zhang, Z. Role of Papain-Like Cysteine Proteases in Plant Development. Front. Plant Sci. 2018, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.D.; Wunschmann, S.; Pomes, A. Proteases as Th2 adjuvants. Curr. Allergy Asthma Rep. 2007, 7, 363–367. [Google Scholar] [CrossRef]
- Levast, B.; Awate, S.; Babiuk, L.; Mutwiri, G.; Gerdts, V.; van Drunen Littel-van den Hurk, S. Vaccine Potentiation by Combination Adjuvants. Vaccines 2014, 2, 297–322. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Laupeze, B.; Di Pasquale, A.; Hergli, N.; Collignon, C.; Garcon, N. Adjuvant system AS01: Helping to overcome the challenges of modern vaccines. Expert Rev. Vaccines 2017, 16, 55–63. [Google Scholar] [CrossRef]
- Garcon, N.; Chomez, P.; Van Mechelen, M. GlaxoSmithKline Adjuvant Systems in vaccines: Concepts, achievements and perspectives. Expert Rev. Vaccines 2007, 6, 723–739. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 2009, 183, 6186–6197. [Google Scholar] [CrossRef]
- Nadeem, A.Y.; Shehzad, A.; Islam, S.U.; Al-Suhaimi, E.A.; Lee, Y.S. Mosquirix RTS, S/AS01 Vaccine Development, Immunogenicity, and Efficacy. Vaccines 2022, 10, 713. [Google Scholar] [CrossRef]
- Garcon, N.; Di Pasquale, A. From discovery to licensure, the Adjuvant System story. Hum. Vaccin Immunother. 2017, 13, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, M.E.; Remich, S.A.; Ogutu, B.R.; Waitumbi, J.N.; Otieno, L.; Apollo, S.; Cummings, J.F.; Kester, K.E.; Ockenhouse, C.F.; Stewart, A.; et al. Evaluation of RTS,S/AS02A and RTS,S/AS01B in adults in a high malaria transmission area. PLoS ONE 2009, 4, e6465. [Google Scholar] [CrossRef] [PubMed]
- Romanowski, B.; Schwarz, T.F.; Ferguson, L.; Peters, K.; Dionne, M.; Behre, U.; Schulze, K.; Hillemanns, P.; Suryakiran, P.; Thomas, F.; et al. Sustained immunogenicity of the HPV-16/18 AS04-adjuvanted vaccine administered as a two-dose schedule in adolescent girls: Five-year clinical data and modeling predictions from a randomized study. Hum. Vaccines Immunother. 2016, 12, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Konno, R.; Tamura, S.; Dobbelaere, K.; Yoshikawa, H. Efficacy of human papillomavirus 16/18 AS04-adjuvanted vaccine in Japanese women aged 20 to 25 years: Interim analysis of a phase 2 double-blind, randomized, controlled trial. Int. J. Gynecol. Cancer 2010, 20, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Macias Parra, M.; Sierra, V.Y.; Salas Cespedes, A.; Granados, M.A.; Luque, A.; Karkada, N.; Castrejon Alba, M.M.; Romano-Mazzotti, L.; Borys, D.; et al. Long-term Immunogenicity and Safety of the AS04-adjuvanted Human Papillomavirus-16/18 Vaccine in Four- to Six-year-old Girls: Three-year Follow-up of a Randomized Phase III Trial. Pediatr. Infect. Dis. J. 2019, 38, 1061–1067. [Google Scholar] [CrossRef]
- Folschweiller, N.; Teixeira, J.; Joshi, S.; Goldani, L.Z.; Supparatpinyo, K.; Basu, P.; Chotpitayasunondh, T.; Chetchotisakd, P.; Ruxrungtham, K.; Roteli-Martins, C.; et al. Immunogenicity and safety of the AS04-HPV-16/18 and HPV-6/11/16/18 human papillomavirus vaccines in asymptomatic young women living with HIV aged 15–25 years: A phase IV randomized comparative study. EClinicalMedicine 2020, 23, 100353. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Adjuvant(s) |
