BKCa Channels as Targets for Cardioprotection



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Localization of BKCa Channels
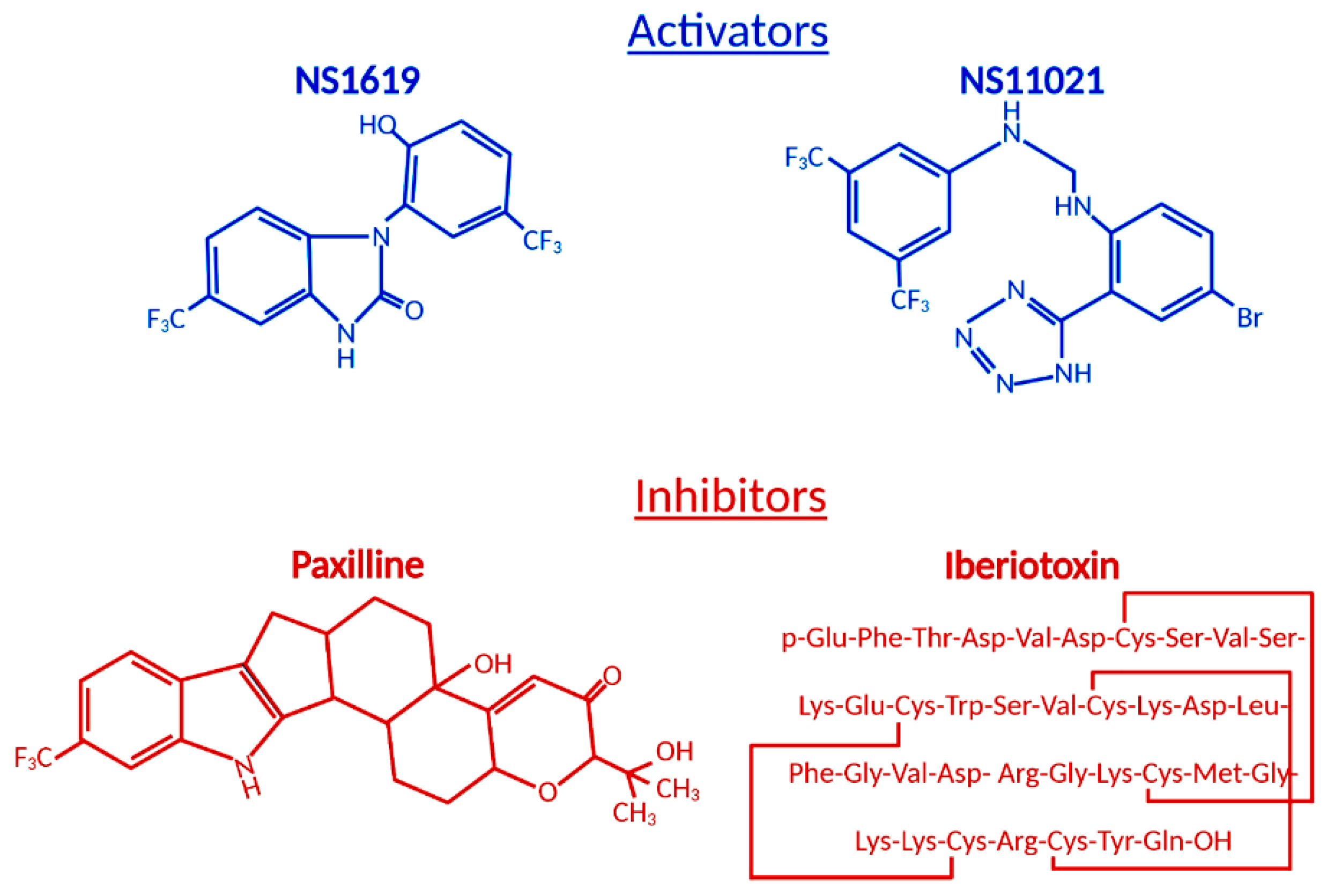
3. Role of BKCa Channel Agonists
4. Role of BKCa Channel Antagonists
5. Genetic Animal Models
6. BKCa as a Therapeutic Target for Cardioprotection
7. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Latorre, R.; Vergara, C.; Hidalgo, C. Reconstitution in planar lipid bilayers of a Ca2+-dependent K+ channel from transverse tubule membranes isolated from rabbit skeletal muscle. Proc. Natl. Acad. Sci. USA 1982, 79, 805–809. [Google Scholar] [CrossRef]
- Pallotta, B.S.; Magleby, K.L.; Barrett, J.N. Single channel recordings of Ca2+-activated K+ currents in rat muscle cell culture. Nature 1981, 293, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Marty, A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature 1981, 291, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Toro, L.; Li, M.; Zhang, Z.; Singh, H.; Wu, Y.; Stefani, E. MaxiK channel and cell signalling. Pflügers Arch. Eur. J. Physiol. 2014, 466, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Lu, R.; Bopassa, J.C.; Meredith, A.L.; Stefani, E.; Toro, L. MitoBK(Ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc. Natl. Acad. Sci. USA 2013, 110, 10836–10841. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jie, W.; Huang, L.; Wei, P.; Li, S.; Luo, Z.; Friedman, A.K.; Meredith, A.L.; Han, M.H.; Zhu, X.H.; et al. Nuclear BK channels regulate gene expression via the control of nuclear calcium signaling. Nat. Neurosci. 2014, 17, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes. Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef]
- Jing, G.; Wang, J.J.; Zhang, S.X. ER stress and apoptosis: A new mechanism for retinal cell death. Exp. Diabetes. Res. 2012, 2012, 589589. [Google Scholar] [CrossRef]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.P. BK Channels Alleviate Lysosomal Storage Diseases by Providing Positive Feedback Regulation of Lysosomal Ca2+ Release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef]
- Singh, H.; Stefani, E.; Toro, L. Intracellular BK(Ca) (iBK(Ca)) channels. J. Physiol. 2012, 590, 5937–5947. [Google Scholar] [CrossRef]
