
Prediction of Novel Drug Targets and Vaccine Candidates against Human Lice (Insecta), Acari (Arachnida), and Their Associated Pathogens


 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Proteomes Retrieval of Vectors, Host, and Pathogens
2.2. Removal of Duplicate Sequences
2.3. Non-Host Homologous Proteins
2.4. Identification of Essential Proteins
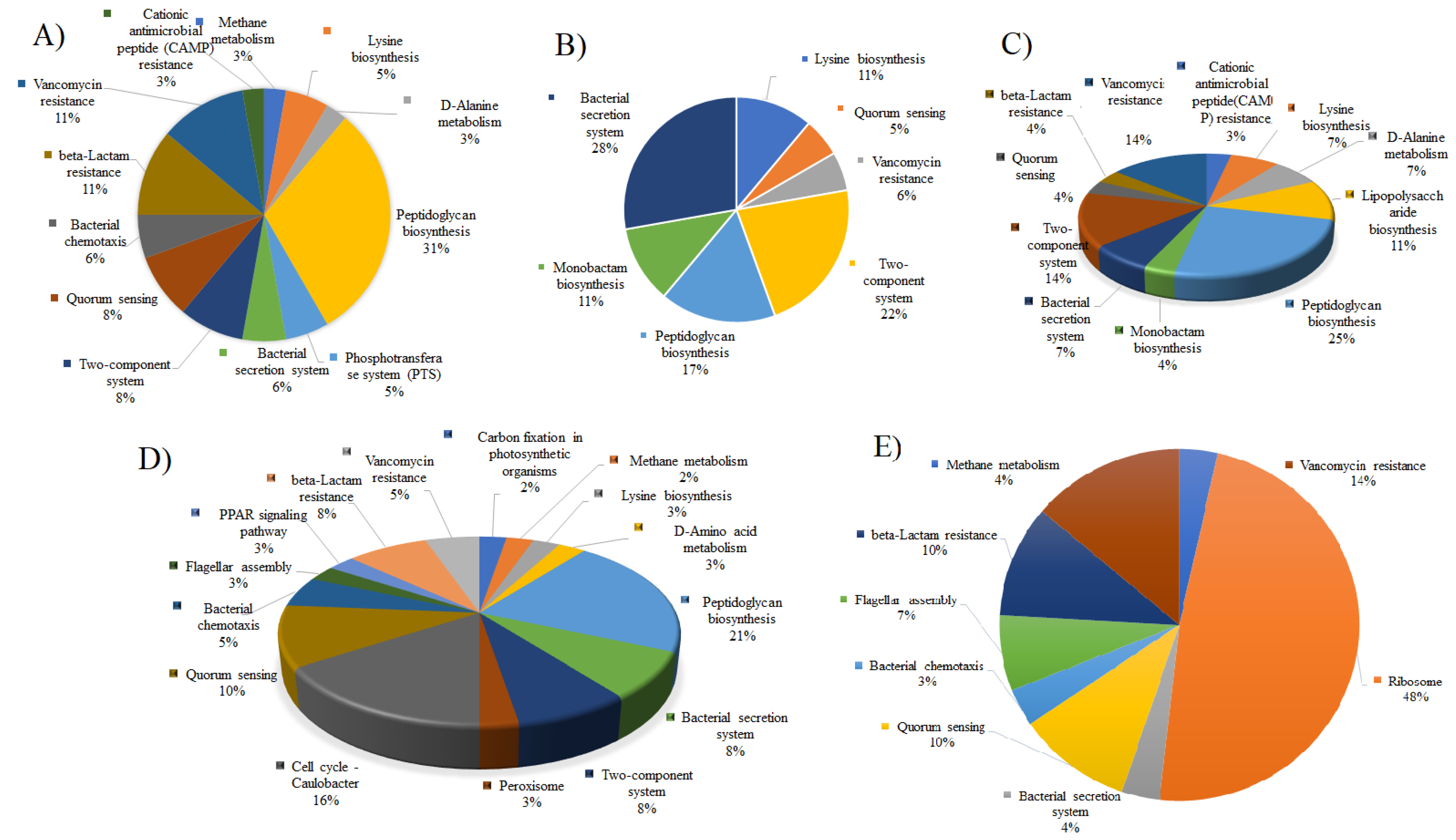
2.5. Analysis of Metabolic Pathway
2.6. Druggability Evaluation of Essential and Unique Proteins
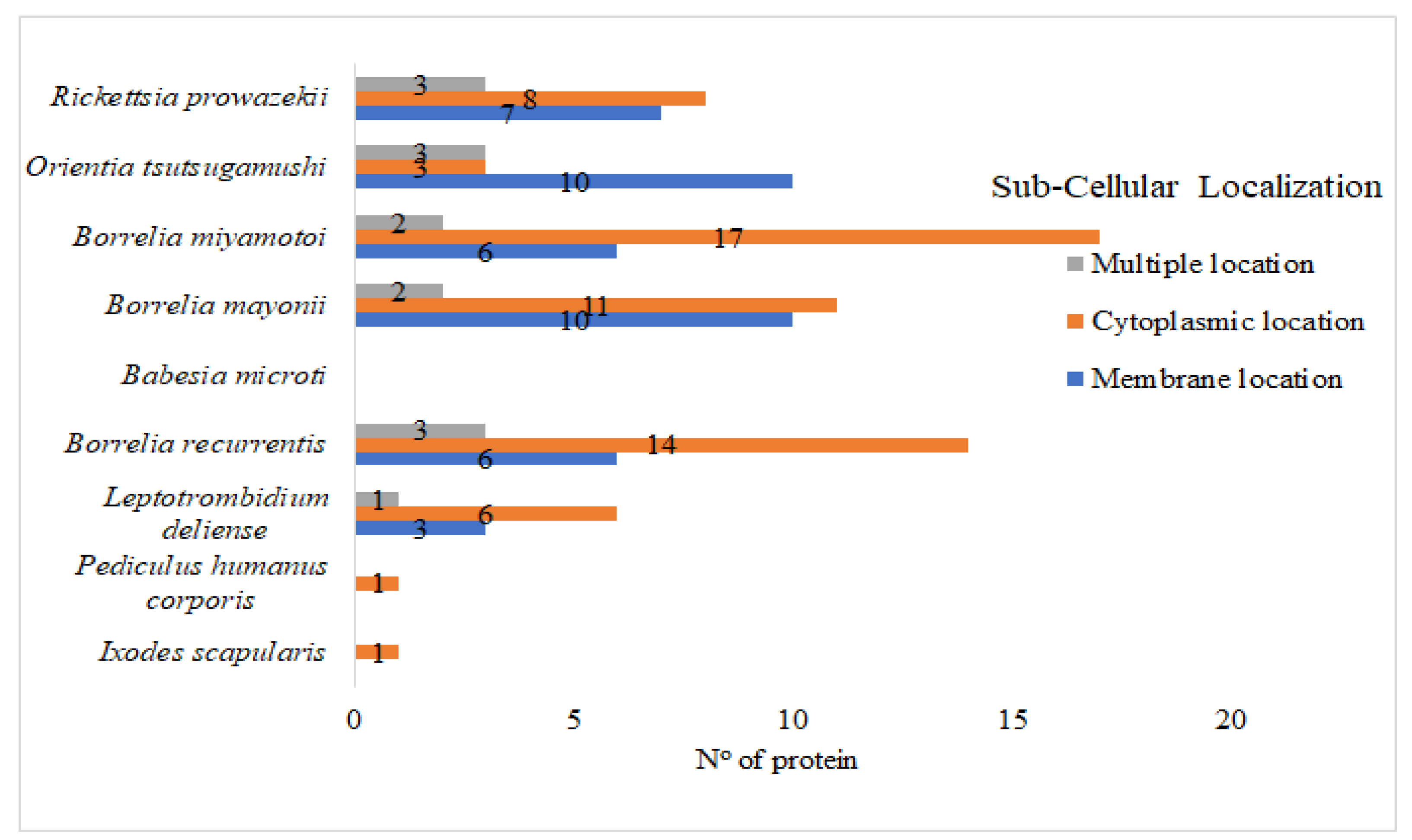
2.7. Subcellular Localization
2.8. Virulence of Non-Host Homologous Unique Target Proteins
2.9. Protein Functional Family Classification
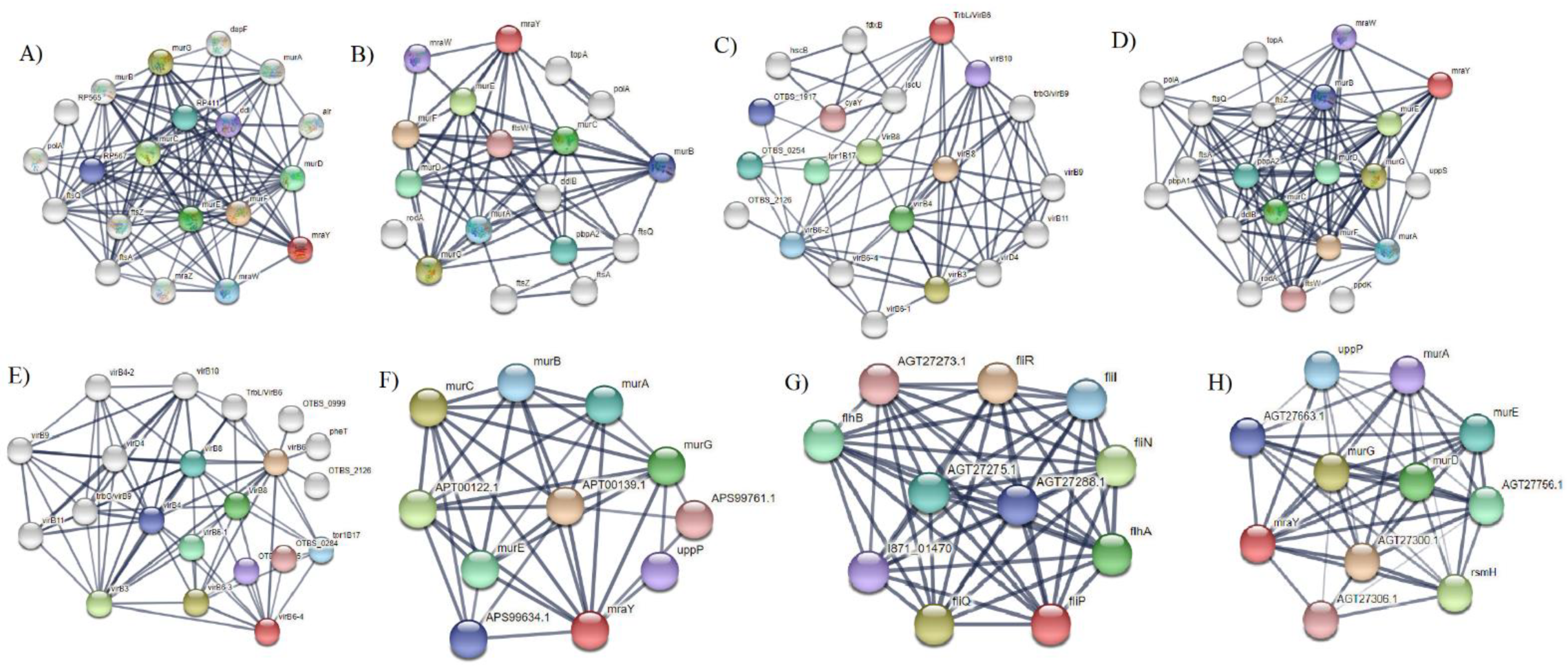
2.10. Protein–Protein Interaction of Selected Membrane Proteins
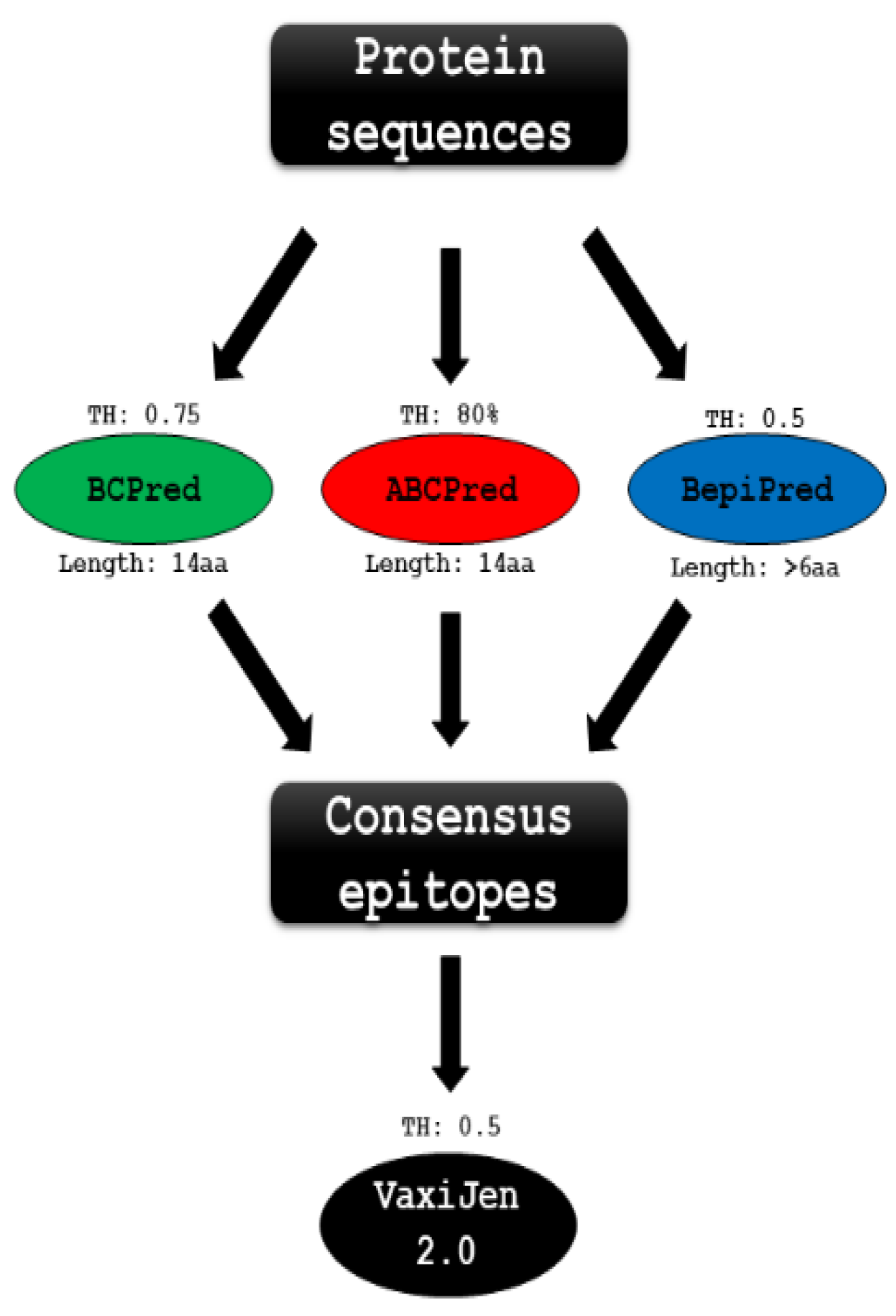
2.11. B-Cell Linear Epitopes Prediction
2.12. D Structure Prediction
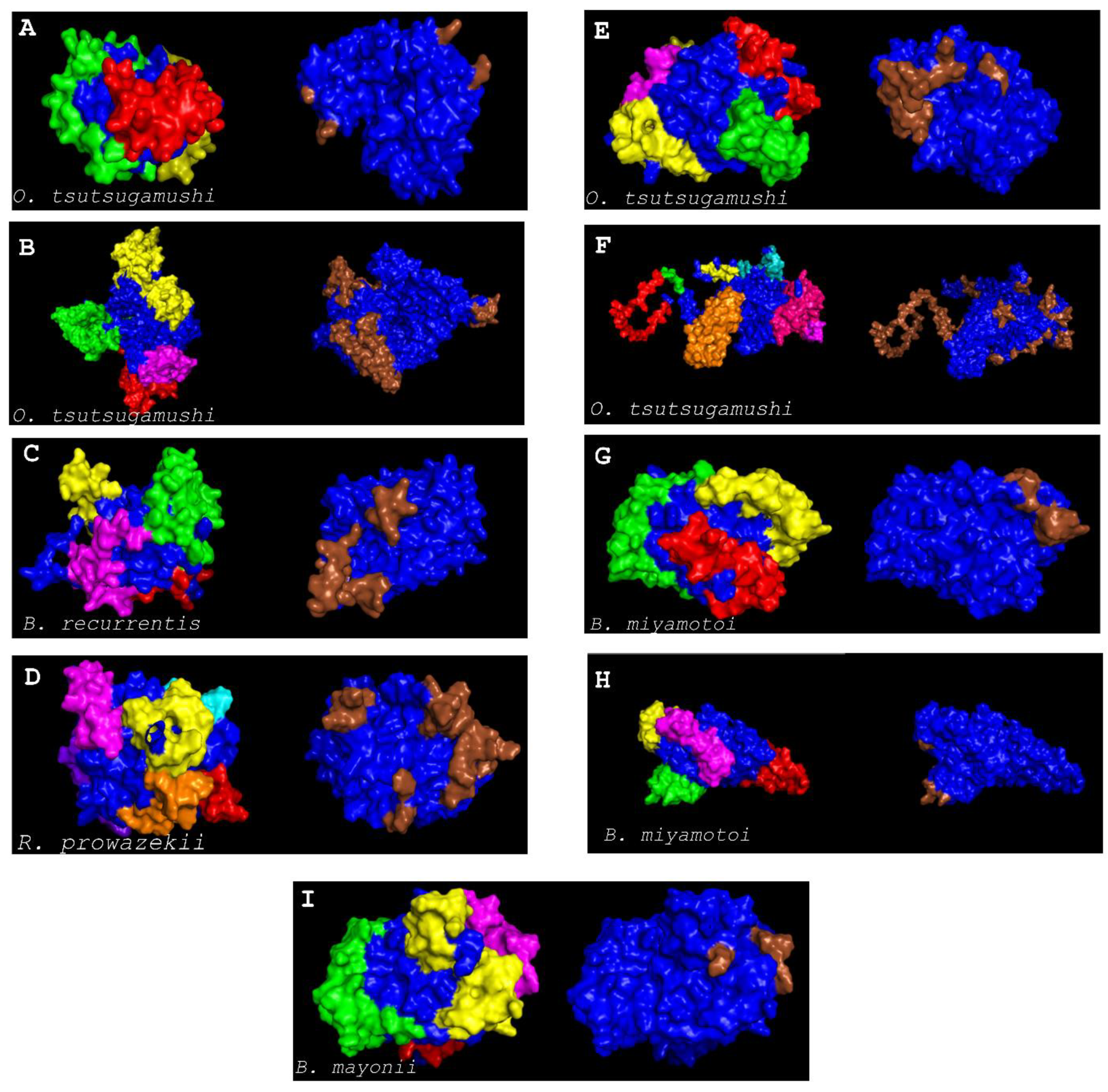
2.13. Discontinuous B-Cell Epitope Prediction
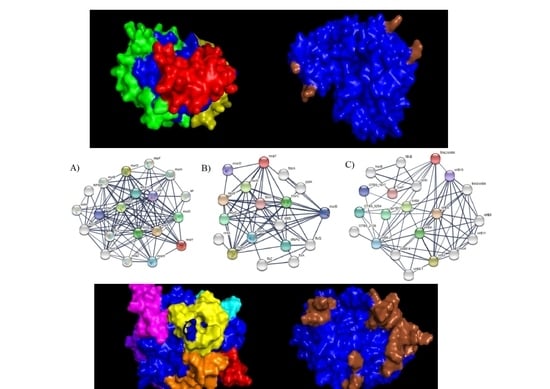
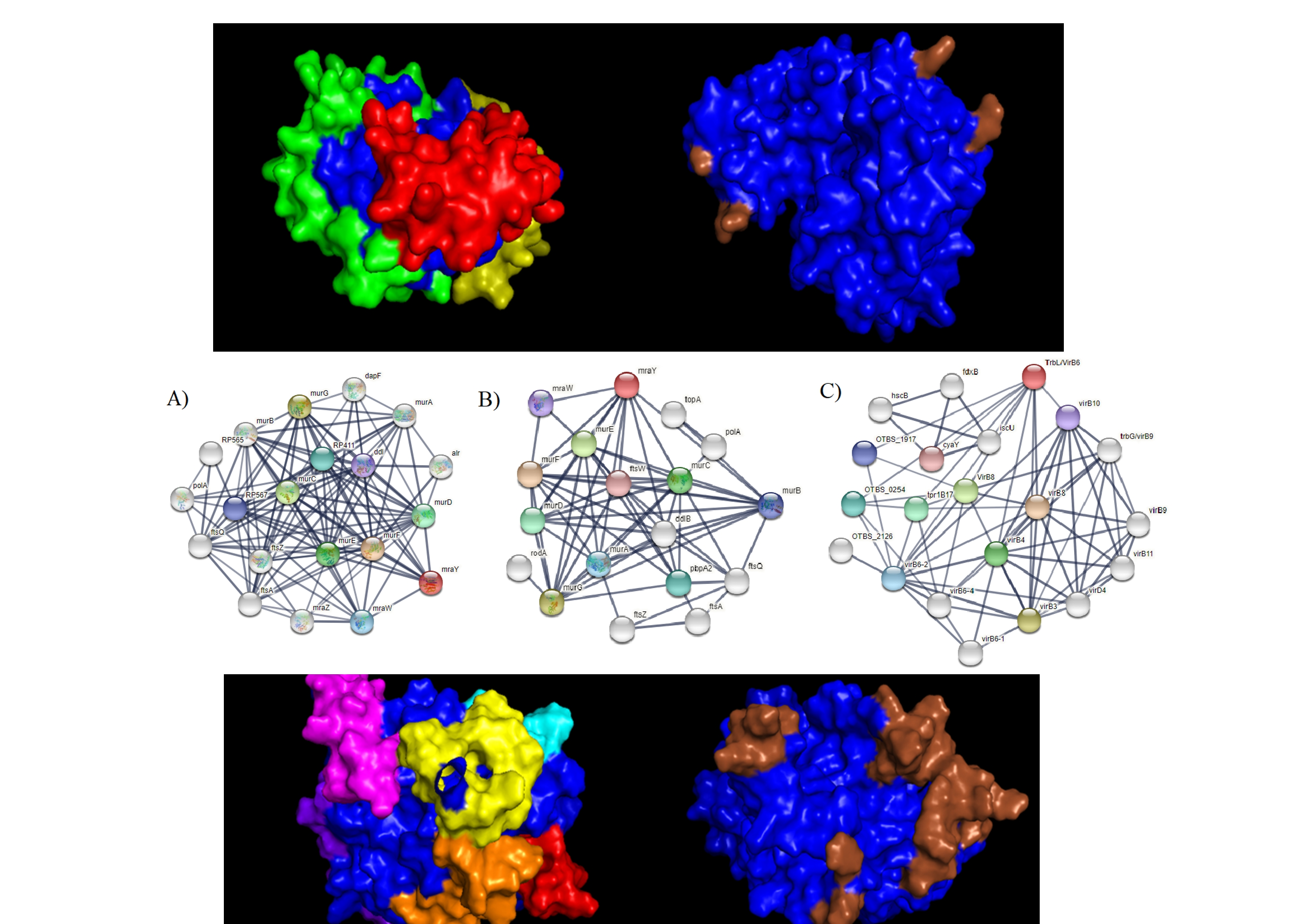
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ogden, N. Changing Geographic Ranges of Ticks and Tick-Borne Pathogens: Drivers, Mechanisms and Consequences for Pathogen Diversity. Front. Cell. Infect. Microbiol. 2013, 3, 46. [Google Scholar] [CrossRef]
- Ali, A.; Mulenga, A.; Vaz, I.S., Jr. Tick and Tick-Borne Pathogens: Molecular and Immune Targets for Control Strategies. Front. Physiol. 2020, 11, 744. [Google Scholar] [CrossRef]
- Parola, P.; Raoult, D. Ticks and Tickborne Bacterial Diseases in Humans: An Emerging Infectious Threat. Clin. Infect. Dis. 2001, 32, 897–928. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, J.; Gershenzon, J.; Baldwin, I.T.; Kessler, A. Attracting Friends to Feast on Foes: Engineering Terpene Emission to Make Crop Plants More Attractive to Herbivore Enemies. Curr. Opin. Biotechnol. 2003, 14, 169–176. [Google Scholar] [CrossRef]
