Clinical Development of a Cytomegalovirus DNA Vaccine: From Product Concept to Pivotal Phase 3 Trial

{kind=link}
Abstract
:1. Introduction
1.1. DNA Vaccine Historical Context
1.2. CMV Background, Unmet Medical Needs, and Previous Vaccine Development Efforts
2. CMV DNA Vaccine Product Development
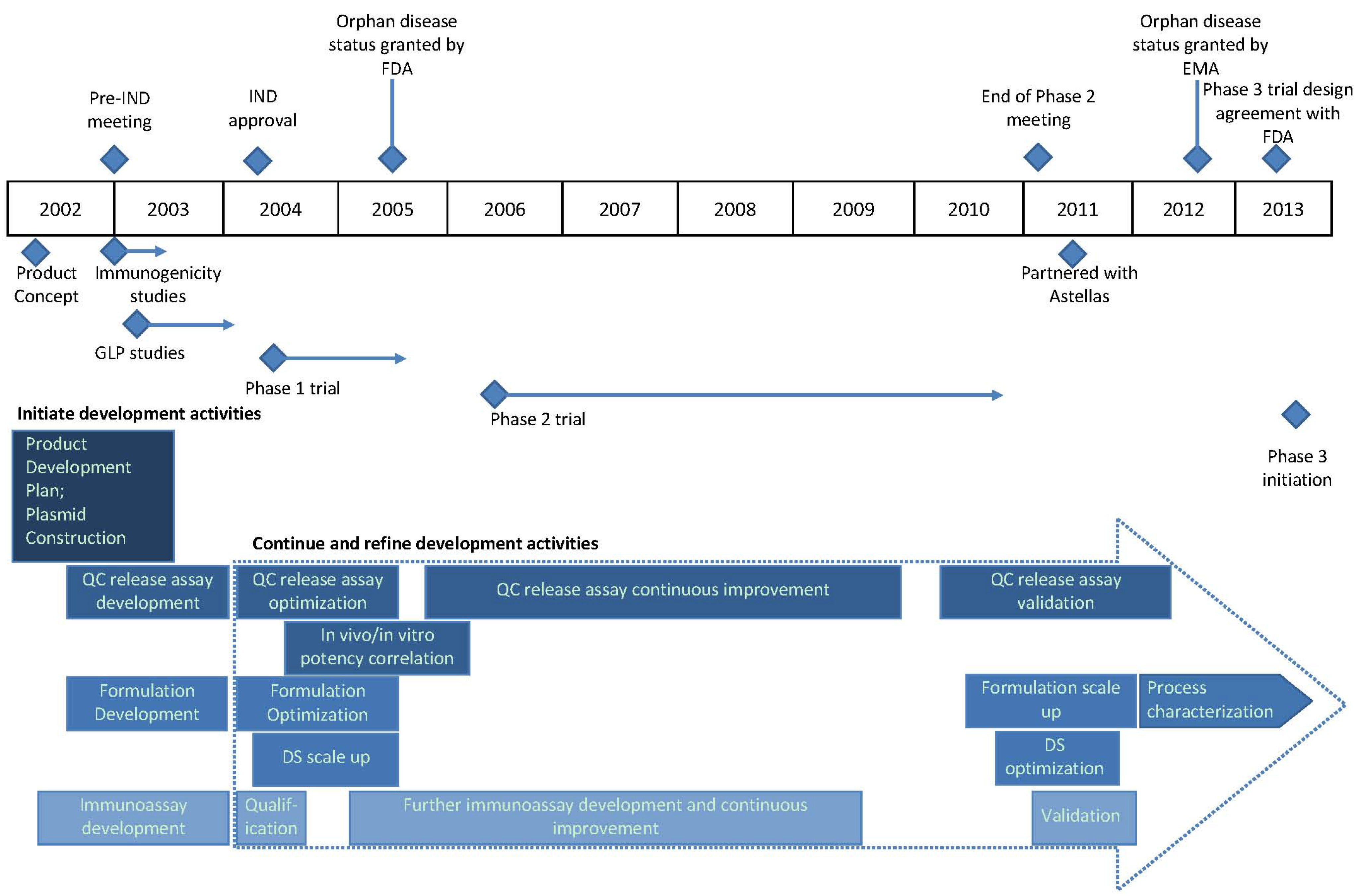
2.1. Functional Areas
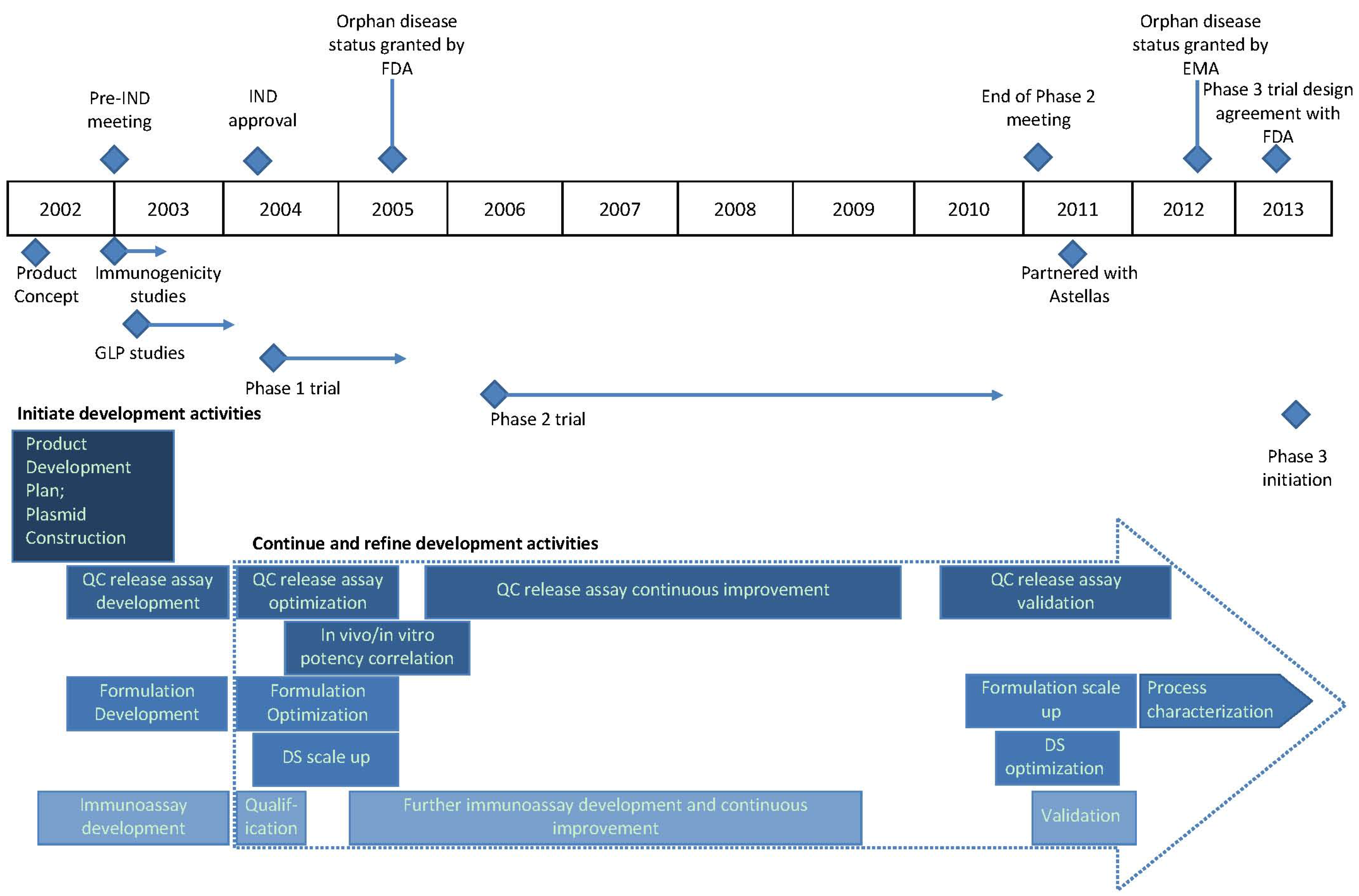
2.2. Vaccine for HCT Recipients as Initial Target Indication
2.3. Antigen Selection
2.4. Plasmid and Genetic Construct Designs
2.5. Formulation
3. Nonclinical Studies
3.1. In Vivo Immunogenicity and Expression Studies
3.2. Safety-Toxicology Study
3.3. Biodistribution/Integration Study
4. Manufacturing, Formulation/Fill/Finish, and Product Release
4.1. Manufacturing of Bulk Drug Substances
4.2. Formulation/Fill/Finish and Release of Drug Product
5. Clinical Trials
5.1. Phase 1
5.1.1. Trial Design and Safety in Normal Healthy Subjects
5.1.2. Immunogenicity Findings
5.2. Phase 2
5.2.1. Trial Design and Safety in HCT Recipients
5.2.2. Efficacy
5.2.3. Immunogenicity Findings
5.3. Phase 3 Trial in HCT Recipients
6. Process Improvements, Manufacturing Scale-Up, and Validation of Assays
7. Timeline Summary of ASP0113 Development History
8. Conclusions—Additional CMV DNA Vaccine Opportunities
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar]
- Tang, D.C.; DeVit, M.; Johnston, S.A. Genetic immunization is a simple method for eliciting an immune response. Nature 1992, 356, 152–154. [Google Scholar] [CrossRef]
- Ulmer, J.B.; Donnelly, J.J.; Parker, S.E.; Rhodes, G.H.; Felgner, P.L.; Dwarki, V.J.; Gromkowski, S.H.; Deck, R.R.; DeWitt, C.M.; Friedman, A.; et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science 1993, 259, 1745–1749. [Google Scholar]
