SIRT1/SIRT3 Modulates Redox Homeostasis during Ischemia/Reperfusion in the Aging Heart




Abstract
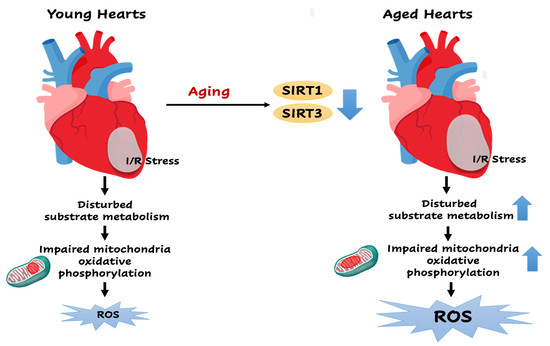
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Role of Longevity Gene Sirtuins in Myocardial I/R
3. Reactive Oxygen Species in Age-Related Ischemic Heart Disease
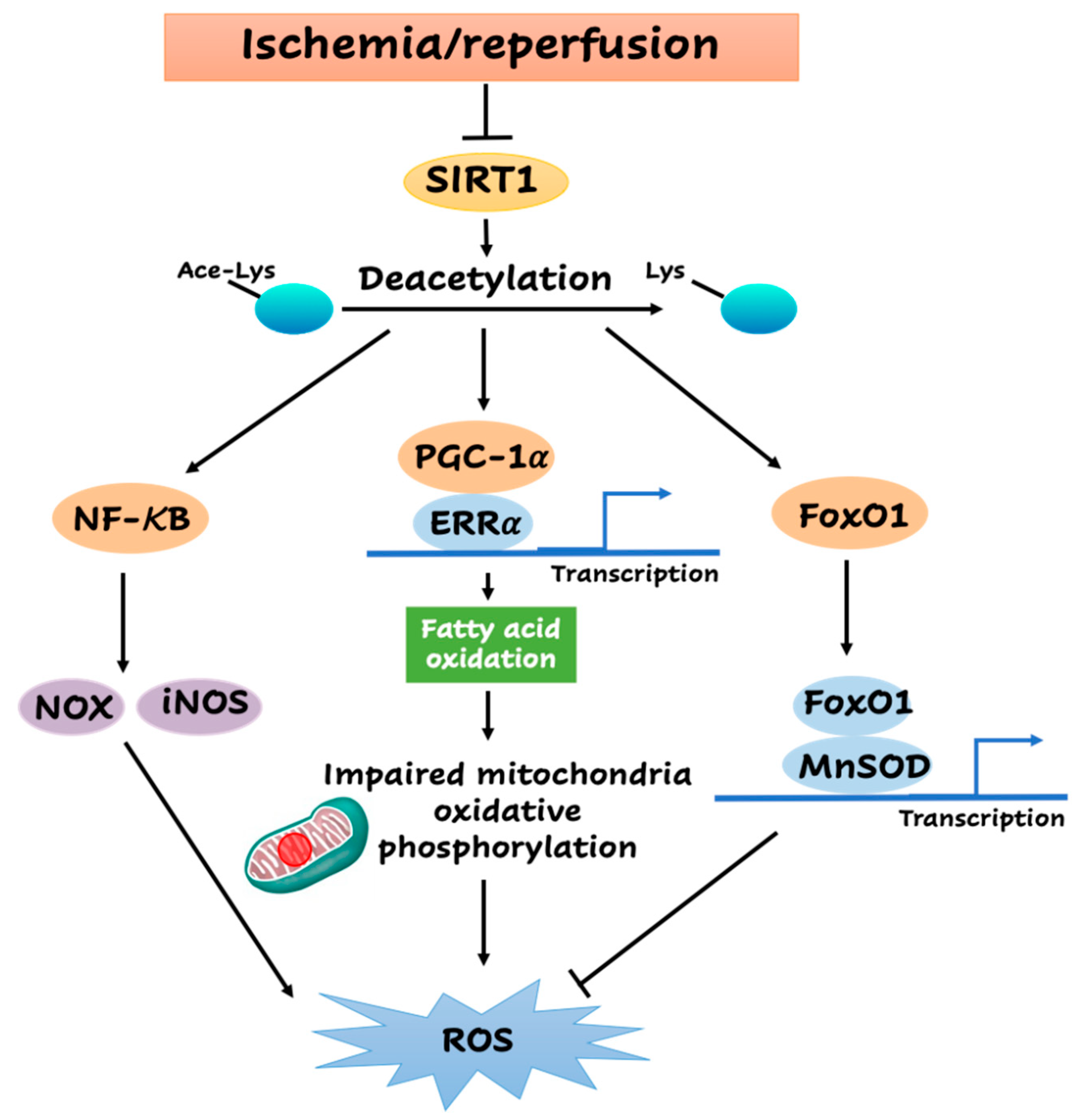
4. Role of SIRT1 in the Redox Homeostasis during Myocardial I/R
5. Role of SIRT3 in the Metabolic Homeostasis during Myocardial I/R
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Akhtar, S. Ischemic heart disease. Anesthesiol. Clin. 2006, 24, 461–485. [Google Scholar] [CrossRef]
- Fan, Q.; Chen, M.; Fang, X.; Lau, W.B.; Xue, L.; Zhao, L.; Zhang, H.; Liang, Y.H.; Bai, X.; Niu, H.Y.; et al. Aging might augment reactive oxygen species (ROS) formation and affect reactive nitrogen species (RNS) level after myocardial ischemia/reperfusion in both humans and rats. Age (Dordr) 2013, 35, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Mitochondria: Are they the seat of senescence? Aging Cell 2004, 3, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortuno-Sahagun, D.; Pallas, M.; Rojas-Mayorquin, A.E. Oxidative stress in aging: Advances in proteomic approaches. Oxid. Med. Cell Longev. 2014, 2014, 573208. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, R.A.H.; Santos, D.; Haigis, M.C. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol. Med. 2017, 23, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.I.; Guarente, L. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [Green Version]
- Lagunas-Rangel, F.A. Current role of mammalian sirtuins in DNA repair. DNA Repair (Amst.) 2019, 80, 85–92. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD(+) in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef]
- Mendes, K.L.; Lelis, D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, H.; Wang, J.; Zhong, L.; Chen, X.; Zhang, R.; Wang, H. Glucose restriction delays senescence and promotes proliferation of HUVECs via the AMPK/SIRT1-FOXA3-Beclin1 pathway. Exp. Gerontol. 2020, 111053. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.H.; Hwang, Y.; Kim, E. PARP1 Impedes SIRT1-Mediated Autophagy during Degeneration of the Retinal Pigment Epithelium under Oxidative Stress. Mol. Cells 2020, 43, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Li, M.; Wang, Z.; Yang, Z.; Zhang, J.; Fang, H.; He, Z.; Wang, X. Depression of Mitochondrial Function in the Rat Skeletal Muscle Model of Myofascial Pain Syndrome Is through Down-Regulation of the AMPK-PGC-1α-SIRT3 Axis. J. Pain Res. 2020, 13, 1747–1756. [Google Scholar] [CrossRef]
- Caballero, E.P.; Mariz-Ponte, N.; Rigazio, C.S.; Santamaría, M.H.; Corral, R.S. Honokiol attenuates oxidative stress-dependent heart dysfunction in chronic Chagas disease by targeting AMPK/NFE2L2/SIRT3 signaling pathway. Free Radic. Biol. Med. 2020, 156, 113–124. [Google Scholar] [CrossRef]
- Wang, C.H.; Wei, Y.H. Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 5266. [Google Scholar] [CrossRef]
- Xin, T.; Lu, C. SirT3 activates AMPK-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging (Albany NY) 2020, 12, 16224–16231. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.M.; Chapa-Dubocq, X.; Rullan, P.J.; Jang, S.; Javadov, S. High Sensitivity of SIRT3 Deficient Hearts to Ischemia-Reperfusion Is Associated with Mitochondrial Abnormalities. Front. Pharmacol. 2017, 8, 275. [Google Scholar] [CrossRef]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1602–H1609. [Google Scholar] [CrossRef] [Green Version]
