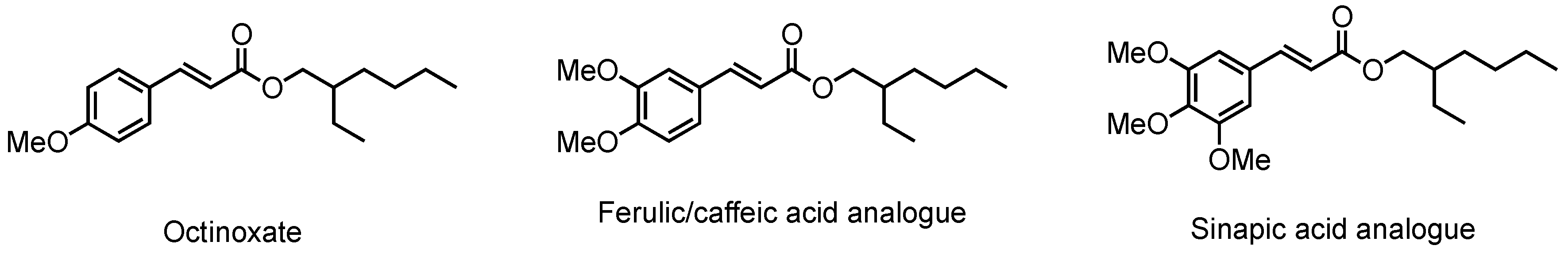
Sinapic Acid Esters: Octinoxate Substitutes Combining Suitable UV Protection and Antioxidant Activity




Abstract
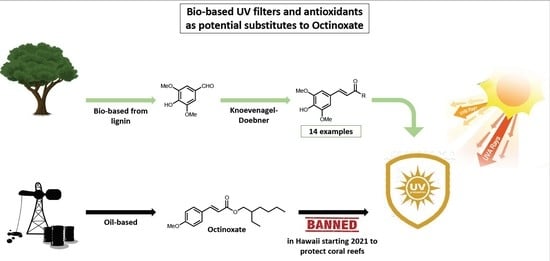
:
1. Introduction
2. Materials and Methods
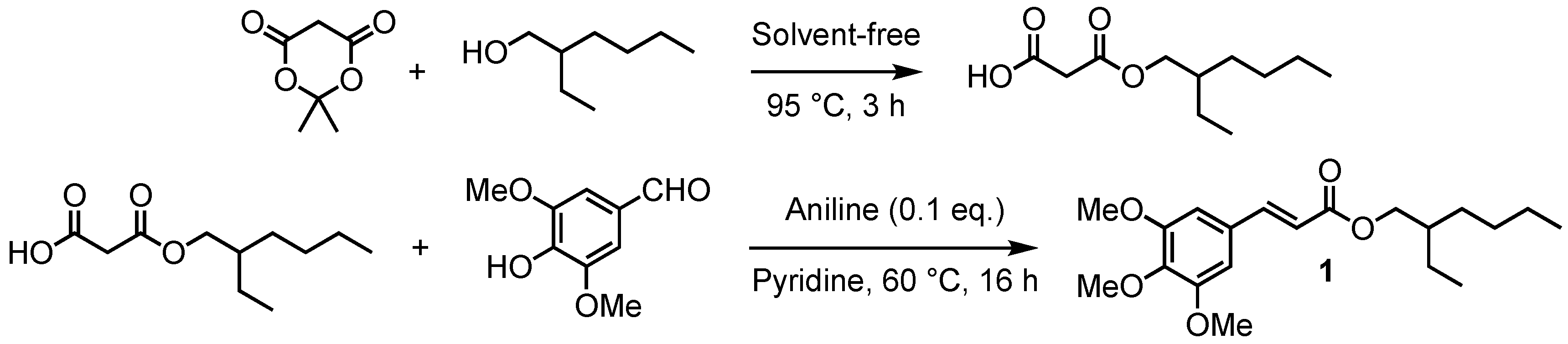
2.1. Chemical Synthesis
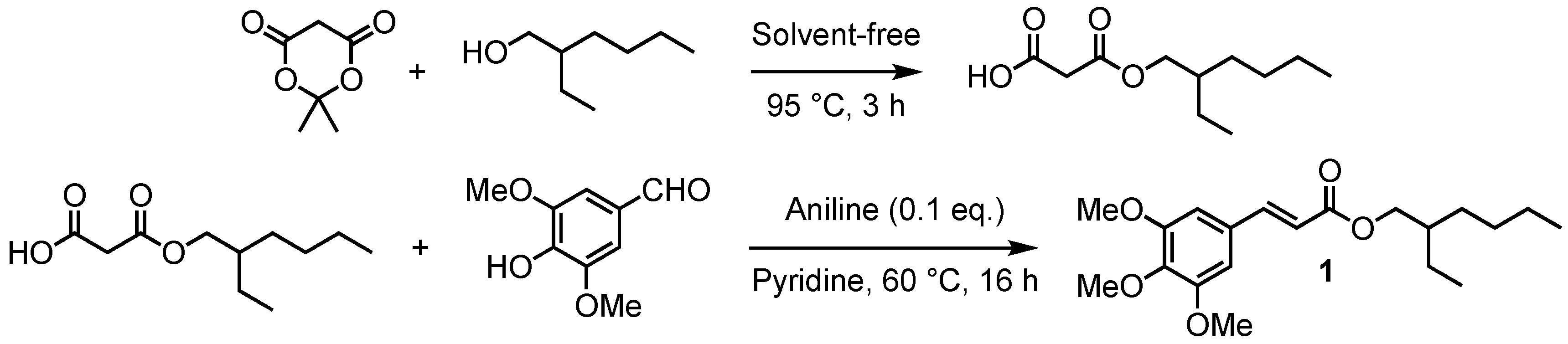
2.1.1. Malonate Mono-Esters General Procedures
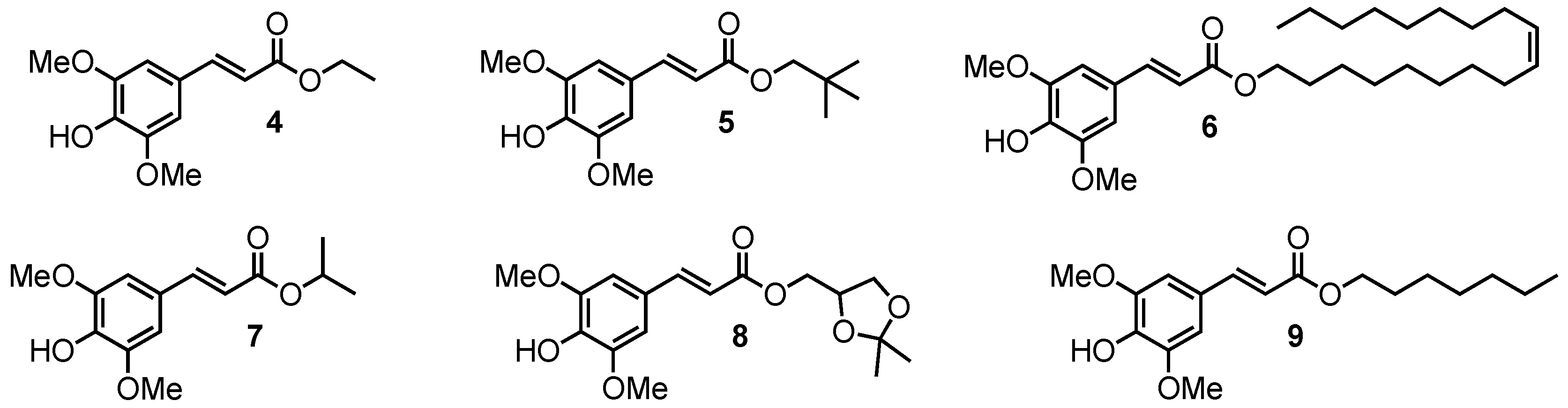
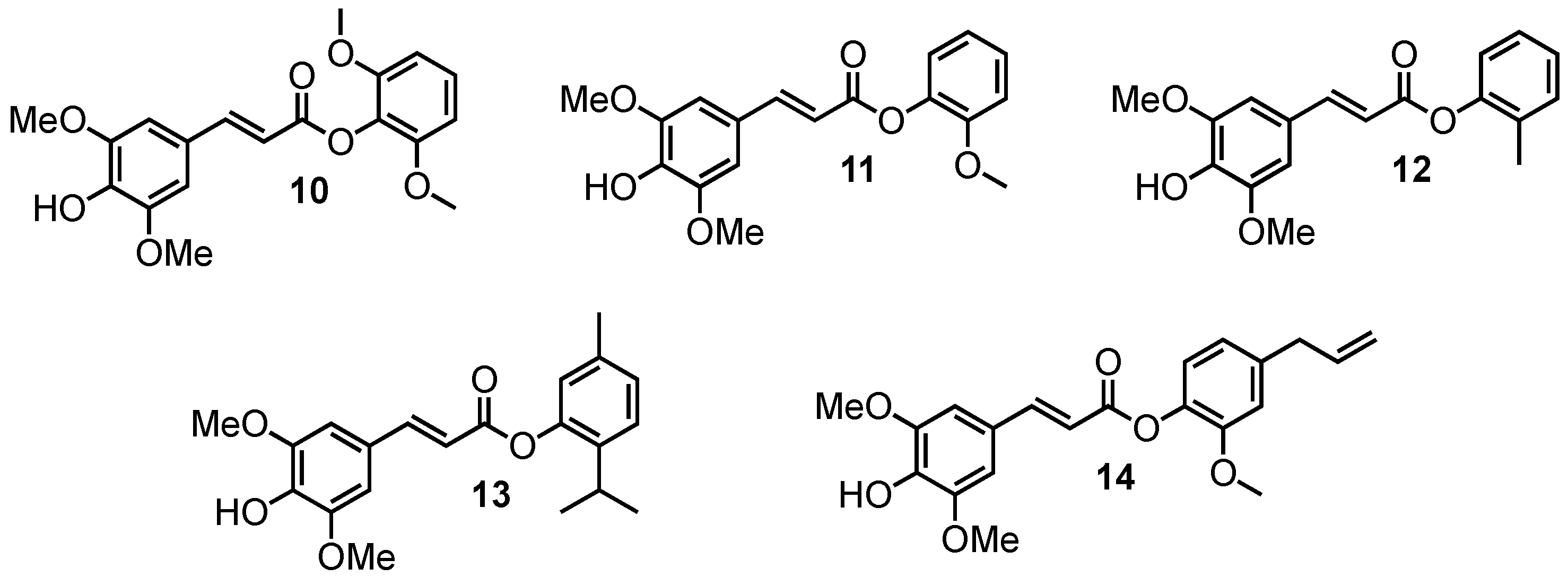
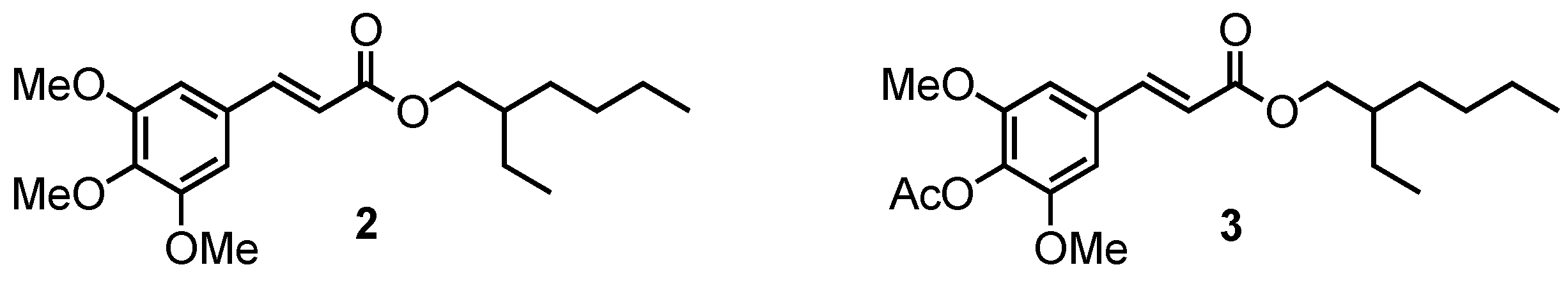
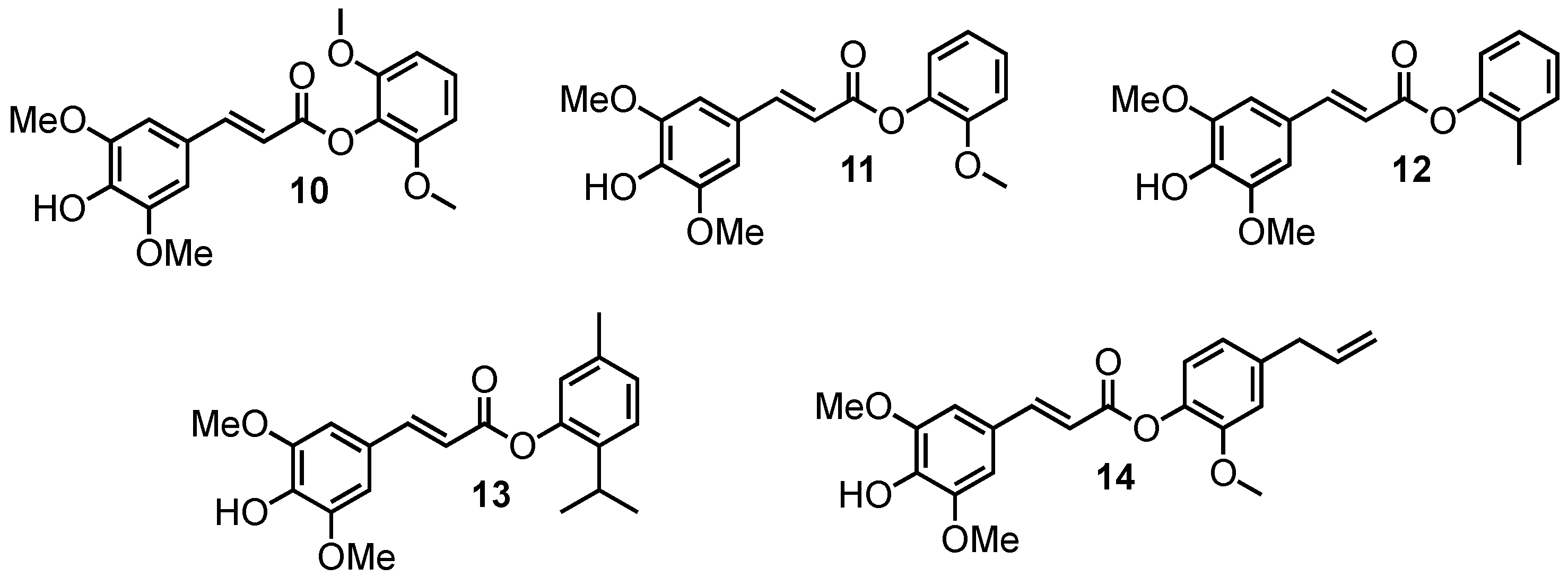
2.1.2. Sinapate Esters
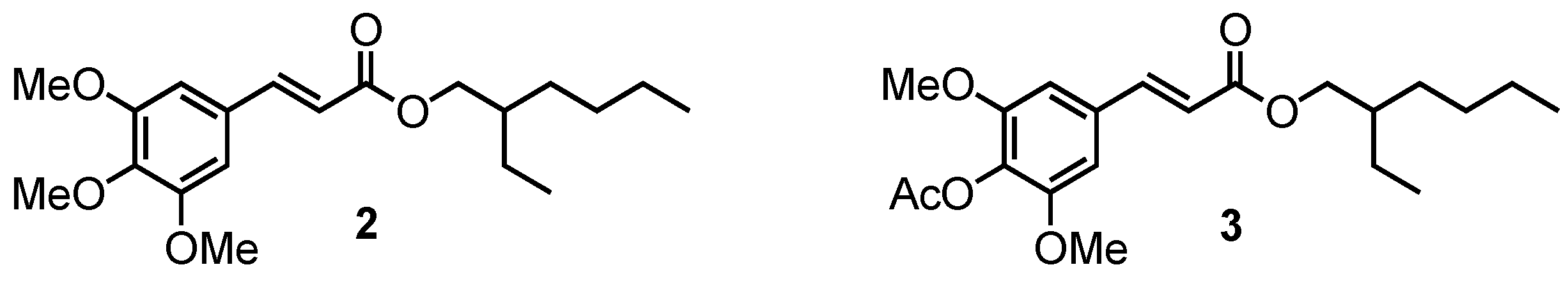
2.1.3. Methylation of (4)