- Lai, M.H.; Wu, Y.; Gao, Z.; Anderson, M.E.; Dalziel, J.E.; Meredith, A.L. BK channels regulate sinoatrial node firing rate and cardiac pacing in vivo. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1327–H1338. [Google Scholar] [CrossRef] [PubMed]
- Lifshitz, L.M.; Carmichael, J.D.; Lai, F.A.; Sorrentino, V.; Bellve, K.; Fogarty, K.E.; ZhuGe, R. Spatial organization of RYRs and BK channels underlying the activation of STOCs by Ca(2+) sparks in airway myocytes. J. Gen. Physiol. 2011, 138, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Meredith, A.L.; Wiler, S.W.; Miller, B.H.; Takahashi, J.S.; Fodor, A.A.; Ruby, N.F.; Aldrich, R.W. BK calcium-activated potassium channels regulate circadian behavioral rhythms and pacemaker output. Nat. Neurosci. 2006, 9, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.E.; Zvara, P.; Meredith, A.L.; Aldrich, R.W.; Nelson, M.T. Erectile dysfunction in mice lacking the large-conductance calcium-activated potassium (BK) channel. J. Physiol. 2005, 567, 545–556. [Google Scholar] [CrossRef]
- Heppner, T.J.; Bonev, A.D.; Nelson, M.T. Ca(2+)-activated K+ channels regulate action potential repolarization in urinary bladder smooth muscle. Am. J. Physiol. 1997, 273, C110–C117. [Google Scholar] [CrossRef]
- Rao, S.G.; Bednarczyk, P.; Towheed, A.; Shah, K.; Karekar, P.; Ponnalagu, D.; Jensen, H.N.; Addya, S.; Reyes, B.A.S.; van Bockstaele, E.J.; et al. BKCa (Slo) Channel Regulates Mitochondrial Function and Lifespan in Drosophila melanogaster. Cells 2019, 9, 945. [Google Scholar]
- Bailey, C.S.; Moldenhauer, H.J.; Park, S.M.; Keros, S.; Meredith, A.L. KCNMA1-linked channelopathy. J. Gen. Physiol. 2019, 151, 1173–1189. [Google Scholar] [CrossRef]
- Du, X.; Carvalho-de-Souza, J.L.; Wei, C.; Carrasquel-Ursulaez, W.; Lorenzo, Y.; Gonzalez, N.; Kubota, T.; Staisch, J.; Hain, T.; Petrossian, N.; et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc. Natl. Acad. Sci. USA 2020, 117, 6023–6034. [Google Scholar] [CrossRef]
- Kcnma1 Channelopathy International Advocacy Foundation. Available online: https://www.kciaf.org/ (accessed on 14 August 2020).