- Sonenshine, D.E.; Anderson, J.M.; Roe, R.M. Mouthparts and Digestive System; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Phasomkusolsil, S.; Tanskul, P.; Ratanatham, S.; Watcharapichat, P.; Phulsuksombati, D.; Frances, S.P.; Lerdthusnee, K.; Linthicum, K.J. Influence of Orientia tsutsugamushi Infection on the Developmental Biology of Leptotrombidium imphalum and Leptotrombidium chiangraiensis (Acari: Trombiculidae). J. Med. Entomol. 2014, 49, 1270–1275. [Google Scholar] [CrossRef]
- Frances, S.P. Potential for Horizontal Transmission of Orientia tsutsugamushi by Chigger Mites (Acari: Trombiculidae). Int. J. Acarol. 2005, 31, 75–82. [Google Scholar] [CrossRef]
- Mullen, G.R.; OConnor, B.M. Mites (Acari). In Medical and Veterinary Entomology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 533–602. [Google Scholar]
- van der Geest, L.P.; Elliot, S.L.; Breeuwer, J.A.; Beerling, E.A. Diseases of Mites. Exp. Appl. Acarol. 2000, 24, 497–560. [Google Scholar] [CrossRef]
- Sangaré, A.K.; Boutellis, A.; Drali, R.; Socolovschi, C.; Barker, S.C.; Diatta, G.; Rogier, C.; Olive, M.-M.; Doumbo, O.K.; Raoult, D. Detection of Bartonella Quintana in African Body and Head Lice. Am. J. Trop. Med. Hyg. 2014, 91, 294–301. [Google Scholar] [CrossRef]
- Barker, S.C. Phylogeny and Classification, Origins, and Evolution of Host Associations of Lice. Int. J. Parasitol. 1994, 24, 1285–1291. [Google Scholar] [CrossRef]
- Light, J.E.; Toups, M.A.; Reed, D.L. What’s in a Name: The Taxonomic Status of Human Head and Body Lice. Mol. Phylogenet. Evol. 2008, 47, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Brouqui, P. Arthropod-Borne Diseases Associated with Political and Social Disorder. Annu. Rev. Entomol. 2011, 56, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Badiaga, S.; Brouqui, P. Human Louse-Transmitted Infectious Diseases. Clin. Microbiol. Infect. 