- MacGregor, R.R.; Boyer, J.D.; Ugen, K.E.; Lacy, K.E.; Gluckman, S.J.; Bagarazzi, M.L.; Chattergoon, M.A.; Baine, Y.; Higgins, T.J.; Ciccarelli, R.B.; et al. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: Safety and host response. J. Infect. Dis. 1998, 178, 92–100. [Google Scholar] [CrossRef]
- Calarota, S.; Bratt, G.; Nordlund, S.; Hinkula, J.; Leandersson, A.C.; Sandstrom, E.; Wahren, B. Cellular cytotoxic response induced by DNA vaccination in HIV-1-infected patients. Lancet 1998, 351, 1320–1325. [Google Scholar] [CrossRef]
- Wang, R.; Doolan, D.L.; Le, T.P.; Hedstrom, R.C.; Coonan, K.M.; Charoenvit, Y.; Jones, T.R.; Hobart, P.; Margalith, M.; Ng, J.; et al. Induction of antigen-specific cytotoxic T lymphocytes in humans by a malaria DNA vaccine. Science 1998, 282, 476–480. [Google Scholar] [CrossRef]
- Liu, M.A. DNA vaccines: An historical perspective and view to the future. Immunol. Rev. 2011, 239, 62–84. [Google Scholar] [CrossRef]
- Ferraro, B.; Morrow, M.P.; Hutnick, N.A.; Shin, T.H.; Lucke, C.E.; Weiner, D.B. Clinical applications of DNA vaccines: Current progress. Clin. Infect. Dis. 2011, 53, 296–302. [Google Scholar] [CrossRef]
- Boeckh, M.; Geballe, A.P. Cytomegalovirus: Pathogen, paradigm, and puzzle. J. Clin. Invest. 2011, 121, 1673–1680. [Google Scholar] [CrossRef]
- Plotkin, S.; Orenstein, W.; Offit, P. Vaccines, 6th ed.; Elsevier: Edinburgh, UK, 2013. [Google Scholar]
- Staras, S.A.; Dollard, S.C.; Radford, K.W.; Flanders, W.D.; Pass, R.F.; Cannon, M.J. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin. Infect. Dis. 2006, 43, 1143–1151. [Google Scholar] [CrossRef]