- Magleby, K.L. Gating mechanism of BK (Slo1) channels: So near, yet so far. J. Gen. Physiol. 2003, 121, 81–96. [Google Scholar] [CrossRef]
- Horrigan, F.T.; Aldrich, R.W. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 2002, 120, 267–305. [Google Scholar] [CrossRef]
- Stefani, E.; Ottolia, M.; Noceti, F.; Olcese, R.; Wallner, M.; Latorre, R.; Toro, L. Voltage-controlled gating in a large conductance Ca2+-sensitive K+channel (hslo). Proc. Natl. Acad. Sci. USA 1997, 94, 5427–5431. [Google Scholar] [CrossRef] [PubMed]
- Meera, P.; Wallner, M.; Song, M.; Toro, L. Large conductance voltage- and calcium-dependent K+ channel, a distinct member of voltage-dependent ion channels with seven N-terminal transmembrane segments (S0-S6), an extracellular N terminus, and an intracellular (S9-S10) C terminus. Proc. Natl. Acad. Sci. USA 1997, 94, 14066–14071. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Leonetti, M.D.; Pico, A.R.; Hsiung, Y.; MacKinnon, R. Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science 2010, 329, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.H.; Aldrich, R.W. Role of the beta1 subunit in large-conductance Ca(2+)-activated K(+) channel gating energetics. Mechanisms of enhanced Ca(2+) sensitivity. J. Gen. Physiol. 2000, 116, 411–432. [Google Scholar] [CrossRef]
- Li, Q.; Yan, J. Modulation of BK Channel Function by Auxiliary Beta and Gamma Subunits. Int. Rev. Neurobiol. 2016, 128, 51–90. [Google Scholar]
- Gonzalez-Perez, V.; Lingle, C.J. Regulation of BK Channels by Beta and Gamma Subunits. Annu. Rev. Physiol. 2019, 81, 113–137. [Google Scholar] [CrossRef]
- Lam, A.; Karekar, P.; Shah, K.; Hariharan, G.; Fleyshman, M.; Kaur, H.; Singh, H.; Rao, S.G. Drosophila Voltage-Gated Calcium Channel alpha1-Subunits Regulate Cardiac Function in the Aging Heart. Sci. Rep. 2018, 8, 6910. [Google Scholar] [CrossRef]
- Ponnalagu, D.; Singh, H. Insights into the Role of Mitochondrial Ion Channels in Inflammatory Response. Front. Physiol. 2020, 11, 258. [Google Scholar] [CrossRef]
- Ponnalagu, D.; Singh, H. Anion Channels of Mitochondria. Handb. Exp. Pharmacol. 2017, 240, 71–101. [Google Scholar]
- Ponnalagu, D.; Rao, S.G.; Farber, J.; Xin, W.; Hussain, A.T.; Shah, K.; Tanda, S.; Berryman, M.; Edwards, J.C.; Singh, H. Molecular identity of cardiac mitochondrial chloride intracellular channel proteins. Mitochondrion 2016, 27, 6–14. [Google Scholar] [CrossRef]
- Martin, L.J.; Lau, E.; Singh, H.; Vergnes, L.; Tarling, E.J.; Mehrabian, M.; Mungrue, I.; Xiao, S.; Shih, D.; Castellani, L.; et al. ABCC6 localizes to the mitochondria-associated membrane. Circ. Res. 2012, 111, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.G.; Patel, N.J.; Singh, H. Intracellular Chloride Channels: Novel Biomarkers in Diseases. Front. Physiol. 2020, 11, 96. [Google Scholar]
- Patel, N.H.; Johannesen, J.; Shah, K.; Goswami, S.K.; Patel, N.J.; Ponnalagu, D.; Kohut, A.R.; Singh, H. Inhibition of BKCa negatively alters cardiovascular function. Physiol. Rep. 2018, 6, e13748. [Google Scholar] [CrossRef] [PubMed]
- Koster, O.F.; Szigeti, G.P.; Beuckelmann, D.J. Characterization of a [Ca2+]i-dependent current in human atrial and ventricular cardiomyocytes in the absence of Na+ and K+. Cardiovasc. Res. 1999, 41, 175–187. [Google Scholar] [CrossRef][Green Version]
- Brugada, R.; Tapscott, T.; Czernuszewicz, G.Z.; Marian, A.J.; Iglesias, A.; Mont, L.; Brugada, J.; Girona, J.; Domingo, A.; Bachinski, L.L.; et al. Identification of a genetic locus for familial atrial fibrillation. N. Engl. J. Med. 1997, 336, 905–911. [Google Scholar] [CrossRef]
- Dopico, A.M.; Bukiya, A.N.; Jaggar, J.H. Calcium- and voltage-gated BK channels in vascular smooth muscle. Pflugers Arch. Eur. J. Physiol. 2018, 470, 1271–1289. [Google Scholar] [CrossRef]
- Xu, W.; Liu, Y.; Wang, S.; McDonald, T.; van Eyk, J.E.; Sidor, A.; O’Rourke, B. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science 2002, 298, 1029–1033. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Nadtochiy, S.M.; Urciuoli, W.R.; Smith, C.O.; Grunnet, M.; Nehrke, K.; Brookes, P.S. A non-cardiomyocyte autonomous mechanism of cardioprotection involving the SLO1 BK channel. PeerJ 2013, 1, e48. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef]