2012, 18, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Roux, V. The Body Louse as a Vector of Reemerging Human Diseases. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1999, 29, 888–911. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Fernando Parizi, L.; Garcia Guizzo, M.; Tirloni, L.; Seixas, A.; da Silva Vaz, I., Jr.; Termignoni, C. Immunoprotective Potential of a Rhipicephalus (Boophilus) Microplus Metalloprotease. Vet. Parasitol. 2015, 207, 107–114. [Google Scholar] [CrossRef]
- Parizi, L.F.; Ali, A.; Tirloni, L.; Oldiges, D.P.; Sabadin, G.A.; Coutinho, M.L.; Seixas, A.; Logullo, C.; Termignoni, C.; da Silva Vaz, I., Jr. Peptidase Inhibitors in Tick Physiology. Med. Vet. Entomol. 2018, 32, 129–144. [Google Scholar] [CrossRef]
- Tabor, A.E.; Ali, A.; Rehman, G.; Rocha Garcia, G.; Zangirolamo, A.F.; Malardo, T.; Jonsson, N.N. Cattle Tick Rhipicephalus microplus-Host Interface: A Review of Resistant and Susceptible Host Responses. Front. Cell. Infect. Microbiol. 2017, 7, 506. [Google Scholar] [CrossRef]
- Penzhorn, B.L.; Oosthuizen, M.C. Babesia Species of Domestic Cats: Molecular Characterization Has Opened Pandora’s Box. Front. Vet. Sci. 2020, 7, 134. [Google Scholar] [CrossRef]
- Fanelli, A. A Historical Review of Babesia spp. Associated with Deer in Europe: Babesia divergens/Babesia divergens-like, Babesia capreoli, Babesia venatorum, Babesia Cf. Odocoilei. Vet. Parasitol. 2021, 109433. [Google Scholar] [CrossRef]
- Onyiche, T.E.; Răileanu, C.; Fischer, S.; Silaghi, C. Global Distribution of Babesia Species in Questing Ticks: A Systematic Review and Meta-Analysis Based on Published Literature. Pathogens 2021, 10, 230. [Google Scholar] [CrossRef]
- Spielman, A.; Clifford, C.M.; Piesman, J.; Corwin, M.D. Human Babesiosis on Nantucket Island, USA: Description of the Vector, Ixodes (Ixodes) Dammini, n. sp.(Acarina: Ixodidae). J. Med. Entomol. 