- Marty, F.M.; Ljungman, P.; Papanicolaou, G.A.; Winston, D.J.; Chemaly, R.F.; Strasfeld, L.; Young, J.A.; Rodriguez, T.; Maertens, J.; Schmitt, M.; et al. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: A phase 3, double-blind, placebo-controlled, randomised trial. Lancet Infect. Dis. 2011, 11, 284–292. [Google Scholar] [CrossRef]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M. L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O’Beirne, J.; et al. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Schleiss, M.R.; Berencsi, K.; Gonczol, E.; Dickey, M.; Khoury, P.; Cadoz, M.; Meric, C.; Zahradnik, J.; Duliege, A.M.; et al. Effect of previous or simultaneous immunization with canarypox expressing cytomegalovirus (CMV) glycoprotein B (gB) on response to subunit gB vaccine plus MF59 in healthy CMV-seronegative adults. J. Infect. Dis. 2002, 185, 686–690. [Google Scholar] [CrossRef]
- Berencsi, K.; Gyulai, Z.; Gonczol, E.; Pincus, S.; Cox, W.I.; Michelson, S.; Kari, L.; Meric, C.; Cadoz, M.; Zahradnik, J.; et al. A canarypox vector-expressing cytomegalovirus (CMV) phosphoprotein 65 induces long-lasting cytotoxic T cell responses in human CMV-seronegative subjects. J. Infect. Dis. 2001, 183, 1171–1179. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Reap, E.A.; Katen, K.; Watson, A.; Smith, K.; Norberg, P.; Olmsted, R.A.; Hoeper, A.; Morris, J.; Negri, S.; et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine 2009, 28, 484–493. [Google Scholar] [CrossRef]
- Soares, H.P.; Lutzky, J. Velimogene aliplasmid. Expert Opin. Biol. Ther. 2010, 10, 841–851. [Google Scholar] [CrossRef]
- Junghanss, C.; Storb, R.; Maris, M.B.; Carter, R.A.; Sandmaier, B.M.; Maloney, D.G.; McSweeney, P.A.; Corey, L.; Boeckh, M. Impact of unrelated donor status on the incidence and outcome of cytomegalovirus infections after non-myeloablative allogeneic stem cell transplantation. Br. J. Haematol. 2003, 123, 662–670. [Google Scholar] [CrossRef]