- Stowe, D.F.; Aldakkak, M.; Camara, A.K.; Riess, M.L.; Heinen, A.; Varadarajan, S.G.; Jiang, M.T. Cardiac mitochondrial preconditioning by Big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H434–H440. [Google Scholar] [CrossRef]
- Soltysinska, E.; Bentzen, B.H.; Barthmes, M.; Hattel, H.; Thrush, A.B.; Harper, M.E.; Qvortrup, K.; Larsen, F.J.; Schiffer, T.A.; Losa-Reyna, J.; et al. KCNMA1 encoded cardiac BK channels afford protection against ischemia-reperfusion injury. PLoS ONE 2014, 9, e103402. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jiang, M.T.; Su, J.; Hutchins, W.; Konorev, E.; Baker, J.E. Mitochondrial big conductance KCa channel and cardioprotection in infant rabbit heart. J. Cardiovasc. Pharmacol. 2007, 50, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Saito, T.; Saegusa, N.; Nakaya, H. Mitochondrial Ca2+-activated K+ channels in cardiac myocytes: A mechanism of the cardioprotective effect and modulation by protein kinase A. Circulation 2005, 111, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ruttiger, L.; Sausbier, M.; Zimmermann, U.; Winter, H.; Braig, C.; Engel, J.; Knirsch, M.; Arntz, C.; Langer, P.; Hirt, B.; et al. Deletion of the Ca2+-activated potassium (BK) alpha-subunit but not the BKbeta1-subunit leads to progressive hearing loss. Proc. Natl. Acad. Sci. USA 2004, 101, 12922–12927. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.K.; Ponnalagu, D.; Hussain, A.T.; Shah, K.; Karekar, P.; Rao, S.G.; Meredith, A.L.; Khan, M.; Singh, H. Expression and Activation of BKCa Channels in Mice Protects Against Ischemia-Reperfusion Injury of Isolated Hearts by Modulating Mitochondrial Function. Front. Cardiovasc. Med. 2018, 5, 194. [Google Scholar] [CrossRef] [PubMed]
- Frankenreiter, S.; Bednarczyk, P.; Kniess, A.; Bork, N.I.; Straubinger, J.; Koprowski, P.; Wrzosek, A.; Mohr, E.; Logan, A.; Murphy, M.P.; et al. cGMP-Elevating Compounds and Ischemic Conditioning Provide Cardioprotection Against Ischemia and Reperfusion Injury via Cardiomyocyte-Specific BK Channels. Circulation 2017, 136, 2337–2355. [Google Scholar] [CrossRef]
- Brenner, R.; Perez, G.J.; Bonev, A.D.; Eckman, D.M.; Kosek, J.C.; Wiler, S.W.; Patterson, A.J.; Nelson, M.T.; Aldrich, R.W. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 2000, 407, 870–876. [Google Scholar] [CrossRef]
- Brayden, J.E.; Nelson, M.T. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 1992, 256, 532–535. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Osadchii, O.; Jespersen, T.; Hansen, R.S.; Olesen, S.P.; Grunnet, M. Activation of big conductance Ca(2+)-activated K (+) channels (BK) protects the heart against ischemia-reperfusion injury. Pflugers Arch. 2009, 457, 979–988. [Google Scholar] [CrossRef]
- Balderas, E.; Zhang, J.; Stefani, E.; Toro, L. Mitochondrial BKCa channel. Front. Physiol. 2015, 6, 104. [Google Scholar] [CrossRef]
- Gollasch, M.; Ried, C.; Bychkov, R.; Luft, F.C.; Haller, H. K+ currents in human coronary artery vascular smooth muscle cells. Circ. Res. 1996, 78, 676–688. [Google Scholar] [CrossRef]
- Leblanc, N.; Wan, X.; Leung, P.M. Physiological role of Ca(2+)-activated and voltage-dependent K+ currents in rabbit coronary myocytes. Am. J. Physiol. 1994, 266, C1523–C1537. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Cheng, H.; Rubart, M.; Santana, L.F.; Bonev, A.D.; Knot, H.J.; Lederer, W.J. Relaxation of arterial smooth muscle by calcium sparks. Science 1995, 270, 633–637. [Google Scholar] [CrossRef] [PubMed]
- McManus, O.B.; Helms, L.M.; Pallanck, L.; Ganetzky, B.; Swanson, R.; Leonard, R.J. Functional role of the beta subunit of high conductance calcium-activated potassium channels. Neuron 1995, 14, 645–650. [Google Scholar] [CrossRef]