1979, 15, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Burgdorfer, W.; Barbour, A.G.; Hayes, S.F.; Benach, J.L.; Grunwaldt, E.; Davis, J.P. Lyme Disease-a Tick-Borne Spirochetosis? Science 1982, 216, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, R.; Jain, K.; Bennett, A.; Briese, T.; Lipkin, W.I. Assessment of Polymicrobial Infections in Ticks in New York State. Vector Borne Zoonotic Dis. 2010, 10, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Pritt, B.S.; Mead, P.S.; Johnson, D.K.H.; Neitzel, D.F.; Respicio-Kingry, L.B.; Davis, J.P.; Schiffman, E.; Sloan, L.M.; Schriefer, M.E.; Replogle, A.J.; et al. Identification of a Novel Pathogenic Borrelia Species Causing Lyme Borreliosis with Unusually High Spirochaetaemia: A Descriptive Study. Lancet Infect. Dis. 2016, 16, 556–564. [Google Scholar] [CrossRef]
- Gupta, R.S. Distinction between Borrelia and Borreliella Is More Robustly Supported by Molecular and Phenotypic Characteristics than All Other Neighbouring Prokaryotic Genera: Response to Margos’ et al. “The Genus Borrelia Reloaded” (PLoS ONE 13(12): E0208432). PLoS ONE 2019, 14, e0221397. [Google Scholar] [CrossRef]
- Scott, J.D.; Pesapane, R.R. Detection of Anaplasma phagocytophilum, Babesia odocoilei, Babesia sp., Borrelia burgdorferi Sensu Lato, and Hepatozoon Canis in Ixodes scapularis Ticks Collected in Eastern Canada. Pathogens 2021, 10, 1265. [Google Scholar] [CrossRef]
- Amineni, U.; Pradhan, D.; Marisetty, H. In Silico Identification of Common Putative Drug Targets in Leptospira interrogans. J. Chem. Biol. 2010, 3, 165–173. [Google Scholar] [CrossRef]
- Georrge, J.J.; Umrania, V. In Silico Identification of Putative Drug Targets in Klebsiella pneumonia MGH78578. Indian J. Biotechnol. 2011, 10, 432–439. [Google Scholar]
