
A Cloned Recombinant Vesicular Stomatitis Virus-Vectored Marburg Vaccine, PHV01, Protects Guinea Pigs from Lethal Marburg Virus Disease

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Ethics and Biosafety Statement
2.2. Viruses and Cells
2.3. VSV-Based Vaccines
2.4. Guinea Pig Study Design
2.5. VSV and MARV RNA Quantification
2.6. IgG ELISAs
2.7. Plaque Reduction Neutralization Test (PRNT)
2.8. Statistical Analyses
3. Results
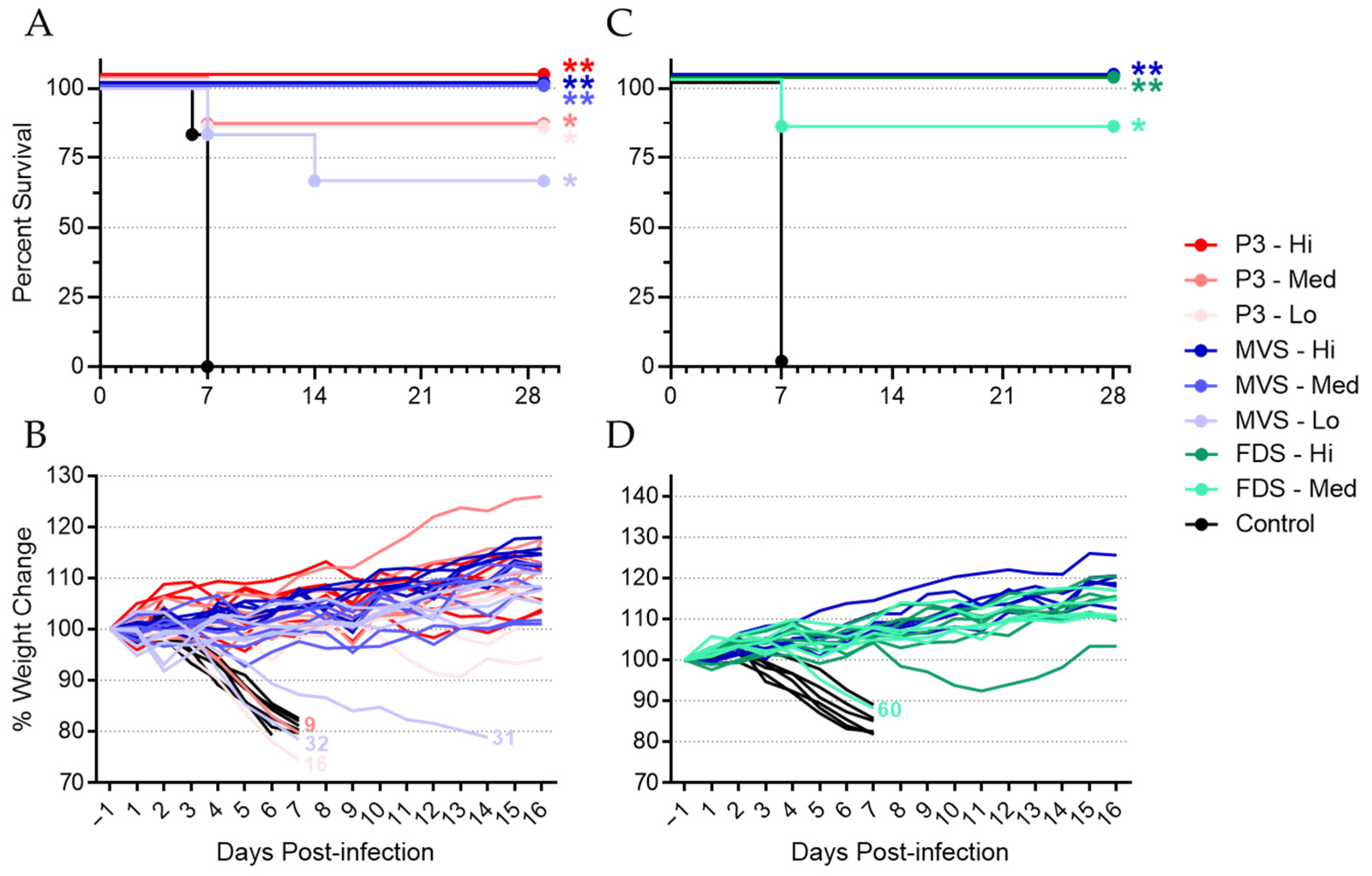
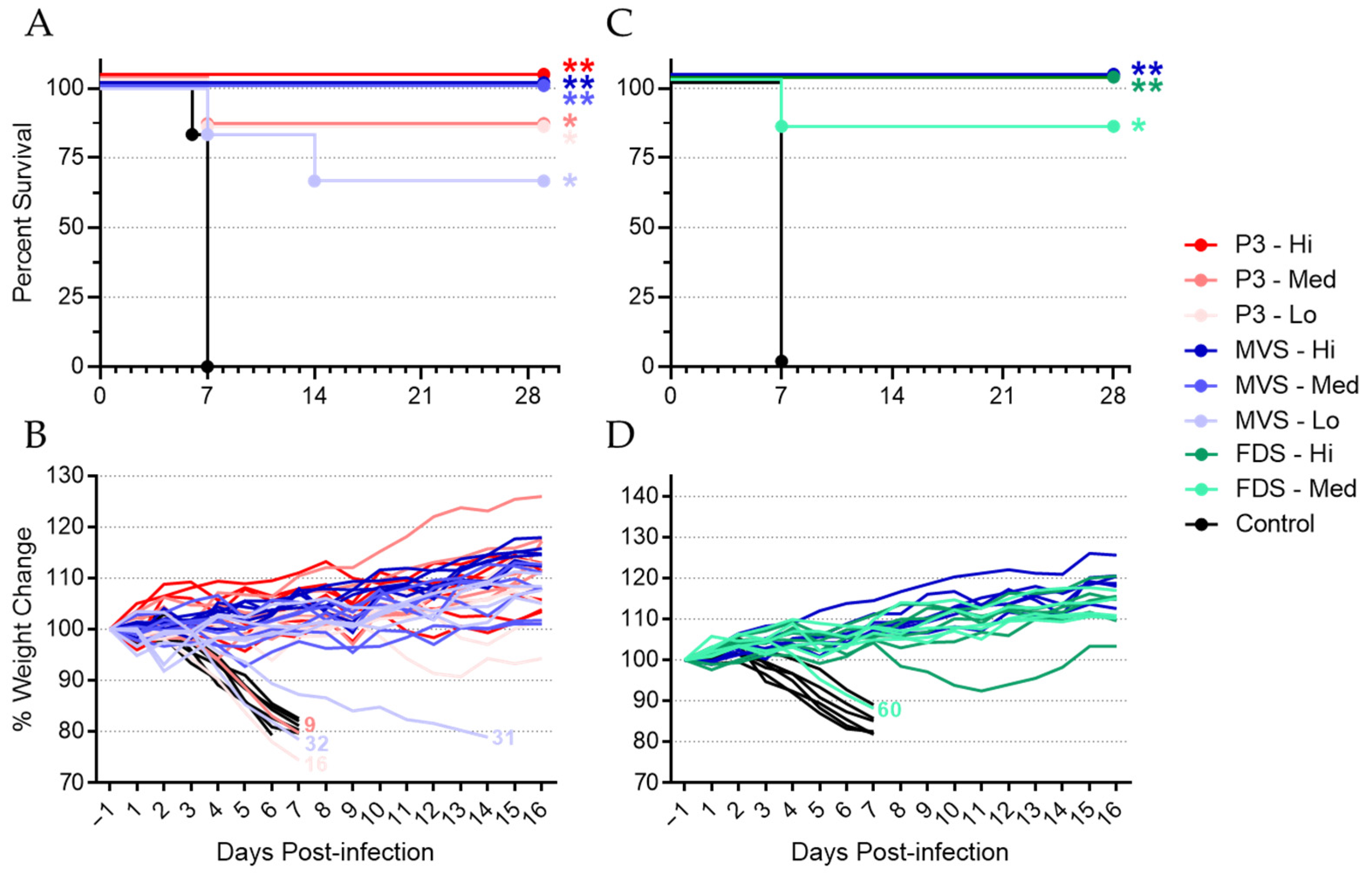
3.1. Vaccination with PHV01 Protects Guinea Pigs from Lethal GPA-MARV/Ang Infection
3.2. Vaccinated Animals Exhibited Transient Vaccinemia
3.3. The Majority of Vaccinated Animals Exhibit No MARV Viremia
3.4. Vaccinated Animals Mount a Robust Humoral Response to MARV GP
3.5. Vaccinated Animals Exhibit a Neutralizing Antibody Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuhn, J.H.; Amarasinghe, G.K.; Perry, D.L. Filoviridae. In Fields Virology: Emerging Viruses; Howley, P.M., Knipe, D.M., Whelan, S., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2021. [Google Scholar]