- Lavu, S.; Boss, O.; Elliott, P.J.; Lambert, P.D. Sirtuins—Novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 2008, 7, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Singh, S.; Rizvi, S.I. Age-dependent altered redox homeostasis in the chronodisrupted rat model and moderation by melatonin administration. Chronobiol. Int. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Y.; Liu, L.; Jiang, Z.; Li, X.; Tong, R.; He, J.; Shi, J. Research progress on sirtuins family members and cell senescence. Eur. J. Med. Chem. 2020, 193, 112207. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [Green Version]
- Maurer, B.; Rumpf, T.; Scharfe, M.; Stolfa, D.A.; Schmitt, M.L.; He, W.; Verdin, E.; Sippl, W.; Jung, M. Inhibitors of the NAD(+)-Dependent Protein Desuccinylase and Demalonylase Sirt5. ACS Med. Chem. Lett. 2012, 3, 1050–1053. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927–945. [Google Scholar] [CrossRef]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [Green Version]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellizzi, D.; Dato, S.; Cavalcante, P.; Covello, G.; Di Cianni, F.; Passarino, G.; Rose, G.; De Benedictis, G. Characterization of a bidirectional promoter shared between two human genes related to aging: SIRT3 and PSMD13. Genomics 2007, 89, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellizzi, D.; Rose, G.; Cavalcante, P.; Covello, G.; Dato, S.; De Rango, F.; Greco, V.; Maggiolini, M.; Feraco, E.; Mari, V.; et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 2005, 85, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Quan, N.; Sun, W.; Chen, X.; Cates, C.; Rousselle, T.; Zhou, X.; Zhao, X.; Li, J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc. Res. 2018, 114, 805–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Sun, W.; Ren, D.; Zhang, J.; He, Z.; Fedorova, J.; Sun, X.; Han, F.; Li, J. SIRT1 agonism modulates cardiac NLRP3 inflammasome through pyruvate dehydrogenase during ischemia and reperfusion. Redox Biol. 2020, 34, 101538. [Google Scholar] [CrossRef]
- Han, D.; Wang, J.; Ma, S.; Chen, Y.; Cao, F. SIRT1 as a Promising Novel Therapeutic Target for Myocardial Ischemia Reperfusion Injury and Cardiometabolic Disease. Curr. Drug Targets 2017, 18, 1746–1753. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Kirshenbaum, L.A.; Imai, S.; Vatner, S.F.; Sadoshima, J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 2004, 95, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Shi, B.; Ma, M.; Wu, X.; Lin, X. The novel relationship between Sirt3 and autophagy in myocardial ischemia-reperfusion. J. Cell Physiol. 2019, 234, 5488–5495. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Young, I.S.; Woodside, J.V. Antioxidants in health and disease. J. Clin. Pathol. 2001, 54, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Lenaz, G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol. 2012, 942, 93–136. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Sazanov, L.A.; Hinchliffe, P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 2006, 311, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.J.; Buckingham, J.A.; Boysen, H.M.; Brand, M.D. Diphenyleneiodonium acutely inhibits reactive oxygen species production by mitochondrial complex I during reverse, but not forward electron transport. Biochim. Biophys. Acta 2008, 1777, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miwa, S.; St-Pierre, J.; Partridge, L.; Brand, M.D. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic. Biol. Med. 2003, 35, 938–948. [Google Scholar] [CrossRef]