- Ali, A.; Ahmad, S.; Wadood, A.; Rehman, A.U.; Zahid, H.; Qayash Khan, M.; Nawab, J.; Rahman, Z.U.; Alouffi, A.S. Modeling Novel Putative Drugs and Vaccine Candidates against Tick-Borne Pathogens: A Subtractive Proteomics Approach. Vet. Sci. 2020, 7, 129. [Google Scholar] [CrossRef]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-Centered Information at NCBI. Nucleic Acids Res. 2007, 35, D26–D31. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V. DrugBank 3.0: A Comprehensive Resource for ‘Omics’ Research on Drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-S.; Chen, Y.-C.; Lu, C.-H.; Hwang, J.-K. Prediction of Protein Subcellular Localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Cai, X.-H.; Zhang, Q.; Shi, S.-Y.; Ding, D.-F. Searching for Potential Drug Targets in Two-Component and Phosphorelay Signal-Transduction Systems Using Three-Dimensional Cluster Analysis. Acta Biochim. Biophys. Sin. 2005, 37, 293–302. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- EL-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting Linear B-Cell Epitopes Using String Kernels. J. Mol. Recognit. Interdiscip. J. 2008, 21, 243–255. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of Continuous B-Cell Epitopes in an Antigen Using Recurrent Neural Network. Proteins Struct. Funct. Bioinforma. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y. Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A New Structure-Based Tool for the Prediction of Antibody Epitopes. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Haste Andersen, P.; Nielsen, M.; Lund, O.L.E. Prediction of Residues in Discontinuous B-Cell Epitopes Using Protein 3D Structures. Protein Sci. 2006, 15, 2558–2567. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T. A Novel Genomics Approach for the Identification of Drug Targets in Pathogens, with Special Reference to Pseudomonas aeruginosa. Silico Biol. 2004, 4, 355–360. [Google Scholar]
- Zhang, R.; Lin, Y. DEG 5.0, a Database of Essential Genes in Both Prokaryotes and Eukaryotes. Nucleic Acids Res. 2009, 37, D455–D458. [Google Scholar] [CrossRef]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef]