- Mehedi, M.; Groseth, A.; Feldmann, H.; Ebihara, H. Clinical aspects of Marburg hemorrhagic fever. Future Virol. 2011, 6, 1091–1106. [Google Scholar] [CrossRef] [Green Version]
- Brauburger, K.; Hume, A.J.; Mühlberger, E.; Olejnik, J. Forty-five years of marburg virus research. Viruses 2012, 4, 1878–1927. [Google Scholar] [CrossRef] [Green Version]
- Towner, J.S.; Khristova, M.L.; Sealy, T.K.; Vincent, M.J.; Erickson, B.R.; Bawiec, D.A.; Hartman, A.L.; Comer, J.A.; Zaki, S.R.; Ströher, U.; et al. Marburgvirus Genomics and Association with a Large Hemorrhagic Fever Outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar] [CrossRef] [Green Version]
- Geisbert, T.W.; Strong, J.E.; Feldmann, H. Considerations in the Use of Nonhuman Primate Models of Ebola Virus and Marburg Virus Infection. J. Infect. Dis. 2015, 212, S91–S97. [Google Scholar] [CrossRef] [Green Version]
- Guinea Declares End of Marburg Virus Disease Outbreak|WHO|Regional Office for Africa. Available online: https://www.afro.who.int/news/guinea-declares-end-marburg-virus-disease-outbreak (accessed on 27 September 2021).