|---|---|
| Mineral Compounds | Aluminum salt * |
| Calcium salt | |
| Microbial/Bacterial Products | Flagellin |
| Lipopolysaccharide | |
| Cholera toxin (CT) | |
| Bacillus Calmette-Guérin (BCG) | |
| Emulsions | CFA/IFA |
| Montanides | |
| MF59 * | |
| GLA-SE | |
| TiterMax | |
| RIBI | |
| Immunostimulatory complexes | Cytokines |
| Chemokines | |
| Particulate | Imidazoquinolines |
| Virus-like particles/Virosomes | |
| Synthetic polynucleotides (e.g., CpG *) | |
| Liposomes | |
| Polysaccharides | |
| Polymeric nanoparticle adjuvants | |
| Glycosphingolipids (NK agonists) | |
| Tesoactive | Saponin-based |
| Protease | Papain |
| Combination Adjuvant Systems (AS) | AS01 *, AS02, AS03 *, AS04 * |
| Adjuvant Class | Immune Response(s) | Experimental Vaccine Details | Development Stage | Ref. |
|---|---|---|---|---|
| Mineral Adjuvants | ||||
| Aluminium salt (phosphate or hydroxide, Alum) | Enhanced cytokine, chemokine, antibody, and Th2 immune response | PiCoVacc (NCT04456595) | Phase 3 | [38] |
| BG505 SOSIP.664 gp140 (NTC04177355) | Phase 1 | [39] | ||
| Respiratory syncytial virus (RSV) (NCT01905215) | Phase 1 | [40] | ||
| RSV F vaccine (NCT01704365) | Phase 2 | [41] | ||
| RSV F vaccine (NCT02247726) | Phase 3 | [42] | ||
| HIV AIDSVAX B/E (NCT00006327) | Phase 3 | [43] | ||
| SCB-2019 (NCT04405908) | Phase 1 | [44] | ||
| Calcium salt | Herpes simplex virus (HSV)-2 vaccine | Pre-clinical | [45,46] | |
| Human enterovirus-71 virus (HEV-71) | [47] | |||
| Newcastle disease virus (RDVF) vaccine | [48] | |||
| Foot and mouth disease virus (FMDV) DNA vaccine | [49] | |||
| Microbial/Bacterial Products—Flagellin | ||||
| Flagellin | Enhanced Th1 and Th2 immune responses (toll-like receptor [TLR] 5 agonist); pattern recognition receptor (PRR) activation; strong mucosal IgA/Th2/Th17 responses | VAX-102 (NCT00603811); | Phase 1 | [25,50] |
| VAX-125 (NCT00966238 and NCT00730457) | Phase 1 | [51,52] | ||
| VAX2012Q (NCT02015494) | Phase 1 | [53] | ||
| VAX2012Q (NCT02434276) | Phase 2 | [25] | ||
| Plague the flagellin-F1-V recombinant fusion protein (NCT01381744) | Phase 1 | [54] | ||
| Microbial/Bacterial Products—Lipopolysaccharides | ||||
| Glucopyranosyl lipid adjuvant-aqueous formulation (GLA-AF) | Enhanced Th1 immune responses (TLR1/2 agonist) | Influenza H5-VLP vaccine (NCT01657929) | Phase 1 | [55] |
| Microbial/Bacterial Products—Cholera toxin | ||||
| Cholera toxin (CT) | Enhanced Th1 and Th2 immune response | Recombinant Naegleria fowleri (rNfa1) protein-based vaccine | Pre-clinical | [56] |
| Malaria (Plasmodium vivax) ookinete surface protein (OSP), Pvs25 (AdPvs25) | [57] | |||
| HIV Envelope (Env) | [58] | |||
| Microbial/Bacterial Products—Bacillus Calmette-Guérin (BCG) | ||||
| Bacillus Calmette-Guérin (BCG) | Enhanced Th1 immune responses | Tuberculosis AERAS-404 and BCG revaccination with recombinant protein vaccines (H4:IC31 and H56:IC31) (NCT02378207; NCT02075203) | Phase 1/2 | [59,60,61] |
| COVID-19 vaccine (NCT04384549; NCT04328441; NCT04327206) | Phase 3 | [62,63] | ||