2.1.4. Acetylation of (4)
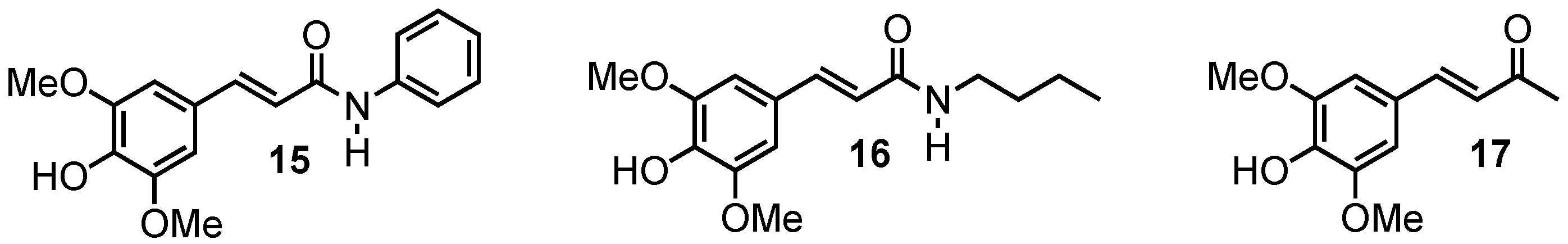
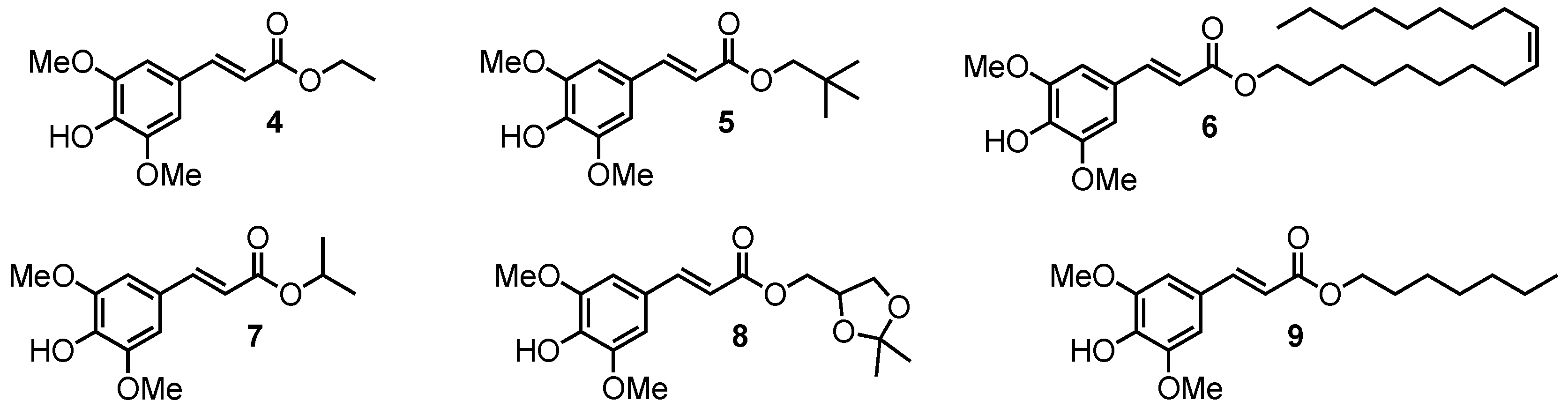
2.1.5. Amide Derivatives
2.1.6. Ketone Derivative
2.1.7. UV Analysis & Loss of Absorbance (LoA)
2.1.8. DPPH Inhibition
3. Results and Discussion
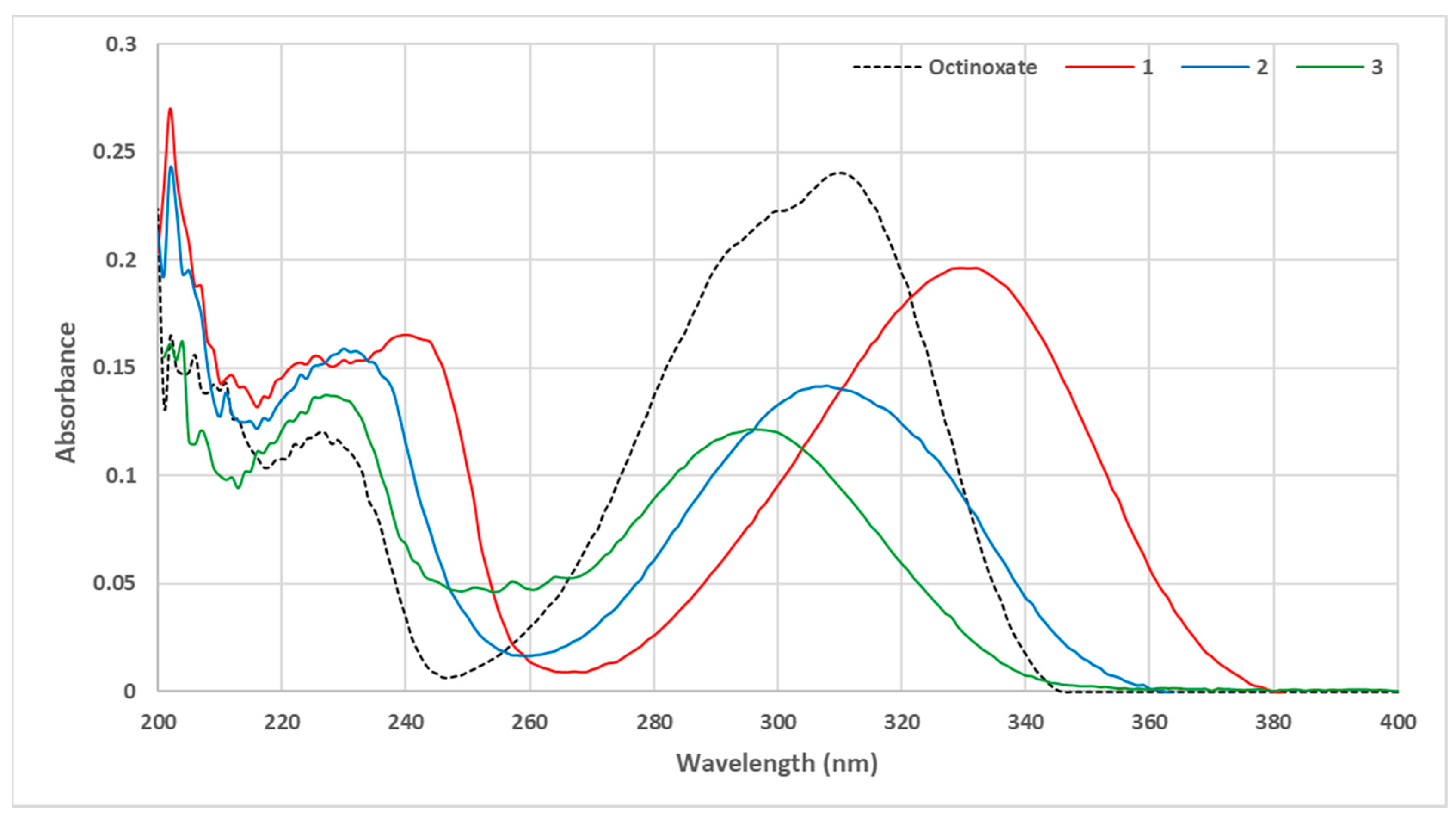
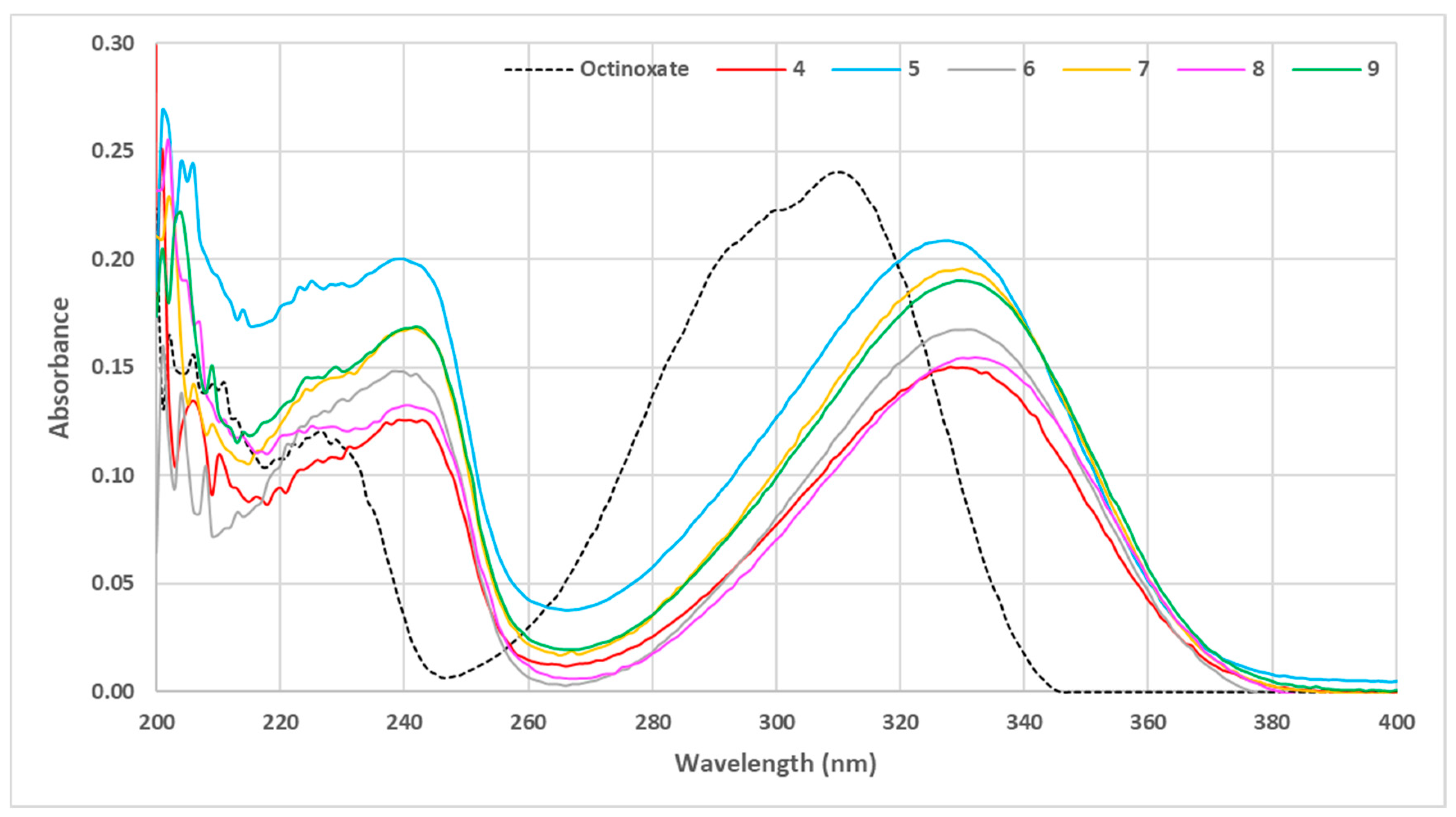
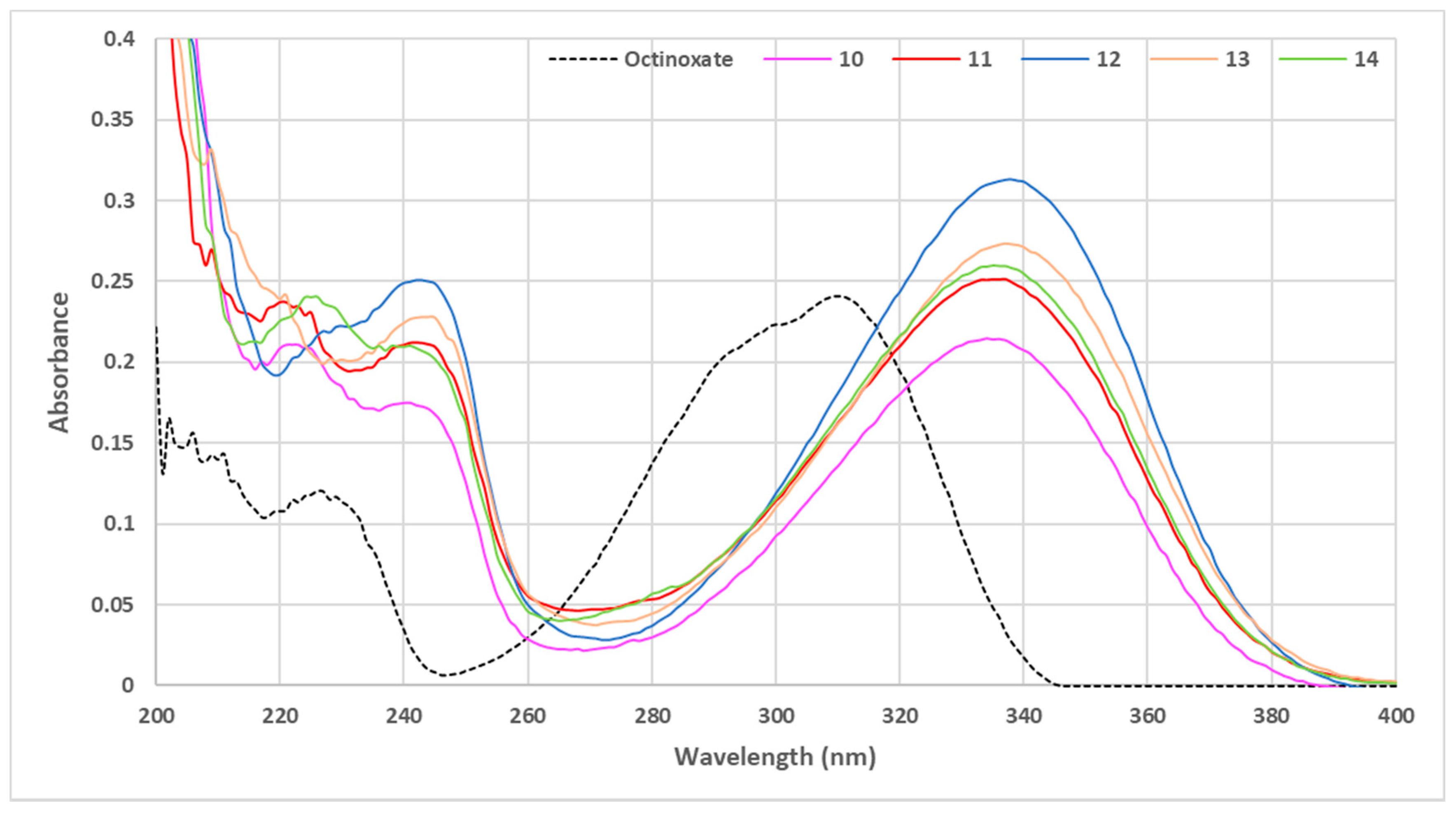
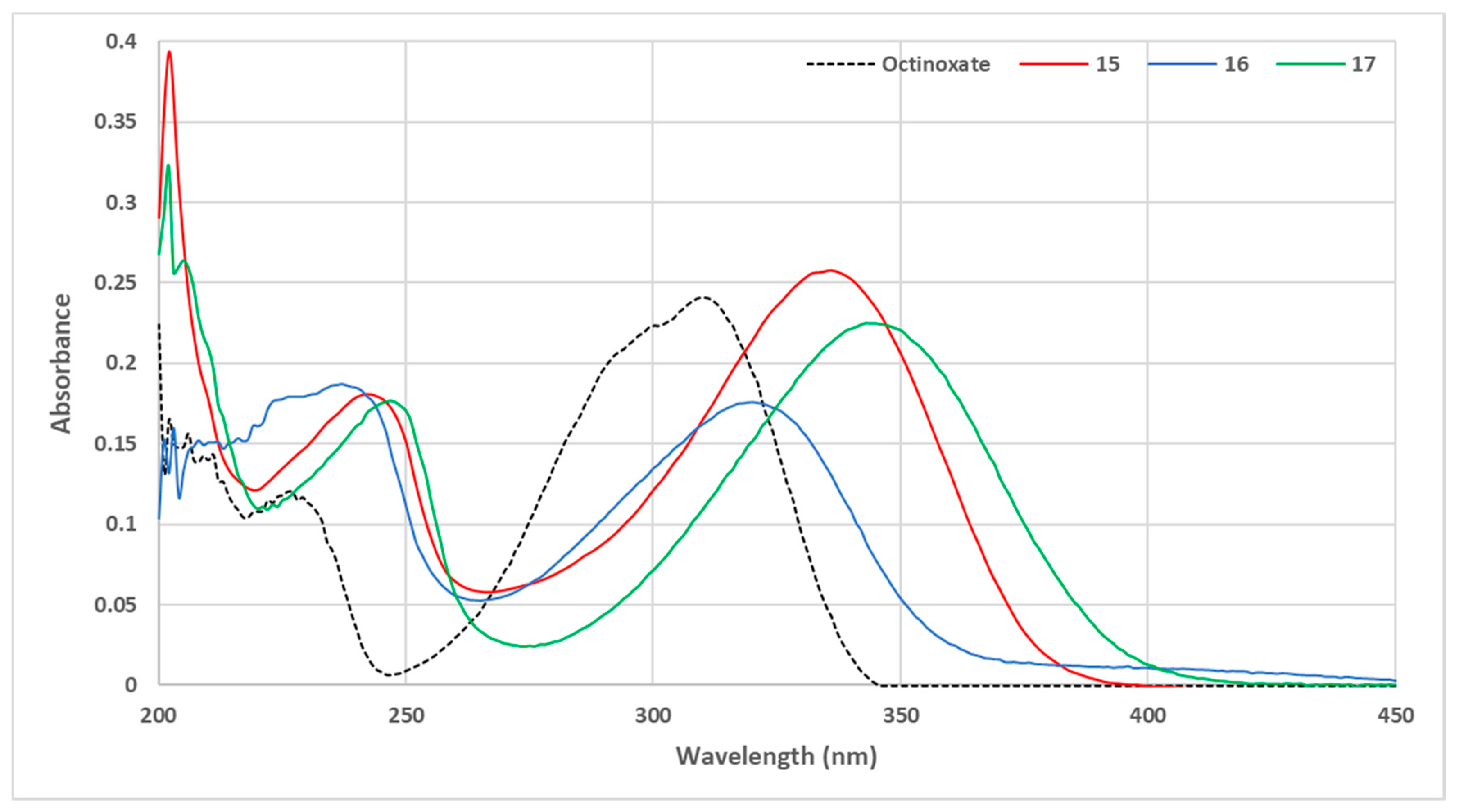
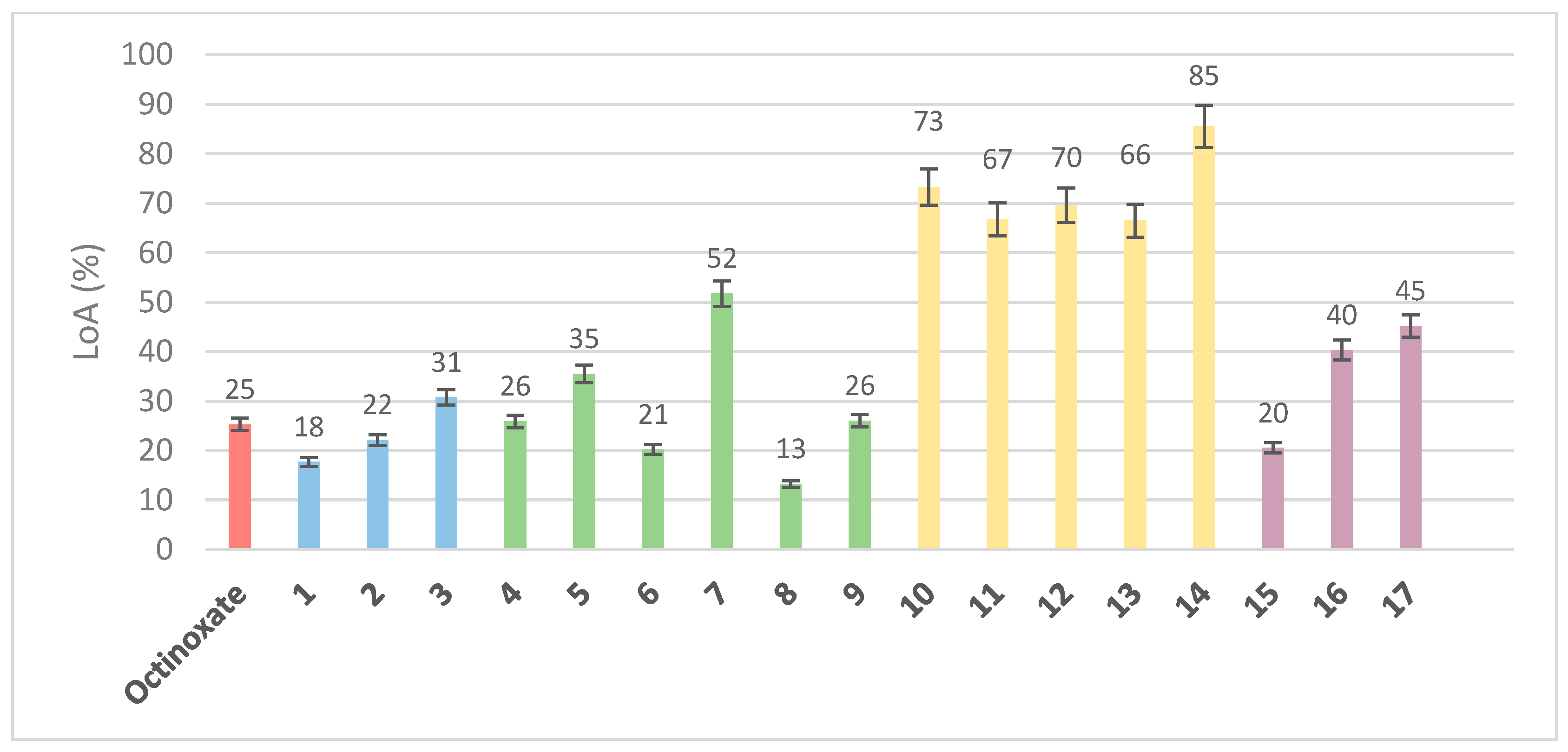
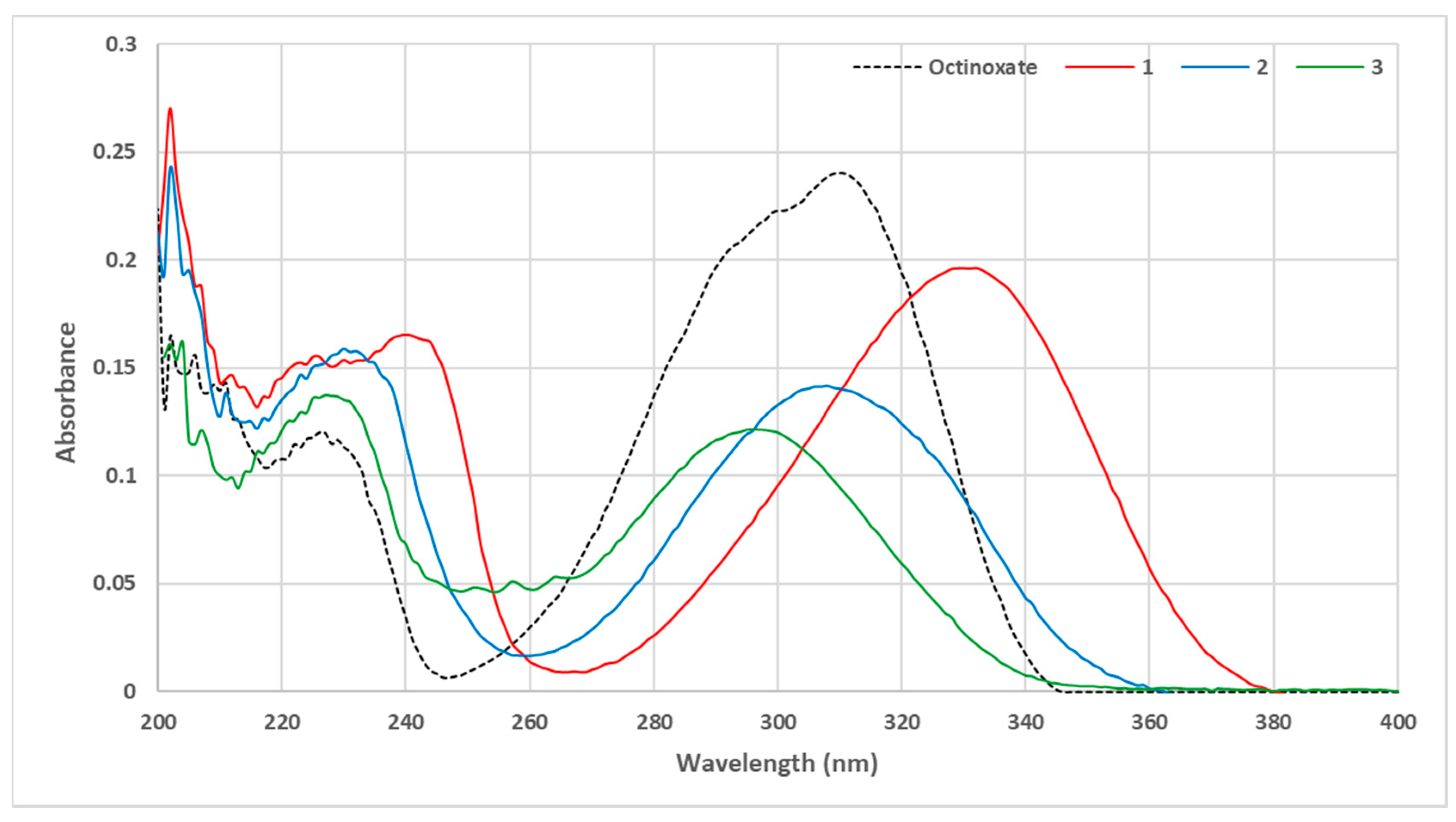
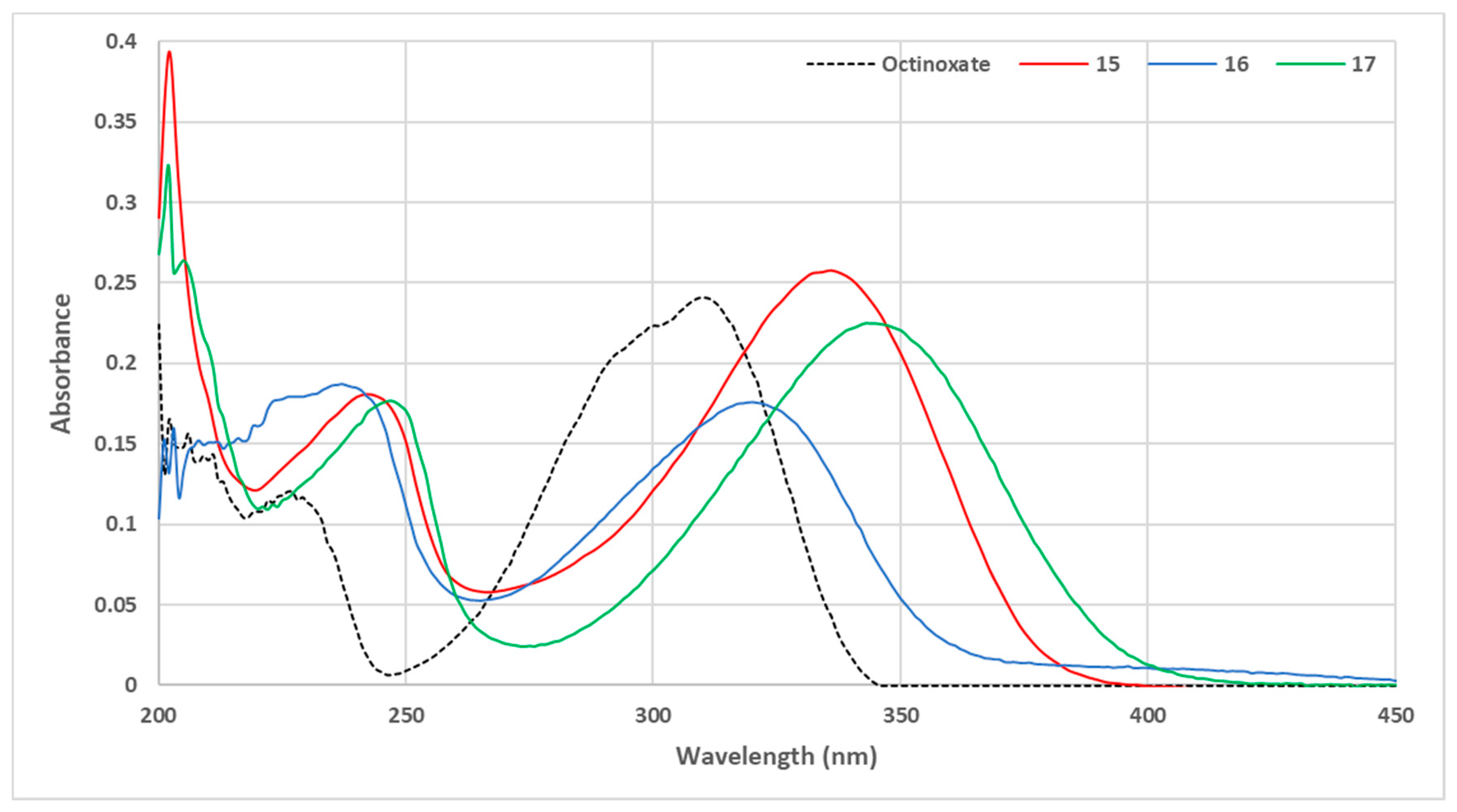
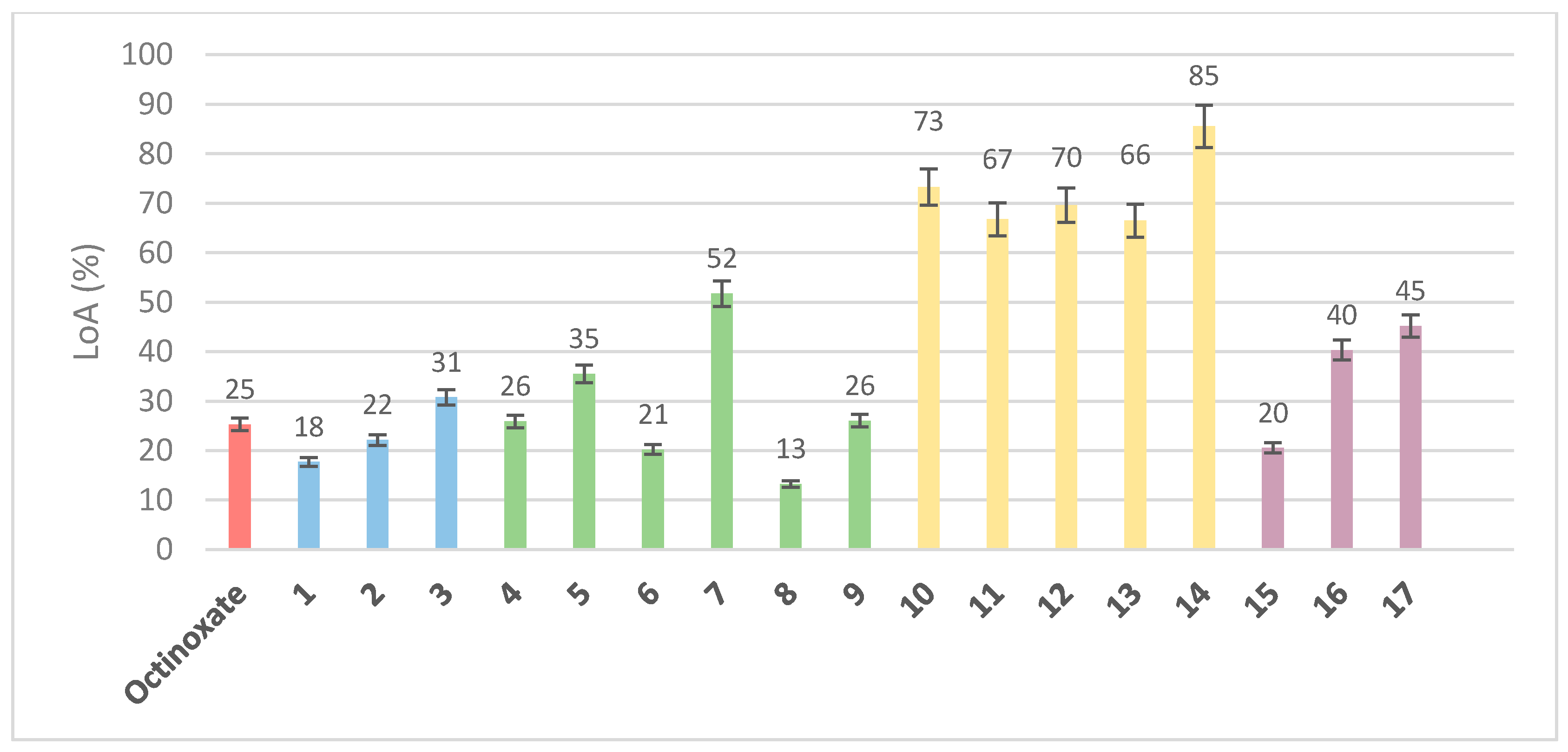
3.1. Loss of Absorbance (LoA)