- World-Health-Organization Prioritizing Diseases for Research and Development in Emergency Contexts. Available online: https://www.who.int/activities/prioritizing-diseases-for-research-and-development-in-emergency-context (accessed on 28 March 2022).
- Reynolds, P.; Marzi, A. Ebola and Marburg virus vaccines. Virus Genes 2017, 53, 501–515. [Google Scholar] [CrossRef]
- Garbutt, M.; Liebscher, R.; Wahl-Jensen, V.; Jones, S.; Möller, P.; Wagner, R.; Volchkov, V.; Klenk, H.-D.; Feldmann, H.; Ströher, U. Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses. J. Virol. 2004, 78, 5458–5465. [Google Scholar] [CrossRef] [Green Version]
- Ollmann Saphire, E. A Vaccine against Ebola Virus. Cell 2020, 181, 6. [Google Scholar] [CrossRef]
- Matassov, D.; Mire, C.E.; Latham, T.; Geisbert, J.B.; Xu, R.; Ota-Setlik, A.; Agans, K.N.; Kobs, D.J.; Wendling, M.Q.S.; Burnaugh, A.; et al. Single-Dose Trivalent VesiculoVax Vaccine Protects Macaques from Lethal Ebolavirus and Marburgvirus Challenge. J. Virol. 2018, 92, e01190-17. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.M.; Feldmann, H.; Ströher, U.; Geisbert, J.B.; Fernando, L.; Grolla, A.; Klenk, H.D.; Sullivan, N.J.; Volchkov, V.E.; Fritz, E.A.; et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 2005, 11, 786–790. [Google Scholar] [CrossRef]
- Mire, C.E.; Geisbert, J.B.; Agans, K.N.; Satterfield, B.A.; Versteeg, K.M.; Fritz, E.A.; Feldmann, H.; Hensley, L.E.; Geisbert, T.W. Durability of a vesicular stomatitis virus-based marburg virus vaccine in nonhuman primates. PLoS ONE 2014, 9, e94355. [Google Scholar] [CrossRef]
- Mire, C.E.; Miller, A.D.; Carville, A.; Westmoreland, S.V.; Geisbert, J.B.; Mansfield, K.G.; Feldmann, H.; Hensley, L.E.; Geisbert, T.W. Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates. PLoS Negl. Trop. Dis. 2012, 6, e1567. [Google Scholar] [CrossRef] [PubMed]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Ströher, U.; Geisbert, J.B.; Grolla, A.; Fritz, E.A.; Fernando, L.; Kagan, E.; Jahrling, P.B.; Hensley, L.E.; et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: An efficacy assessment. Lancet 2006, 367, 1399–1404. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Geisbert, J.B.; Leung, A.; Johnson, J.C.; Grolla, A.; Feldmann, H. Postexposure treatment of marburg virus infection. Emerg. Infect. Dis. 2010, 16, 1119–1122. [Google Scholar] [CrossRef]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Geisbert, J.B.; Ströher, U.; Hensley, L.E.; Grolla, A.; Fritz, E.A.; Feldmann, F.; Feldmann, H.; Jones, S.M. Cross-Protection against Marburg Virus Strains by Using a Live, Attenuated Recombinant Vaccine. J. Virol. 2006, 80, 9659–9666. [Google Scholar] [CrossRef] [Green Version]
- Marzi, A.; Menicucci, A.R.; Engelmann, F.; Callison, J.; Horne, E.J.; Feldmann, F.; Jankeel, A.; Feldmann, H.; Messaoudi, I. Protection against marburg virus using a recombinant VSV-vaccine depends on T and B cell activation. Front. Immunol. 2019, 10, 3071. [Google Scholar] [CrossRef] [Green Version]
- Marzi, A.; Jankeel, A.; Menicucci, A.R.; Callison, J.; O’Donnell, K.L.; Feldmann, F.; Pinski, A.N.; Hanley, P.W.; Messaoudi, I. Single Dose of a VSV-Based Vaccine Rapidly Protects Macaques From Marburg Virus Disease. Front. Immunol. 2021, 12, 774026. [Google Scholar] [CrossRef]