- Mühlen, S.; Dersch, P. Anti-Virulence Strategies to Target Bacterial Infections. In How to Overcome the Antibiotic Crisis; Springer: Berlin/Heidelberg, Germany, 2015; pp. 147–183. [Google Scholar]
- Xing, X.; Bi, S.; Fan, X.; Jin, M.; Liu, W.; Wang, B. Intranasal Immunization with Multi-Virulence Factors Promotes Mucosal Clearance of Streptococcus suis across Serotypes and Protects against Meningitis in Mice. J. Infect. Dis. 2019, jiz352. [Google Scholar] [CrossRef]
- Solanki, V.; Tiwari, V. Subtractive Proteomics to Identify Novel Drug Targets and Reverse Vaccinology for the Development of Chimeric Vaccine against Acinetobacter baumannii. Sci. Rep. 2018, 8, 1–19. [Google Scholar]
- Wu, C.H.; Huang, H.; Yeh, L.-S.L.; Barker, W.C. Protein Family Classification and Functional Annotation. Comput. Biol. Chem. 2003, 27, 37–47. [Google Scholar] [CrossRef]
- Zheng, C.J.; Zhou, H.; Xie, B.; Han, L.Y.; Yap, C.W.; Chen, Y.Z. TRMP: A Database of Therapeutically Relevant Multiple Pathways. Bioinformatics 2004, 20, 2236–2241. [Google Scholar] [CrossRef][Green Version]
- Ali, A.; Khan, S.; Ali, I.; Karim, S.; da Silva Vaz, I., Jr.; Termignoni, C. Probing the Functional Role of Tick Metalloproteases. Physiol. Entomol. 2015, 40, 177–188. [Google Scholar] [CrossRef]
- Belete, T.M. Novel Targets to Develop New Antibacterial Agents and Novel Alternatives to Antibacterial Agents. Hum. Microbiome J. 2019, 11, 100052. [Google Scholar] [CrossRef]
- Liu, C.; Sun, D.; Zhu, J.; Liu, W. Two-Component Signal Transduction Systems: A Major Strategy for Connecting Input Stimuli to Biofilm Formation. Front. Microbiol. 2019, 9. [Google Scholar] [CrossRef]
- Deutscher, J.; Francke, C.; Postma, P.W. How Phosphotransferase System-Related Protein Phosphorylation Regulates Carbohydrate Metabolism in Bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 939–1031. [Google Scholar] [CrossRef]