- Gyulai, Z.; Endresz, V.; Burian, K.; Pincus, S.; Toldy, J.; Cox, W.I.; Meric, C.; Plotkin, S.; Gonczol, E.; Berencsi, K. Cytotoxic T lymphocyte (CTL) responses to human cytomegalovirus pp65, IE1-Exon4, gB, pp150, and pp28 in healthy individuals: Reevaluation of prevalence of IE1-specific CTLs. J. Infect. Dis. 2000, 181, 1537–1546. [Google Scholar] [CrossRef]
- Kern, F.; Bunde, T.; Faulhaber, N.; Kiecker, F.; Khatamzas, E.; Rudawski, I.M.; Pruss, A.; Gratama, J.W.; Volkmer-Engert, R.; Ewert, R.; et al. Cytomegalovirus (CMV) phosphoprotein 65 makes a large contribution to shaping the T cell repertoire in CMV-exposed individuals. J. Infect. Dis. 2002, 185, 1709–1716. [Google Scholar] [CrossRef]
- Britt, W.J.; Vugler, L.; Butfiloski, E.J.; Stephens, E.B. Cell surface expression of human cytomegalovirus (HCMV) gp55-116 (gB): Use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J. Virol. 1990, 64, 1079–1085. [Google Scholar]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef]
- Peggs, K.S.; Verfuerth, S.; Pizzey, A.; Khan, N.; Guiver, M.; Moss, P.A.; Mackinnon, S. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet 2003, 362, 1375–1377. [Google Scholar] [CrossRef]
- Einsele, H.; Roosnek, E.; Rufer, N.; Sinzger, C.; Riegler, S.; Loffler, J.; Grigoleit, U.; Moris, A.; Rammensee, H.G.; Kanz, L.; et al. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002, 99, 3916–3922. [Google Scholar] [CrossRef]
- Einsele, H.; Kapp, M.; Grigoleit, G.U. CMV-specific T cell therapy. Blood Cells Mol. Dis. 2008, 40, 71–75. [Google Scholar] [CrossRef]
- Selinsky, C.; Luke, C.; Wloch, M.; Geall, A.; Hermanson, G.; Kaslow, D.; Evans, T. A DNA-based vaccine for the prevention of human cytomegalovirus-associated diseases. Hum. Vaccine Immunother. 2005, 1, 16–23. [Google Scholar]