- Woolsey, C.; Geisbert, J.B.; Matassov, D.; Agans, K.N.; Borisevich, V.; Cross, R.W.; Deer, D.J.; Fenton, K.A.; Eldridge, J.H.; Mire, C.E.; et al. Postexposure Efficacy of Recombinant Vesicular Stomatitis Virus Vectors Against High and Low Doses of Marburg Virus Variant Angola in Nonhuman Primates. J. Infect. Dis. 2018, 218, S582–S587. [Google Scholar] [CrossRef] [Green Version]
- Preparing for the next pandemic. Nat. Med. 2021, 27, 357. [CrossRef]
- Wong, G.; Cao, W.G.; He, S.H.; Zhang, Z.R.; Zhu, W.J.; Moffat, E.; Ebihara, H.; Embury-Hyatt, C.; Qiu, X.G. Development and characterization of a guinea pig model for Marburg virus. Zool. Res. 2018, 39, 32–41. [Google Scholar] [CrossRef]
- Liu, G.; Cao, W.; Salawudeen, A.; Zhu, W.; Emeterio, K.; Safronetz, D.; Banadyga, L. Vesicular stomatitis virus: From agricultural pathogen to vaccine vector. Pathogens 2021, 10, 1092. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Geisbert, J.B.; Reed, D.S.; Feldmann, F.; Grolla, A.; Ströher, U.; Fritz, E.A.; Hensley, L.E.; Jones, S.M.; et al. Vesicular stomatitis virus-based vaccines protect nonhuman primates against aerosol challenge with Ebola and Marburg viruses. Vaccine 2008, 26, 6894–6900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisbert, T.W.; Geisbert, J.B.; Leung, A.; Daddario-DiCaprio, K.M.; Hensley, L.E.; Grolla, A.; Feldmann, H. Single-Injection Vaccine Protects Nonhuman Primates against Infection with Marburg Virus and Three Species of Ebola Virus. J. Virol. 2009, 83, 7296–7304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawęska, J.T.; Storm, N.; Markotter, W.; Di Paola, N.; Wiley, M.R.; Palacios, G.; van Vuren, P.J. Shedding of marburg virus in naturally infected egyptian rousette bats, South Africa, 2017. Emerg. Infect. Dis. 2020, 26, 3051–3055. [Google Scholar] [CrossRef] [PubMed]
- Pawęska, J.T.; Jansen van Vuren, P.; Kemp, A.; Storm, N.; Grobbelaar, A.A.; Wiley, M.R.; Palacios, G.; Markotter, W. Marburg virus infection in egyptian rousette bats, South Africa, 2013–2014. Emerg. Infect. Dis. 2018, 24, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, I.V.; Niezgoda, M.; Franka, R.; Agwanda, B.; Markotter, W.; Breiman, R.F.; Shieh, W.J.; Zaki, S.R.; Rupprecht, C.E. Marburg virus in fruit bat, Kenya. Emerg. Infect. Dis. 2010, 16, 352–354. [Google Scholar] [CrossRef]
- Kajihara, M.; Hang’Ombe, B.M.; Changula, K.; Harima, H.; Isono, M.; Okuya, K.; Yoshida, R.; Mori-Kajihara, A.; Eto, Y.; Orba, Y.; et al. Marburgvirus in Egyptian fruit bats, Zambia. Emerg. Infect. Dis. 2019, 25, 1577–1580. [Google Scholar] [CrossRef] [Green Version]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Reeder Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef]
- Amman, B.R.; Carroll, S.A.; Reed, Z.D.; Sealy, T.K.; Balinandi, S.; Swanepoel, R.; Kemp, A.; Erickson, B.R.; Comer, J.A.; Campbell, S.; et al. Seasonal Pulses of Marburg Virus Circulation in Juvenile Rousettus aegyptiacus Bats Coincide with Periods of Increased Risk of Human Infection. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Amman, B.R.; Bird, B.H.; Bakarr, I.A.; Bangura, J.; Schuh, A.J.; Johnny, J.; Sealy, T.K.; Conteh, I.; Koroma, A.H.; Foday, I.; et al. Isolation of Angola-like Marburg virus from Egyptian rousette bats from West Africa. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Timen, A. Response to Imported Case of Marburg Hemorrhagic Fever, the Netherlands. Emerg. Infect. Dis. 2009, 15, 1171–1175. [Google Scholar] [CrossRef]