- Grimm, P.R.; Sansom, S.C. BK channels and a new form of hypertension. Kidney Int. 2010, 78, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Imlach, W.L.; Finch, S.C.; Miller, J.H.; Meredith, A.L.; Dalziel, J.E. A role for BK channels in heart rate regulation in rodents. PLoS ONE 2010, 5, e8698. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Nargeot, J. Genesis and regulation of the heart automaticity. Physiol. Rev. 2008, 88, 919–982. [Google Scholar] [CrossRef]
- Vandael, D.H.; Marcantoni, A.; Mahapatra, S.; Caro, A.; Ruth, P.; Zuccotti, A.; Knipper, M.; Carbone, E. Ca(v)1.3 and BK channels for timing and regulating cell firing. Mol. Neurobiol. 2010, 42, 185–198. [Google Scholar] [CrossRef]
- Fakler, B.; Adelman, J.P. Control of K(Ca) channels by calcium nano/microdomains. Neuron 2008, 59, 873–881. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Wei, A.C.; Grunnet, M.; O’Rourke, B. Energetic performance is improved by specific activation of K+ fluxes through K(Ca) channels in heart mitochondria. Biochim. Biophys. Acta. 2010, 1797, 71–80. [Google Scholar] [CrossRef]
- Xu, C.; Chen, B.; Wang, W.; Tian, Y.; Zhao, H.; Jiang, B.; Gao, B.; Qin, S.; Yue, M.; Qi, G. Detecting residual ischemia and identifying coronary artery disease after myocardial infarction using dobutamine technetium-99m-MIBI SPECT. Chin. Med. J. 2000, 113, 579–583. [Google Scholar] [PubMed]
- Tang, Q.Y.; Qi, Z.; Naruse, K.; Sokabe, M. Characterization of a functionally expressed stretch-activated BKca channel cloned from chick ventricular myocytes. J. Membr. Biol. 2003, 196, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yu, Y.; Wu, X.; Liu, S.; Liu, B.; Du, J.; Li, B.; Jiang, L.; Feng, X. A Role of BK Channel in Regulation of Ca(2+) Channel in Ventricular Myocytes by Substrate Stiffness. Biophys. J. 2017, 112, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.C.; Agula, H.; Zhang, W.; Wang, F.; Sokabe, M.; Li, L.M. Membrane stretch and cytoplasmic Ca2+ independently modulate stretch-activated BK channel activity. J. Biomech. 2010, 43, 3015–3019. [Google Scholar] [CrossRef]
- Li, H.; Xu, J.; Shen, Z.S.; Wang, G.M.; Tang, M.; Du, X.R.; Lv, Y.T.; Wang, J.J.; Zhang, F.F.; Qi, Z.; et al. The neuropeptide GsMTx4 inhibits a mechanosensitive BK channel through the voltage-dependent modification specific to mechano-gating. J. Biol. Chem. 2019, 294, 11892–11909. [Google Scholar] [CrossRef]
- Banerjee, I.; Fuseler, J.W.; Price, R.L.; Borg, T.K.; Baudino, T.A. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1883–H1891. [Google Scholar] [CrossRef]
- Li, G.R.; Sun, H.Y.; Chen, J.B.; Zhou, Y.; Tse, H.F.; Lau, C.P. Characterization of multiple ion channels in cultured human cardiac fibroblasts. PLoS ONE 2009, 4, e7307. [Google Scholar] [CrossRef]
- Wang, Y.J.; Sung, R.J.; Lin, M.W.; Wu, S.N. Contribution of BK(Ca)-channel activity in human cardiac fibroblasts to electrical coupling of cardiomyocytes-fibroblasts. J. Membr. Biol. 2006, 213, 175–185. [Google Scholar] [CrossRef]
- Wang, Y.J.; Lin, M.W.; Wu, S.N.; Sung, R.J. The activation by estrogen receptor agonists of the BK(Ca)-channel in human cardiac fibroblasts. Biochem. Pharmacol. 2007, 73, 1347–1357. [Google Scholar] [CrossRef]
- Chilton, L.; Ohya, S.; Freed, D.; George, E.; Drobic, V.; Shibukawa, Y.; Maccannell, K.A.; Imaizumi, Y.; Clark, R.B.; Dixon, I.M.; et al. K+ currents regulate the resting membrane potential, proliferation, and contractile responses in ventricular fibroblasts and myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2931–H2939. [Google Scholar] [CrossRef]
- Vasquez, C.; Mohandas, P.; Louie, K.L.; Benamer, N.; Bapat, A.C.; Morley, G.E. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ. Res. 2010, 107, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Yang, H.; Lee, U.S. Molecular mechanisms of BK channel activation. Cell Mol. Life Sci. 2009, 66, 852–875. [Google Scholar] [CrossRef] [PubMed]
- Augustynek, B.; Kunz, W.S.; Szewczyk, A. Guide to the Pharmacology of Mitochondrial Potassium Channels. Handb. Exp. Pharmacol. 2017, 240, 103–127. [Google Scholar] [PubMed]