- Endresz, V.; Kari, L.; Berencsi, K.; Kari, C.; Gyulai, Z.; Jeney, C.; Pincus, S.; Rodeck, U.; Meric, C.; Plotkin, S.A.; et al. Induction of human cytomegalovirus (HCMV)-glycoprotein B (gB)-specific neutralizing antibody and phosphoprotein 65 (pp65)-specific cytotoxic T lymphocyte responses by naked DNA immunization. Vaccine 1999, 17, 50–58. [Google Scholar] [CrossRef]
- Yao, Z.Q.; Gallez-Hawkins, G.; Lomeli, N.A.; Li, X.; Molinder, K.M.; Diamond, D.J.; Zaia, J.A. Site-directed mutation in a conserved kinase domain of human cytomegalovirus-pp65 with preservation of cytotoxic T lymphocyte targeting. Vaccine 2001, 19, 1628–1635. [Google Scholar] [CrossRef]
- Triozzi, P.L.; Stevens, V.C.; Aldrich, W.; Powell, J.; Todd, C.W.; Newman, M.J. Effects of a beta-human chorionic gonadotropin subunit immunogen administered in aqueous solution with a novel nonionic block copolymer adjuvant in patients with advanced cancer. Clin. Cancer Res. 1997, 3, 2355–2362. [Google Scholar]
- Casimiro, D.R.; Chen, L.; Fu, T.M.; Evans, R.K.; Caulfield, M.J.; Davies, M.E.; Tang, A.; Chen, M.; Huang, L.; Harris, V.; et al. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J. Virol. 2003, 77, 6305–6313. [Google Scholar]
- Shiver, J.W.; Fu, T.M.; Chen, L.; Casimiro, D.R.; Davies, M.E.; Evans, R.K.; Zhang, Z.Q.; Simon, A.J.; Trigona, W.L.; Dubey, S.A.; et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 2002, 415, 331–335. [Google Scholar]
- Evans, R.K. Characterization and biological evaluation of a microparticle adjuvant formulation for plasmid DNA vaccines. J. Pharm. Sci. 2004, 93, 1924–1939. [Google Scholar]
- Inactive Ingredient Search for Approved Drug Products. Available online: http://www.accessdata.fda.gov/ (accessed on 12 April 2013).