| Emulsion—Freund’s Complete/Incomplete Adjuvants | ||||
| Incomplete Freund’s adjuvant (IFA) | Enhanced Th1-or mixed Th1/Th17 and Th1/Th2 type immune responses | HIV-1 immunogen emulsified in IFA adjuvant, which was conducted on 25 participates (NCT00381875) | Phase 1 | [64] |
| Emulsion—Montanide | ||||
| ISA-51 | Enhanced antigen uptake by antigen-presenting cells and potent stimulator of adjuvant core response genes, and cytokine, chemokine and Th2 (antibody) responses | Mosquito borne disease anopheles gambiae saliva vaccine (NCT03055000) | Phase 1 | [65] |
| Emulsion—MF59® | ||||
| Squalene in oil-in-water emulsion (Squalene + Tween 80 + Span 85) | Enhanced Th1-or mixed Th1/Th17 and Th1/Th2-type immune responses | Seasonal influenza aIIV3; Fluad® (NCT04576702) | Phase 2 | [66] |
| SARS-CoV-2 Sclamp antigen combined with MF59® (NCT04495933) | Phase 1 | [67] | ||
| Emulsion—GLA-SE | ||||
| Glucopyranosyl lipid adjuvant (GLA) in combination with squalene (SE) | Strong Th1 immune response | Schistosoma Sm14 antigen combined with GLA-SE adjuvant (NCT01154049) | Phase 1 | [68] |
| Tuberculosis ID93 antigen combined with GLA-SE adjuvant (NCT01599897) | [69] | |||
| Emulsion—TiterMax | ||||
| TiterMax® | Enhanced Th2 immune response | Schistosoma mansoni venom allergen-like protein with TiterMax adjuvant | Pre-clinical | [12] |
| Emulsion—RIBI Adjuvant System | ||||
| RIBI adjuvant system (RAS) | Enhanced Th2 immune response | Recombinant influenza viral nucleoprotein combined with RAS® system | Pre-clinical | [70] |
| Mycobacterium paratuberculosis 85B antigen of MPT combined with RIBI adjuvant system | [71] | |||
| Immunostimulatory Complexes—Cytokines | ||||
| Granulocyte-macrophage colony-stimulating factor (GM-CSF) | Enhanced Th1/Th2/CD8+ T and mucosal IgA responses; immune responses, activation of dendritic cells; increased migration and antigen presentation to CD4+ T cells; cross-priming of CD8+ T cells; generation of Th1-biased CD4+ T cells | BRII-179 combined with IFN-α (ACTRN12619001210167). | Phase 1 | [72] |
| HIV Vacc-C5, containing residues 489–511 from the HIV-1 virus C5 domain, with GM-CSF adjuvant (NCT01627678) | Phase 1/2 | [73] | ||
| Dendritic cells loaded with S-protein from SARS-CoV-2 alongside GM-CSF (NCT04386252) | Phase 1 | [74] | ||
| Type 1 interferon (IFN) | BRII-179 combined with IFN-α (NCT04749368) | Phase 2 | [72] | |
| Interleukins (IL-1, IL-15, IL-2, and Il-18) | Influenza A viruses (H1N1 and H3N2) adjuvanted with INF-α (NCT00436046). | Phase 1 | [75] | |
| Immunostimulatory Complexes—Chemokines | ||||
| Chemokine CCL3 | Secretion of mucosal IgA and CTL | HIV-1 Gag antigen with murine chemokine CCL3 | Pre-clinical | [76] |
| Particulate—Imidazoquinolines | ||||
| Resiquimod (R-848) | Enhanced Th1 and Th2 immune responses (TLR7/8 agonist) and secretion of Type 1 IFN, pro-inflammatory cytokines, antibodies, and CD8+ T cells | Influenza IPR8-R848 | Pre-clinical | [77] |
| Imidazoquinolines | HSV RSV vaccine candidate | [78] | ||
| Particulate—Virus-like Particles/Virosomes | ||||
| Virus-like particles (VLPs) | Enhanced Th1, Th2, and CD8+ T cell immune response | Nine-valent human papilloma virus vaccine (V503-020) (NCT02114385) | Phase 3 | [79] |