- Olesen, S.P.; Munch, E.; Watjen, F.; Drejer, J. NS 004--an activator of Ca(2+)-dependent K+ channels in cerebellar granule cells. Neuroreport 1994, 5, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Olesen, S.P.; Munch, E.; Moldt, P.; Drejer, J. Selective activation of Ca(2+)-dependent K+ channels by novel benzimidazolone. Eur. J. Pharmacol. 1994, 251, 53–59. [Google Scholar] [CrossRef]
- Redel, A.; Lange, M.; Jazbutyte, V.; Lotz, C.; Smul, T.M.; Roewer, N.; Kehl, F. Activation of mitochondrial large-conductance calcium-activated K+ channels via protein kinase A mediates desflurane-induced preconditioning. Anesth. Analg. 2008, 106, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yin, C.; Xi, L.; Kukreja, R.C. Opening of Ca2+-activated K+ channels triggers early and delayed preconditioning against I/R injury independent of NOS in mice. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2070–H2077. [Google Scholar] [CrossRef]
- Cao, C.M.; Xia, Q.; Gao, Q.; Chen, M.; Wong, T.M. Calcium-activated potassium channel triggers cardioprotection of ischemic preconditioning. J. Pharmacol. Exp. Ther. 2005, 312, 644–650. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, S.Z.; Cao, C.M.; Bruce, I.C.; Xia, Q. The mitochondrial permeability transition pore and the Ca2+-activated K+ channel contribute to the cardioprotection conferred by tumor necrosis factor-alpha. Cytokine 2005, 32, 199–205. [Google Scholar] [CrossRef]
- Shintani, Y.; Node, K.; Asanuma, H.; Sanada, S.; Takashima, S.; Asano, Y.; Liao, Y.; Fujita, M.; Hirata, A.; Shinozaki, Y.; et al. Opening of Ca2+-activated K+ channels is involved in ischemic preconditioning in canine hearts. J. Mol. Cell Cardiol. 2004, 37, 1213–1218. [Google Scholar] [CrossRef]
- Park, W.S.; Kang, S.H.; Son, Y.K.; Kim, N.; Ko, J.H.; Kim, H.K.; Ko, E.A.; Kim, C.D.; Han, J. The mitochondrial Ca2+-activated K+ channel activator, NS 1619 inhibits L-type Ca2+ channels in rat ventricular myocytes. Biochem. Biophys. Res. Commun. 2007, 362, 31–36. [Google Scholar] [CrossRef]
- Saleh, S.N.; Angermann, J.E.; Sones, W.R.; Leblanc, N.; Greenwood, I.A. Stimulation of Ca2+-gated Cl- currents by the calcium-dependent K+ channel modulators NS1619 [1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-2H-benzi midazol-2-one] and isopimaric acid. J. Pharmacol. Exp. Ther. 2007, 321, 1075–1084. [Google Scholar] [CrossRef]
- Edwards, G.; Niederste-Hollenberg, A.; Schneider, J.; Noack, T.; Weston, A.H. Ion channel modulation by NS 1619, the putative BKCa channel opener, in vascular smooth muscle. Br. J. Pharmacol. 1994, 113, 1538–1547. [Google Scholar] [CrossRef]
- Holland, M.; Langton, P.D.; Standen, N.B.; Boyle, J.P. Effects of the BKCa channel activator, NS1619, on rat cerebral artery smooth muscle. Br. J. Pharmacol. 1996, 117, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Lukasiak, A.; Skup, A.; Chlopicki, S.; Lomnicka, M.; Kaczara, P.; Proniewski, B.; Szewczyk, A.; Wrzosek, A. SERCA, complex I of the respiratory chain and ATP-synthase inhibition are involved in pleiotropic effects of NS1619 on endothelial cells. Eur. J. Pharmacol. 2016, 786, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Cancherini, D.V.; Queliconi, B.B.; Kowaltowski, A.J. Pharmacological and physiological stimuli do not promote Ca(2+)-sensitive K+ channel activity in isolated heart mitochondria. Cardiovasc. Res. 2007, 73, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, B.H.; Nardi, A.; Calloe, K.; Madsen, L.S.; Olesen, S.P.; Grunnet, M. The small molecule NS11021 is a potent and specific activator of Ca2+-activated big-conductance K+ channels. Mol. Pharmacol. 2007, 72, 1033–1044. [Google Scholar] [CrossRef]
- Cheng, Y.; Gu, X.Q.; Bednarczyk, P.; Wiedemann, F.R.; Haddad, G.G.; Siemen, D. Hypoxia increases activity of the BK-channel in the inner mitochondrial membrane and reduces activity of the permeability transition pore. Cell Physiol. Biochem. 2008, 22, 127–136. [Google Scholar] [CrossRef]
- Hermann, A.; Sitdikova, G.F.; Weiger, T.M. Oxidative Stress and Maxi Calcium-Activated Potassium (BK) Channels. Biomolecules 2015, 5, 1870–1911. [Google Scholar] [CrossRef]
- Hou, S.; Xu, R.; Heinemann, S.H.; Hoshi, T. The RCK1 high-affinity Ca2+ sensor confers carbon monoxide sensitivity to Slo1 BK channels. Proc. Natl. Acad. Sci. USA 2008, 105, 4039–4043. [Google Scholar] [CrossRef]