- Leggiadro, R.J. Imported Case of Marburg Hemorrhagic Fever—Colorado, 2008. Pediatr. Infect. Dis. J. 2010, 29, 400. [Google Scholar] [CrossRef]
- Dulin, N.; Spanier, A.; Merino, K.; Hutter, J.N.; Waterman, P.E.; Lee, C.; Hamer, M.J. Systematic review of Marburg virus vaccine nonhuman primate studies and human clinical trials. Vaccine 2021, 39, 202–208. [Google Scholar] [CrossRef]
- Marzi, A.; Engelmann, F.; Feldmann, F.; Haberthur, K.; Shupert, W.L.; Brining, D.; Scott, D.P.; Geisbert, T.W.; Kawaoka, Y.; Katze, M.G.; et al. Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc. Natl. Acad. Sci. USA 2013, 110, 1893–1898. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Malherbe, D.C.; Bukreyev, A. Can Ebola Virus Vaccines Have Universal Immune Correlates of protection? Trends Microbiol. 2019, 27, 8–16. [Google Scholar] [CrossRef]
- Meyer, M.; Gunn, B.M.; Malherbe, D.C.; Gangavarapu, K.; Yoshida, A.; Pietzsch, C.; Kuzmina, N.A.; Saphire, E.O.; Collins, P.L.; Crowe, J.E.; et al. Ebola vaccine-induced protection in nonhuman primates correlates with antibody specificity and Fc-mediated effects. Sci. Transl. Med. 2021, 13, 6128. [Google Scholar] [CrossRef]
- Wagstaffe, H.R.; Clutterbuck, E.A.; Bockstal, V.; Stoop, J.N.; Luhn, K.; Douoguih, M.; Shukarev, G.; Snape, M.D.; Pollard, A.J.; Riley, E.M.; et al. Antibody-Dependent Natural Killer Cell Activation after Ebola Vaccination. J. Infect. Dis. 2021, 223, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Gunn, B.M.; Roy, V.; Karim, M.M.; Hartnett, J.N.; Suscovich, T.J.; Goba, A.; Momoh, M.; Sandi, J.D.; Kanneh, L.; Andersen, K.G.; et al. Survivors of ebola virus disease develop polyfunctional antibody responses. J. Infect. Dis. 2020, 221, 156–161. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Liu, G.; Cao, W.; He, S.; Leung, A.; Ströher, U.; Fairchild, M.J.; Nichols, R.; Crowell, J.; Fusco, J.; et al. A Cloned Recombinant Vesicular Stomatitis Virus-Vectored Marburg Vaccine, PHV01, Protects Guinea Pigs from Lethal Marburg Virus Disease. Vaccines 2022, 10, 1004. https://doi.org/10.3390/vaccines10071004
Zhu W, Liu G, Cao W, He S, Leung A, Ströher U, Fairchild MJ, Nichols R, Crowell J, Fusco J, et al. A Cloned Recombinant Vesicular Stomatitis Virus-Vectored Marburg Vaccine, PHV01, Protects Guinea Pigs from Lethal Marburg Virus Disease. Vaccines. 2022; 10(7):1004. https://doi.org/10.3390/vaccines10071004
Chicago/Turabian StyleZhu, Wenjun, Guodong Liu, Wenguang Cao, Shihua He, Anders Leung, Ute Ströher, Michael J. Fairchild, Rick Nichols, Joseph Crowell, Joan Fusco, and et al. 2022. "A Cloned Recombinant Vesicular Stomatitis Virus-Vectored Marburg Vaccine, PHV01, Protects Guinea Pigs from Lethal Marburg Virus Disease" Vaccines 10, no. 7: 1004. https://doi.org/10.3390/vaccines10071004
APA StyleZhu, W., Liu, G., Cao, W., He, S., Leung, A., Ströher, U., Fairchild, M. J., Nichols, R., Crowell, J., Fusco, J., & Banadyga, L. (2022). A Cloned Recombinant Vesicular Stomatitis Virus-Vectored Marburg Vaccine, PHV01, Protects Guinea Pigs from Lethal Marburg Virus Disease. Vaccines, 10(7), 1004. https://doi.org/10.3390/vaccines10071004