- Siebold, C.; Flükiger, K.; Beutler, R.; Erni, B. Carbohydrate Transporters of the Bacterial Phosphoenolpyruvate: Sugar Phosphotransferase System (PTS). FEBS Lett. 2001, 504, 104–111. [Google Scholar] [CrossRef]
- Kushwaha, S.K.; Shakya, M. Protein Interaction Network Analysis—Approach for Potential Drug Target Identification in Mycobacterium tuberculosis. J. Theor. Biol. 2010, 262, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Zhang, L.; Wang, X.; He, Z.-G. Uncovering New Signaling Proteins and Potential Drug Targets through the Interactome Analysis of Mycobacterium tuberculosis. BMC Genom. 2009, 10, 1–10. [Google Scholar] [CrossRef]
- Parvizpour, S.; Pourseif, M.M.; Razmara, J.; Rafi, M.A.; Omidi, Y. Epitope-Based Vaccine Design: A Comprehensive Overview of Bioinformatics Approaches. Drug Discov. Today 2020, 25, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Oscherwitz, J. The Promise and Challenge of Epitope-Focused Vaccines. Hum. Vaccines Immunother. 2016, 12, 2113–2116. [Google Scholar] [CrossRef] [PubMed]
- de Assis, L.M.; Sousa, J.R.; Pinto, N.F.S.; Silva, A.A.; Vaz, A.F.D.M.; Andrade, P.P.; Carvalho, E.M.D.; De Melo, M.A. B-Cell Epitopes of Antigenic Proteins in Leishmania infantum: An in Silico Analysis. Parasite Immunol. 2014, 36, 313–323. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Steps | Ixodes scapularis | Pediculus humanus var. corporis | Leptotrombidium deliense |
|---|---|---|---|
| Host | |||
| Homo sapiens | Homo sapiens | Homo sapiens | |
| Total proteome | 20,467 | 10,775 | 14,667 |
| Duplicates (>60% identical) in CD-HIT | 14,618 | 9726 | 11,328 |
| Non-homologs | 5619 | 2210 | 1872 |
| Essential proteins in DEG | 169 | 120 | 76 |
| Unique metabolic pathway KEGG | 1 | 3 | - |
| Essential Proteins involved KEGG and KAAS | 1 | 1 | 10 |
| Druggability with cutoff E-value 10−5 | 0 | 0 | 1 |
| Steps | Babesia microti str. R1 | Borreliella mayonii | Borrelia miyamotoi | Borrelia recurrentis | Rickettsia prowazekii str. Madrid E | Orientia tsutsugamushi str. Boryong |