3.2. Antioxidant Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pillai, S.; Oresajo, C.; Hayward, J. Ultraviolet radiation and skin aging: Roles of reactive oxygen species, inflammation and protease activation, and strategies for prevention of inflammation-induced matrix degradation—A review. Int. J. Cosmet. Sci. 2005, 27, 17–34. [Google Scholar] [CrossRef] [PubMed]
- De Gruijl, F.R. Skin cancer and solar UV radiation. Eur. J. Cancer 1999, 35, 2003–2009. [Google Scholar] [CrossRef]
- Mohania, D.; Chandel, S.; Kumar, P.; Verma, V.; Digvijay, K.; Tripathi, D.; Choudhury, K.; Mitten, S.K.; Shah, D. Ultraviolet radiations: Skin defense-damage mechanism. Exp. Med. Biol. 2017, 996, 71–87. [Google Scholar] [CrossRef]
- Schneider, S.L.; Lim, H.W. A review of inorganic UV filters zinc oxide and titanium dioxide. Photodermatol. Photoimmunol. Photomed. 2018, 35, 442–446. [Google Scholar] [CrossRef] [Green Version]
- Serpone, N.; Dondi, D.; Albini, A. Inorganic and organic UV filters: Their role and efficacy in sunscreens and suncare products. Inorganica Chim. Acta 2007, 360, 794–802. [Google Scholar] [CrossRef]
- Siller, A.; Blaszak, S.C.; Lazar, M.; Harken, E.O. Update about the effects of the sunscreen ingredients oxybenzone and octinoxate on humans and the environment. Plast. Surg. Nurs. 2018, 38, 158–161. [Google Scholar] [CrossRef]