| Chikungunya virus VRC-CHKVLP059-00-VP (NCT02562482) | Phase 2 | [80] | ||
| Recombinant hemagglutinin trivalent nanoparticle vaccine (tNIV) produced in Sf9 insect cell-recombinant baculovirus platform containing wide-type virus sequences composed of conserved H3N2 epitope, as an influenza vaccine candidate (NCT03293498). | Phase 1/2 | [81] | ||
| Virosomes | Enhanced Th1, Th2, and CD8+ T cell immune response | Combined HIV-1 virulence peptide antigens (gp41, p1) adjuvanted with virosomes (NCT04553016). | Phase 1 | [82] |
| Anti-malaria peptide vaccine derived from the circumsporozoite protein and apical antigen-1 (AMA-1) adjuvanted with virosomes (NCT00408668). | Phase 2 | [83] | ||
| Double stranded (ds)-RNA (e.g., Poly I:C; PolyI:C12U (Ampligen®) | Type I IFN induction; pro-inflammatory cytokine/chemokine/antibody/CD4+/CD8+ T cell responses; mucosal adjuvant inducing Th1 and Th17 immune responses | H5N1 influenza antigen was a formalin-inactivated whole virus (NIBRG14) derived from a recombinant avian virus from H1N5 combined with Poly I: Poly C12 U (Ampligen®) adjuvant (NCT00711295) | Phase 3 | [84] |
| Combined plasma HIV-1 RNA with the TLR-3 agonist Poly-ICLC adjuvant as an HIV vaccine (NCT00207195). | Phase 1 | [85] | ||
| RNA cyclic di-GMP oligodeoxynucleotide (CpG ODN) (e.g., unmethylated CpG DNA, CpG ODN; CpG 1018; CpG 7909; IC31) | PRR activation; potent inducer of ‘adjuvant core response genes’ | Anthrax AVA vaccine (NCT01263691) | Phase 1 | [86] |
| Malaria vaccine (NCT00344539) | Phase 1 | [87] | ||
| Tuberculosis H4:IC31(ASERAS-404) (NCT02066428 and NCT02074956) | Phase 1/2 | [88] | ||
| Particulate—Liposomes | ||||
| Cationic liposomes (e.g., CAF01) | Immunostimulatory depot effect; strong antigen-specific antibody and Th1/Th2 cell responses | COVID-19 mRNA-1273 vaccine—lipid nanoparticle-encapsulated with nucleoside-modified mRNA which encodes the SARS-CoV-2 spike (s) glycoprotein (NCT04470427) | Phase 3 | [89] |
| Reformed liposomes | Tuberculosis hybrid 1(H1) fusion protein adjuvanted with CAF01 liposome (NCT00922363) | Phase 1 | [90] | |
| Chlamydia trachomatis recombinant protein subunit CTH 522 adjuvanted with CAF01 liposome (NCT02787109) | Phase 1 | [91] | ||
| Particulate—Polysaccharides | ||||
| Chitosan | Enhanced Th1 and Th2 immune response and mucosal immune response; site-directed delivery of antigens; source of DAMP (chitosan); mucosal (chitosan); induction of pro-inflammatory cytokines and secretory antibody responses; Th2 and mucosal IgA responses; PRR activation; upregulation of co-stimulatory molecules; activation of complement pathways. | Norwalk virus MPL/chitosan-VLP vaccine (NCT00973284) | Phase 2 | [92] |
| AdvaxTM | Hepatitis B surface antigen (HBsAg) combined with Advax (ACTRN12607000598482) | Phase 1 | [93] | |
| HIV-1 vaccine candidate plus Advax adjuvant (NCT00249106) | Phase 1 | [94] | ||
| Particulate—Polymeric nanoparticles | ||||