|---|---|---|---|---|---|---|
| Host | ||||||
| Homo sapiens | Homo sapiens | Homo sapiens | Homo sapiens | Homo sapiens | Homo sapiens | |
| Total proteome | 3601 | 1133 | 1118 | 1012 | 843 | 1085 |
| Duplicates (>60% identical) in CD-HIT | 3363 | 922 | 906 | 890 | 772 | 731 |
| Non-homologs | 1780 | 704 | 696 | 677 | 492 | 473 |
| Essential proteins in DEG | 106 | 95 | 121 | 119 | 95 | 94 |
| Unique metabolic pathway KEGG | 3 | 15 | 8 | 16 | 21 | 12 |
| Essential Proteins involved KEGG and KAAS | - | 23 | 25 | 23 | 18 | 14 |
| Druggability with cutoff E-value 10−5 | - | 5 | 8 | 6 | 6 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Ahmad, S.; de Albuquerque, P.M.M.; Kamil, A.; Alshammari, F.A.; Alouffi, A.; da Silva Vaz, I., Jr. Prediction of Novel Drug Targets and Vaccine Candidates against Human Lice (Insecta), Acari (Arachnida), and Their Associated Pathogens. Vaccines 2022, 10, 8. https://doi.org/10.3390/vaccines10010008
Ali A, Ahmad S, de Albuquerque PMM, Kamil A, Alshammari FA, Alouffi A, da Silva Vaz I Jr. Prediction of Novel Drug Targets and Vaccine Candidates against Human Lice (Insecta), Acari (Arachnida), and Their Associated Pathogens. Vaccines. 2022; 10(1):8. https://doi.org/10.3390/vaccines10010008
Chicago/Turabian StyleAli, Abid, Shabir Ahmad, Pedro Machado Medeiros de Albuquerque, Atif Kamil, Fahdah Ayed Alshammari, Abdulaziz Alouffi, and Itabajara da Silva Vaz, Jr. 2022. "Prediction of Novel Drug Targets and Vaccine Candidates against Human Lice (Insecta), Acari (Arachnida), and Their Associated Pathogens" Vaccines 10, no. 1: 8. https://doi.org/10.3390/vaccines10010008
APA StyleAli, A., Ahmad, S., de Albuquerque, P. M. M., Kamil, A., Alshammari, F. A., Alouffi, A., & da Silva Vaz, I., Jr. (2022). Prediction of Novel Drug Targets and Vaccine Candidates against Human Lice (Insecta), Acari (Arachnida), and Their Associated Pathogens. Vaccines, 10(1), 8. https://doi.org/10.3390/vaccines10010008