- Narla, S.; Lim, H.W. Sunscreen: FDA regulation, and environmental and health impact. Photochem. Photobiol. Sci. 2020, 19, 66–70. [Google Scholar] [CrossRef]
- Danovaro, R.; Bongiorni, L.; Corinaldesi, C.; Giovannelli, D.; Damiani, E.; Astolfi, P.; Greci, L.; Pusceddu, A. Sunscreens cause coral bleaching by promoting viral infections. Environ. Health Perspect. 2008, 116, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Downs, C.A.; Kramarsky-Winter, E.; Segal, R.; Fauth, J.; Knutson, S.; Bronstein, O.; Ciner, F.R.; Jeger, R.; Lichtenfeld, Y.; Woodley, C.M.; et al. Toxicopathological effects of the sunscreen UV filter, oxybenzone (Benzophenone-3), on Coral Planulae and Cultured primary cells and its environmental contamination in Hawaii and the U.S. Virgin Islands. Arch. Environ. Contam. Toxicol. 2015, 70, 265–288. [Google Scholar] [CrossRef]
- Ouchene, L.; Litvinov, I.V.; Netchiporouk, E. Hawaii and other jurisdictions ban oxybenzone or octinoxate sunscreens based on the confirmed adverse environmental effects of sunscreen ingredients on aquatic environments. J. Cutan. Med. Surg. 2019, 23, 648–649. [Google Scholar] [CrossRef]
- Wang, J.; Pan, L.; Wu, S.; Lu, L.; Xu, Y.; Zhu, Y.; Guo, M.; Zhuang, S. Recent advances on endocrine disrupting effects of UV filters. Int. J. Environ. Res. Public Health 