- Miwa, S.; Brand, M.D. The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim. Biophys. Acta 2005, 1709, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Kundu, A.; Vaze, A.; Sardar, P.; Nagy, A.; Aronow, W.S.; Botkin, N.F. Variant Angina and Aborted Sudden Cardiac Death. Curr. Cardiol. Rep. 2018, 20, 26. [Google Scholar] [CrossRef]
- Morrison, A.; Li, J. PPAR-gamma and AMPK--advantageous targets for myocardial ischemia/reperfusion therapy. Biochem. Pharmacol. 2011, 82, 195–200. [Google Scholar] [CrossRef]
- Costa, R.; Morrison, A.; Wang, J.; Manithody, C.; Li, J.; Rezaie, A.R. Activated protein C modulates cardiac metabolism and augments autophagy in the ischemic heart. J. Thromb. Haemost. 2012, 10, 1736–1744. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Augustine, A.; Sun, D.; Li, L.; Fliegel, L. Activation of the Na(+)/H(+) exchanger in isolated cardiomyocytes through beta-Raf dependent pathways. Role of Thr(653) of the cytosolic tail. J. Mol. Cell. Cardiol. 2016, 99, 65–75. [Google Scholar] [CrossRef]
- Yokoyama, H.; Gunasegaram, S.; Harding, S.E.; Avkiran, M. Sarcolemmal Na+/H+ exchanger activity and expression in human ventricular myocardium. J. Am. Coll. Cardiol. 2000, 36, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Chuang, C.C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. Biomed. Res. Int. 2015, 2015, 864946. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zuo, L. Characterization of oxygen radical formation mechanism at early cardiac ischemia. Cell Death Dis. 2013, 4, e787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: Involvement of reactive oxygen species and cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [Green Version]
- Inserte, J.; Hernando, V.; Garcia-Dorado, D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2012, 96, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Turgeon, R.D.; Pearson, G.J.; Graham, M.M. Pharmacologic Treatment of Patients with Myocardial Ischemia With No Obstructive Coronary Artery Disease. Am. J. Cardiol. 2018, 121, 888–895. [Google Scholar] [CrossRef]
- Bulluck, H.; Yellon, D.M.; Hausenloy, D.J. Reducing myocardial infarct size: Challenges and future opportunities. Heart 2016, 102, 341–348. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Pei, H.; Yang, Y.; Zhao, H.; Li, X.; Yang, D.; Li, D. The Role of Mitochondrial Functional Proteins in ROS Production in Ischemic Heart Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 5470457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrienko, T.N.; Pasdois, P.; Pereira, G.C.; Ovens, M.J.; Halestrap, A.P. The role of succinate and ROS in reperfusion injury—A critical appraisal. J. Mol. Cell. Cardiol. 2017, 110, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solaini, G.; Harris, D.A. Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem. J. 2005, 390, 377–394. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Li, J. Metabolic shifts during aging and pathology. Compr. Physiol. 2015, 5, 667–686. [Google Scholar] [CrossRef] [Green Version]