- Hou, S.; Heinemann, S.H.; Hoshi, T. Modulation of BKCa channel gating by endogenous signaling molecules. Physiol. Bethesda 2009, 24, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Hayabuchi, Y.; Nakaya, Y.; Matsuoka, S.; Kuroda, Y. Effect of acidosis on Ca2+-activated K+ channels in cultured porcine coronary artery smooth muscle cells. Pflugers Arch. 1998, 436, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Xu, R.; Heinemann, S.H.; Hoshi, T. Reciprocal regulation of the Ca2+ and H+ sensitivity in the SLO1 BK channel conferred by the RCK1 domain. Nat. Struct. Mol. Biol. 2008, 15, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef]
- Avdonin, V.; Tang, X.D.; Hoshi, T. Stimulatory action of internal protons on Slo1 BK channels. Biophys. J. 2003, 84, 2969–2980. [Google Scholar] [CrossRef]
- Bolotina, V.M.; Najibi, S.; Palacino, J.J.; Pagano, P.J.; Cohen, R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Brakemeier, S.; Eichler, I.; Knorr, A.; Fassheber, T.; Kohler, R.; Hoyer, J. Modulation of Ca2+-activated K+ channel in renal artery endothelium in situ by nitric oxide and reactive oxygen species. Kidney Int. 2003, 64, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Peers, C.; Bauer, C.C.; Boyle, J.P.; Scragg, J.L.; Dallas, M.L. Modulation of ion channels by hydrogen sulfide. Antioxid. Redox Signal. 2010, 17, 95–105. [Google Scholar] [CrossRef]
- Telezhkin, V.; Brazier, S.P.; Cayzac, S.H.; Wilkinson, W.J.; Riccardi, D.; Kemp, P.J. Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir. Physiol. Neurobiol. 2010, 172, 169–178. [Google Scholar] [CrossRef]
- Santarelli, L.C.; Chen, J.; Heinemann, S.H.; Hoshi, T. The beta1 subunit enhances oxidative regulation of large-conductance calcium-activated K+ channels. J. Gen. Physiol. 2004, 124, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Sitdikova, G.F.; Fuchs, R.; Kainz, V.; Weiger, T.M.; Hermann, A. Phosphorylation of BK channels modulates the sensitivity to hydrogen sulfide (H2S). Front. Physiol. 2014, 5, 431. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.H.; Hlavackova, M.; Kolar, F. Pharmacological activation of mitochondrial BK(Ca) channels protects isolated cardiomyocytes against simulated reperfusion-induced injury. Exp. Biol. Med. 2013, 238, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox modulation of vascular tone: Focus of potassium channel mechanisms of dilation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 671–678. [Google Scholar] [CrossRef]
- Yellen, G. Ionic permeation and blockade in Ca2+-activated K+ channels of bovine chromaffin cells. J. Gen. Physiol. 1984, 84, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, N.; Petersen, O.H. Action of tetraethylammonium on calcium-activated potassium channels in pig pancreatic acinar cells studied by patch-clamp single-channel and whole-cell current recording. J. Membr. Biol. 1985, 86, 139–144. [Google Scholar] [CrossRef]
- Lenaeus, M.J.; Vamvouka, M.; Focia, P.J.; Gross, A. Structural basis of TEA blockade in a model potassium channel. Nat. Struct. Mol. Biol. 2005, 12, 454–459. [Google Scholar] [CrossRef]
- Nardi, A.; Olesen, S.P. BK channel modulators: A comprehensive overview. Curr. Med. Chem. 2008, 15, 1126–1146. [Google Scholar] [CrossRef]
- Miller, C.; Moczydlowski, E.; Latorre, R.; Phillips, M. Charybdotoxin, a protein inhibitor of single Ca2+-activated K+ channels from mammalian skeletal muscle. Nature 1985, 313, 316–318. [Google Scholar] [CrossRef]
- Panyi, G.; Possani, L.D.; de la Vega, R.C.R.; Gaspar, R.; Varga, Z. K+ channel blockers: Novel tools to inhibit T cell activation leading to specific immunosuppression. Curr. Pharm. Des. 2006, 12, 2199–2220. [Google Scholar] [CrossRef]
- Galvez, A.; Gimenez-Gallego, G.; Reuben, J.P.; Roy-Contancin, L.; Feigenbaum, P.; Kaczorowski, G.J.; Garcia, M.L. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J. Biol. Chem. 1990, 265, 11083–11090. [Google Scholar] [PubMed]