| Polymeric nanoparticles e.g., PLA: poly (lactic acid); PLGA: poly (lactic-co-glycolic acid) | Enhanced Th1 and Th2 immune response; depot effect; mucoadhesive; strong antigen specific Th1/Th2, CD8+ T cell a and antibody responses | MPL/chitosan-VLP HIV vaccine (NCT00973284) | Phase 2 | [92,95] |
| HBV antigen (HBsAg) has been conducted into healthy adults (ACTRN12607000598482). | Phase 1 | [93] | ||
| HIV-1 vaccine candidate plus Advax adjuvant (NCT00249106) | Phase 1 | [94] | ||
| Particulate—Glycosphingolipids | ||||
| Glycosphingolipids (α-GalCer) | Pattern recognition receptor activation; Th1/CTL and mucosal IgA responses | Combined HBV protein antigen with synthetic α-GalCer (KRN 7000), intravenously administrated into participants against HBV (NCT00363155). | Phase 2 | [96] |
| Tensoactive—Saponin | ||||
| Matrix-MTM | Induction of cytokines and cellular influx; induction of Th1- or mixed Th1/Th17-, Th1/Th1-type as well as strong antibody (CD8+ T cell) responses | Matrix-M adjuvant combined with the full-length SARS-CoV-2 spike (S) glycoprotein (NCT04368988) | Phase 1/2 | [97] |
| COVID-19 NVX-CoV2373 (EudraCT number, 2020-004123-16) | Phase 3 | [98] | ||
| Immune-stimulating complexes (ISCOMs) | ||||
| QS-21 | H7N9 influenza (NCT01897701) | Phase 1 | [99] | |
| Protease—Papain-like Cysteine Proteases | ||||
| Papain-like cysteine proteases | Enhanced T helper (Th) 1 and Th17 immune responses | Schistosomiasis glyceraldehyde 3-phosphate dehydrogenase (SG3PDH), peroxiredoxin (TPX), and other larval excretory–secretory products (ESP) with Papain | Pre-clinical | [100] |
| Combination Adjuvant Systems | ||||
| AS01(MPL, QS-21 and liposome) | Transient induction of cytokines at the site of injection; increased influx of antigen-loaded monocytes in draining lymph nodes (dLNs) | RTS, S/AS01 malaria (NCT00866619) | Phase 3 | [101] |
| AS02 (3-O-desacyl-4′-monophosphoryl lipid A [MPL] and QS-21) | Malaria FMP2.1/AS02A vaccine (NCT00460525) | Phase 2 | [102] | |
| AS03 (Squalene, polysorbate 80 and α-tocopherol) | SK SARS-CoV-2 recombinant protein nanoparticle vaccine (GBP 510) combined with AS03 (NCT04742738) and (NCT04750343) | Phase 1/2 | [103,104] | |
| Influenza A (H1N1) pdm09 vaccines combined with AS03 (NCT00616928) | Phase 3 | [105,106] | ||
| AS04 (MPL adsorbed onto aluminium hydroxide or aluminium phosphate) | HPV-16/-18 VLP adjuvanted with AS04 (NCT00128661) | Phase 3 | [107] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, J.; Jin, S.; Gilmartin, L.; Toth, I.; Hussein, W.M.; Stephenson, R.J. Advances in Infectious Disease Vaccine Adjuvants. Vaccines 2022, 10, 1120. https://doi.org/10.3390/vaccines10071120
Fan J, Jin S, Gilmartin L, Toth I, Hussein WM, Stephenson RJ. Advances in Infectious Disease Vaccine Adjuvants. Vaccines. 2022; 10(7):1120. https://doi.org/10.3390/vaccines10071120
Chicago/Turabian StyleFan, Jingyi, Shengbin Jin, Lachlan Gilmartin, Istvan Toth, Waleed M. Hussein, and Rachel J. Stephenson. 2022. "Advances in Infectious Disease Vaccine Adjuvants" Vaccines 10, no. 7: 1120. https://doi.org/10.3390/vaccines10071120
APA StyleFan, J., Jin, S., Gilmartin, L., Toth, I., Hussein, W. M., & Stephenson, R. J. (2022). Advances in Infectious Disease Vaccine Adjuvants. Vaccines, 10(7), 1120. https://doi.org/10.3390/vaccines10071120