- Favero, G.; Franceschetti, L.; Buffoli, B.; Moghadasian, M.H.; Reiter, R.J.; Rodella, L.F.; Rezzani, R. Melatonin: Protection against age-related cardiac pathology. Ageing Res. Rev. 2017, 35, 336–349. [Google Scholar] [CrossRef]
- Gates, P.E.; Tanaka, H.; Graves, J.; Seals, D.R. Left ventricular structure and diastolic function with human ageing. Relation to habitual exercise and arterial stiffness. Eur. Heart J. 2003, 24, 2213–2220. [Google Scholar] [CrossRef]
- Arieli, Y.; Gursahani, H.; Eaton, M.M.; Hernandez, L.A.; Schaefer, S. Gender modulation of Ca(2+) uptake in cardiac mitochondria. J. Mol. Cell. Cardiol. 2004, 37, 507–513. [Google Scholar] [CrossRef]
- Adler, A.; Messina, E.; Sherman, B.; Wang, Z.; Huang, H.; Linke, A.; Hintze, T.H. NAD(P)H oxidase-generated superoxide anion accounts for reduced control of myocardial O2 consumption by NO in old Fischer 344 rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1015–H1022. [Google Scholar] [CrossRef] [PubMed]
- Wojtovich, A.P.; Nadtochiy, S.M.; Brookes, P.S.; Nehrke, K. Ischemic preconditioning: The role of mitochondria and aging. Exp. Gerontol. 2012, 47, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Wang, J.; Thomas, D.P.; Tong, C.; Leng, L.; Wang, W.; Merk, M.; Zierow, S.; Bernhagen, J.; Ren, J.; et al. Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation 2010, 122, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, N.; Sun, W.; Wang, L.; Chen, X.; Bogan, J.S.; Zhou, X.; Cates, C.; Liu, Q.; Zheng, Y.; Li, J. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. FASEB J. 2017, 31, 4153–4167. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Free radical theory of aging: An update: Increasing the functional life span. Ann. N. Y. Acad. Sci. 2006, 1067, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef]
- Thoma, A.; Akter-Miah, T.; Reade, R.L.; Lightfoot, A.P. Targeting reactive oxygen species (ROS) to combat the age-related loss of muscle mass and function. Biogerontology 2020, 21, 475–484. [Google Scholar] [CrossRef]
- de Almeida, A.J.P.O.; de Almeida Rezende, M.S.; Dantas, S.H.; de Lima Silva, S.; de Oliveira, J.C.P.L.; de Lourdes Assunção Araújo de Azevedo, F.; Alves, R.M.F.R.; de Menezes, G.M.S.; Dos Santos, P.F.; Gonçalves, T.A.F.; et al. Unveiling the Role of Inflammation and Oxidative Stress on Age-Related Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 1954398. [Google Scholar] [CrossRef]
- Boengler, K.; Schulz, R.; Heusch, G. Loss of cardioprotection with ageing. Cardiovasc. Res. 2009, 83, 247–261. [Google Scholar] [CrossRef]
- Escobales, N.; Nuñez, R.E.; Jang, S.; Parodi-Rullan, R.; Ayala-Peña, S.; Sacher, J.R.; Skoda, E.M.; Wipf, P.; Frontera, W.; Javadov, S. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J. Mol. Cell. Cardiol. 2014, 77, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Lesnefsky, E.J.; Hoppel, C.L. Ischemia-reperfusion injury in the aged heart: Role of mitochondria. Arch. Biochem. Biophys. 2003, 420, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Tocchi, A.; Quarles, E.K.; Basisty, N.; Gitari, L.; Rabinovitch, P.S. Mitochondrial dysfunction in cardiac aging. Biochim. Biophys. Acta 2015, 1847, 1424–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelm, N.Q.; Beare, J.E.; Weber, G.J.; LeBlanc, A.J. Thrombospondin-1 mediates Drp-1 signaling following ischemia reperfusion in the aging heart. FASEB Bioadv. 2020, 2, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, A.; Lopaschuk, G.D. Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim. Biophys. Acta 2016, 1862, 2211–2220. [Google Scholar] [CrossRef]