- Hartikka, J.; Geall, A.; Bozoukova, V.; Kurniadi, D.; Rusalov, D.; Enas, J.; Yi, J.H.; Nanci, A.; Rolland, A. Physical characterization and in vivo evaluation of poloxamer-based DNA vaccine formulations. J. Gene Med. 2008, 10, 770–782. [Google Scholar] [CrossRef]
- Mahajan, R.; Feher, B.; Jones, B.; Jones, D.; Marjerison, L.; Sam, M.; Hartikka, J.; Wloch, M.; Lalor, P.; Kaslow, D.; et al. A TaqMan reverse transcription polymerase chain reaction (RT-PCR) in vitro potency assay for plasmid-based vaccine products. Mol. Biotechnol. 2008, 40, 47–57. [Google Scholar] [CrossRef]
- Wloch, M.K.; Smith, L.R.; Boutsaboualoy, S.; Reyes, L.; Han, C.; Kehler, J.; Smith, H.D.; Selk, L.; Nakamura, R.; Brown, J.M.; et al. Safety and immunogenicity of a bivalent cytomegalovirus DNA vaccine in healthy adult subjects. J. Infect. Dis. 2008, 19, 1634–1642. [Google Scholar]
- Kharfan-Dabaja, M.A.; Boeckh, M.; Wilck, M.B.; Langston, A.A.; Chu, A.H.; Wloch, M.K.; Guterwill, D.F.; Smith, L.R.; Rolland, A.P.; Kenney, R.T. A novel therapeutic cytomegalovirus DNA vaccine in allogeneic haemopoietic stem-cell transplantation: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Infect. Dis. 2012, 12, 290–299. [Google Scholar] [CrossRef]
- Kharfan-Dabaja, M.A.; Boeckh, M.; Wilck, M.B.; Langston, A.A.; Chu, A.H.; Wloch, M.K.; Smith, L.R.; Rolland, A.P.; Kenney, R.T. Reanalysis of TransVax immunogenicity. Lancet Infect. Dis. 2013, 13, 18. [Google Scholar] [CrossRef]
- Go, V.; Pollard, R.B. A cytomegalovirus vaccine for transplantation: Are we closer? J. Infect. Dis. 2008, 19, 1631–1633. [Google Scholar]
- Adler, S.P. Human CMV vaccine trials: What if CMV caused a rash? J. Clin. Virol. 2008, 41, 231–623. [Google Scholar] [CrossRef]
- Public Workshop—The Development and Evaluation of Human Cytomegalovirus Vaccines. Available online: http://www.fda.gov/BiologicsBloodVaccines/NewsEvents/WorkshopsMeetingsConferences/ucm280016.htm (accessed on 23 August 2013).
- Hartikka, J.; Bozoukova, V.; Morrow, J.; Rusalov, D.; Shlapobersky, M.; Wei, Q.; Boutsaboualoy, S.; Ye, M.; Wloch, M.K.; Doukas, J.; et al. Preclinical evaluation of the immunogenicity and safety of plasmid DNA-based prophylactic vaccines for human cytomegalovirus. Hum. Vaccine Immunother. 2012, 8, 1595–1606. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smith, L.R.; Wloch, M.K.; Chaplin, J.A.; Gerber, M.; Rolland, A.P. Clinical Development of a Cytomegalovirus DNA Vaccine: From Product Concept to Pivotal Phase 3 Trial. Vaccines 2013, 1, 398-414. https://doi.org/10.3390/vaccines1040398
Smith LR, Wloch MK, Chaplin JA, Gerber M, Rolland AP. Clinical Development of a Cytomegalovirus DNA Vaccine: From Product Concept to Pivotal Phase 3 Trial. Vaccines. 2013; 1(4):398-414. https://doi.org/10.3390/vaccines1040398
Chicago/Turabian StyleSmith, Larry R., Mary K. Wloch, Jennifer A. Chaplin, Michele Gerber, and Alain P. Rolland. 2013. "Clinical Development of a Cytomegalovirus DNA Vaccine: From Product Concept to Pivotal Phase 3 Trial" Vaccines 1, no. 4: 398-414. https://doi.org/10.3390/vaccines1040398
APA StyleSmith, L. R., Wloch, M. K., Chaplin, J. A., Gerber, M., & Rolland, A. P. (2013). Clinical Development of a Cytomegalovirus DNA Vaccine: From Product Concept to Pivotal Phase 3 Trial. Vaccines, 1(4), 398-414. https://doi.org/10.3390/vaccines1040398