- Candia, S.; Garcia, M.L.; Latorre, R. Mode of action of iberiotoxin, a potent blocker of the large conductance Ca(2+)-activated K+ channel. Biophys. J. 1992, 63, 583–590. [Google Scholar] [CrossRef]
- Nardi, A.; Calderone, V.; Chericoni, S.; Morelli, I. Natural modulators of large-conductance calcium-activated potassium channels. Planta Med. 2003, 69, 885–892. [Google Scholar]
- Knaus, H.G.; McManus, O.B.; Lee, S.H.; Schmalhofer, W.A.; Garcia-Calvo, M.; Helms, L.M.; Sanchez, M.; Giangiacomo, K.; Reuben, J.P.; Smith, A.B., 3rd; et al. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 1994, 33, 5819–5828. [Google Scholar] [CrossRef]
- Sanchez, M.; McManus, O.B. Paxilline inhibition of the alpha-subunit of the high-conductance calcium-activated potassium channel. Neuropharmacology 1996, 35, 963–968. [Google Scholar] [CrossRef]
- Padmanaban, G.; Venkateswar, V.; Rangarajan, P.N. Haem as a multifunctional regulator. Trends Biochem. Sci. 1989, 14, 492–496. [Google Scholar] [CrossRef]
- Hou, S.; Reynolds, M.F.; Horrigan, F.T.; Heinemann, S.H.; Hoshi, T. Reversible binding of heme to proteins in cellular signal transduction. Acc. Chem. Res. 2006, 39, 918–924. [Google Scholar] [CrossRef]
- Horrigan, F.T.; Heinemann, S.H.; Hoshi, T. Heme regulates allosteric activation of the Slo1 BK channel. J. Gen. Physiol. 2005, 126, 7–21. [Google Scholar] [CrossRef]
- Tang, X.D.; Xu, R.; Reynolds, M.F.; Garcia, M.L.; Heinemann, S.H.; Hoshi, T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature 2003, 425, 531–535. [Google Scholar] [CrossRef]
- Jaggar, J.H.; Li, A.; Parfenova, H.; Liu, J.; Umstot, E.S.; Dopico, A.M.; Leffler, C.W. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ. Res. 2005, 97, 805–812. [Google Scholar] [CrossRef]
- Wang, B.; Jaffe, D.B.; Brenner, R. Current understanding of iberiotoxin-resistant BK channels in the nervous system. Front. Physiol. 2014, 5, 382. [Google Scholar] [CrossRef]
- Heinen, A.; Winning, A.; Schlack, W.; Hollmann, M.W.; Preckel, B.; Frassdorf, J.; Weber, N.C. The regulation of mitochondrial respiration by opening of mKCa channels is age-dependent. Eur. J. Pharmacol. 2008, 578, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Meredith, A.L.; Thorneloe, K.S.; Werner, M.E.; Nelson, M.T.; Aldrich, R.W. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J. Biol. Chem. 2004, 279, 36746–36752. [Google Scholar] [CrossRef] [PubMed]
- Kyle, B.D.; Hurst, S.; Swayze, R.D.; Sheng, J.; Braun, A.P. Specific phosphorylation sites underlie the stimulation of a large conductance, Ca(2+)-activated K(+) channel by cGMP-dependent protein kinase. FASEB J. 2013, 27, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Swayze, R.D.; Braun, A.P. A catalytically inactive mutant of type I cGMP-dependent protein kinase prevents enhancement of large conductance, calcium-sensitive K+ channels by sodium nitroprusside and cGMP. J. Biol. Chem. 2001, 276, 19729–19737. [Google Scholar] [CrossRef]
- Zhou, X.B.; Wulfsen, I.; Utku, E.; Sausbier, U.; Sausbier, M.; Wieland, T.; Ruth, P.; Korth, M. Dual role of protein kinase C on BK channel regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 8005–8010. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. American Heart Association Council on, C. Prevention Statistics, and S. Stroke Statistics, Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Olesen, S.P.; Ronn, L.C.; Grunnet, M. BK channel activators and their therapeutic perspectives. Front. Physiol. 2014, 5, 389. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szteyn, K.; Singh, H. BKCa Channels as Targets for Cardioprotection. Antioxidants 2020, 9, 760. https://doi.org/10.3390/antiox9080760
Szteyn K, Singh H. BKCa Channels as Targets for Cardioprotection. Antioxidants. 2020; 9(8):760. https://doi.org/10.3390/antiox9080760
Chicago/Turabian StyleSzteyn, Kalina, and Harpreet Singh. 2020. "BKCa Channels as Targets for Cardioprotection" Antioxidants 9, no. 8: 760. https://doi.org/10.3390/antiox9080760
APA StyleSzteyn, K., & Singh, H. (2020). BKCa Channels as Targets for Cardioprotection. Antioxidants, 9(8), 760. https://doi.org/10.3390/antiox9080760