- Guarente, L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant Biol. 2007, 72, 483–488. [Google Scholar] [CrossRef]
- Tong, C.; Morrison, A.; Mattison, S.; Qian, S.; Bryniarski, M.; Rankin, B.; Wang, J.; Thomas, D.P.; Li, J. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress. FASEB J. 2013, 27, 4332–4342. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.; Seok, S.; Yau, P.; Li, X.; Kemper, B.; Kemper, J.K. Obesity and aging diminish sirtuin 1 (SIRT1)-mediated deacetylation of SIRT3, leading to hyperacetylation and decreased activity and stability of SIRT3. J. Biol. Chem. 2017, 292, 17312–17323. [Google Scholar] [CrossRef] [Green Version]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Winnik, S.; Auwerx, J.; Sinclair, D.A.; Matter, C.M. Protective effects of sirtuins in cardiovascular diseases: From bench to bedside. Eur. Heart J. 2015, 36, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2011, 90, 276–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Xing, Y.; Jiang, L.; Chen, M.; Xu, M.; Yin, Y.; Li, C.; Yang, Z.; Yu, L.; Ma, H. Impaired cardiac SIRT1 activity by carbonyl stress contributes to aging-related ischemic intolerance. PLoS ONE 2013, 8, e74050. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, Y.; Ma, H.; Guan, Y.; Cao, Y.; Tian, Y.; Zhang, Y. Enhancement of Glucose Metabolism via PGC-1alpha Participates in the Cardioprotection of Chronic Intermittent Hypobaric Hypoxia. Front. Physiol. 2016, 7, 219. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.J.; Boudina, S.; Banke, N.H.; Sambandam, N.; Han, X.; Young, D.M.; Leone, T.C.; Gross, R.W.; Lewandowski, E.D.; Abel, E.D.; et al. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H185–H196. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, T.; Biggs, W.H.; Tieu, D.; Boyer, A.D.; Varki, N.M.; Cavenee, W.K.; Arden, K.C. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc. Natl. Acad. Sci. USA 2004, 101, 2975–2980. [Google Scholar] [CrossRef] [Green Version]
- Puthanveetil, P.; Wan, A.; Rodrigues, B. FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc. Res. 2013, 97, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.H.; Liu, X.H.; Hong, X.; Zhao, N.; Xiao, Y.F.; Wang, L.F.; Tang, L.; Jiang, K.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Protects the Heart from Ischemia/Reperfusion Injury through Activating SIRT1/FOXOs-Mediated Antioxidative Stress Pathway. Oxid. Med. Cell Longev. 2016, 2016, 7410257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Li, S.A.; Huang, C.H.; Su, H.H.; Chen, Y.H.; Chang, J.T.; Huang, S.S. Sirt1 Activation by Post-ischemic Treatment With Lumbrokinase Protects against Myocardial Ischemia-Reperfusion Injury. Front. Pharmacol. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Marden, J.J.; Fan, C.; Sanlioglu, S.; Weiss, R.M.; Ritchie, T.C.; Davisson, R.L.; Engelhardt, J.F. Genetic redox preconditioning differentially modulates AP-1 and NF kappa B responses following cardiac ischemia/reperfusion injury and protects against necrosis and apoptosis. Mol. Ther. 2003, 7, 341–353. [Google Scholar] [CrossRef]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef] [Green Version]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M. Regulation of NADPH oxidase subunit p22(phox) by NF-kB in human aortic smooth muscle cells. Arch. Physiol. Biochem. 2007, 113, 163–172. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Browder, W.; Kao, R.L. Early activation of transcription factor NF-kappaB during ischemia in perfused rat heart. Am. J. Physiol. 1999, 276, H543–H552. [Google Scholar] [CrossRef]
- Li, D.; Wang, X.; Huang, Q.; Li, S.; Zhou, Y.; Li, Z. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-kappaB pathway in vivo and in vitro. Redox Biol. 2018, 15, 62–73. [Google Scholar] [CrossRef]
- Thiemermann, C. Inhibition of the activation of nuclear factor kappa B to reduce myocardial reperfusion injury and infarct size. Cardiovasc. Res. 2004, 63, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Shao, L.; Du, Q.; Park, K.S.; Geller, D.A. Identification of a classic cytokine-induced enhancer upstream in the human iNOS promoter. FASEB J. 2007, 21, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Helenius, M.; Kyrylenko, S.; Vehvilainen, P.; Salminen, A. Characterization of aging-associated up-regulation of constitutive nuclear factor-kappa B binding activity. Antioxid. Redox Signal. 2001, 3, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Helenius, M.; Hänninen, M.; Lehtinen, S.K.; Salminen, A. Aging-induced up-regulation of nuclear binding activities of oxidative stress responsive NF-kB transcription factor in mouse cardiac muscle. J. Mol. Cell Cardiol. 1996, 28, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Li, J.; Zhang, Y.; Bu, P. Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS ONE 2015, 10, e0118909. [Google Scholar] [CrossRef]
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance exercise as a countermeasure for aging. Diabetes 2008, 57, 2933–2942. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Liu, C.; Chen, Q.; Liu, N.; Yan, Y.; Liu, B. SIRT3: A New Regulator of Cardiovascular Diseases. Oxid. Med. Cell Longev. 2018, 2018, 7293861. [Google Scholar] [CrossRef] [Green Version]
- Jing, E.; O’Neill, B.T.; Rardin, M.J.; Kleinridders, A.; Ilkeyeva, O.R.; Ussar, S.; Bain, J.R.; Lee, K.Y.; Verdin, E.M.; Newgard, C.B.; et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013, 62, 3404–3417. [Google Scholar] [CrossRef] [Green Version]
- Abo Alrob, O.; Lopaschuk, G.D. Role of CoA and acetyl-CoA in regulating cardiac fatty acid and glucose oxidation. Biochem. Soc. Trans. 2014, 42, 1043–1051. [Google Scholar] [CrossRef]
- Zhang, X.; Ji, R.; Liao, X.; Castillero, E.; Kennel, P.J.; Brunjes, D.L.; Franz, M.; Mobius-Winkler, S.; Drosatos, K.; George, I.; et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 2018, 137, 2052–2067. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozden, O.; Park, S.H.; Wagner, B.A.; Song, H.Y.; Zhu, Y.; Vassilopoulos, A.; Jung, B.; Buettner, G.R.; Gius, D. SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radic. Biol. Med. 2014, 76, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, T.B.; Olson, M.S. Regulation of pyruvate dehydrogenase complex in ischemic rat heart. Am. J. Physiol. 1984, 246, H858–H864. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Fang, Y.H.; Kubler, M.M.; Donnino, M.W.; Sharp, W.W. Enhanced pyruvate dehydrogenase activity improves cardiac outcomes in a murine model of cardiac arrest. PLoS ONE 2017, 12, e0185046. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, E.D.; White, L.T. Pyruvate dehydrogenase influences postischemic heart function. Circulation 1995, 91, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Draper, J.A.; Davidson, M.T.; Williams, A.S.; Narowski, T.M.; Slentz, D.H.; Ilkayeva, O.R.; Stevens, R.D.; Wagner, G.R.; Najjar, R.; et al. Respiratory Phenomics across Multiple Models of Protein Hyperacylation in Cardiac Mitochondria Reveals a Marginal Impact on Bioenergetics. Cell Rep. 2019, 26, 1557–1572.e8. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Cimen, H.; Han, M.J.; Yang, Y.; Tong, Q.; Koc, H.; Koc, E.C. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 2010, 49, 304–311. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Newman, J.C.; Wang, M.Z.; Ho, L.; Verdin, E. Mitochondrial sirtuins: Regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 2012, 23, 467–476. [Google Scholar] [CrossRef]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [Green Version]
- Ku, H.J.; Ahn, Y.; Lee, J.H.; Park, K.M.; Park, J.W. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic. Biol. Med. 2015, 80, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.L.; Giguere, V. Phosphatases at the heart of FoxO metabolic control. Cell Metab. 2008, 7, 101–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, G.; Chen, Y.; Zhang, H.; Zhou, W. Trans sodium crocetinate alleviates ischemia/reperfusion-induced myocardial oxidative stress and apoptosis via the SIRT3/FOXO3a/SOD2 signaling pathway. Int. Immunopharmacol. 2019, 71, 361–371. [Google Scholar] [CrossRef]
- Kurundkar, D.; Kurundkar, A.R.; Bone, N.B.; Becker, E.J., Jr.; Liu, W.; Chacko, B.; Darley-Usmar, V.; Zmijewski, J.W.; Thannickal, V.J. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight 2019, 4, e120722. [Google Scholar] [CrossRef]
- Winnik, S.; Gaul, D.S.; Siciliani, G.; Lohmann, C.; Pasterk, L.; Calatayud, N.; Weber, J.; Eriksson, U.; Auwerx, J.; van Tits, L.J.; et al. Mild endothelial dysfunction in Sirt3 knockout mice fed a high-cholesterol diet: Protective role of a novel C/EBP-β-dependent feedback regulation of SOD2. Basic Res. Cardiol. 2016, 111, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.B.; Rinaldi, C.; Lu, Y.; Amorim, J.A.; Sinclair, D.A. Molecular and Cellular Characterization of SIRT1 Allosteric Activators. Methods Mol. Biol. 2019, 1983, 133–149. [Google Scholar] [CrossRef]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Ren, D.; Fedorova, J.; He, Z.; Li, J. SIRT1/SIRT3 Modulates Redox Homeostasis during Ischemia/Reperfusion in the Aging Heart. Antioxidants 2020, 9, 858. https://doi.org/10.3390/antiox9090858
Zhang J, Ren D, Fedorova J, He Z, Li J. SIRT1/SIRT3 Modulates Redox Homeostasis during Ischemia/Reperfusion in the Aging Heart. Antioxidants. 2020; 9(9):858. https://doi.org/10.3390/antiox9090858
Chicago/Turabian StyleZhang, Jingwen, Di Ren, Julia Fedorova, Zhibin He, and Ji Li. 2020. "SIRT1/SIRT3 Modulates Redox Homeostasis during Ischemia/Reperfusion in the Aging Heart" Antioxidants 9, no. 9: 858. https://doi.org/10.3390/antiox9090858
APA StyleZhang, J., Ren, D., Fedorova, J., He, Z., & Li, J. (2020). SIRT1/SIRT3 Modulates Redox Homeostasis during Ischemia/Reperfusion in the Aging Heart. Antioxidants, 9(9), 858. https://doi.org/10.3390/antiox9090858