2016, 13, 782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenstadt, A.; Keren, Y. Process for the Preparation of Octyl Methoxy Cinnamate. U.S. Patent 5187303A, 16 February 1993. [Google Scholar]
- Li, F.; Meng, J. Preparing Method and Application of p-Methoxy Cinnamate Ester. China Patent 106831415A, 8 August 2012. [Google Scholar]
- Hu, W.; Meng, J. Process for the Preparation of Sun Screening Agent 2-Ethylhexyl p-Methoxycinnamate. China Patent 105503596A, 20 April 2016. [Google Scholar]
- Lin, D.; Yang, Q.-C.; Wang, Y.-F.; Zhang, M.-F. High efficient synthesis of octyl 4-methoxy cinnamate. Huaxue Yu Shengwu Gongcheng 2012, 29, 47–49. [Google Scholar] [CrossRef]
- Hamasaka, G.; Ichii, S.; Uozumi, Y. A Palladium NNC-pincer complex as an efficient catalyst precursor for the Mizoroki−Heck reaction. Adv. Synth. Catal. 2018, 360, 1833–1840. [Google Scholar] [CrossRef]
- Sharma, Y.O.; Degani, M.S. The Heck reaction of aryl bromides: A green protocol for synthesis of 2-ethylhexyl-4-methoxy cinnamate. Green Chem. Lett. Rev. 2010, 3, 201–204. [Google Scholar] [CrossRef]
- Wang, Y.F.; Yang, Q.C.; Zhang, B.J.; Lin, D.; Zhang, M.J. An efficient protocol for synthesis of 4-methoxycinnamate in ionic liquid. J. Indian Chem. Soc. 2013, 90, 489–492. [Google Scholar]
- Schutyser, W.; Renders, T.; Bosch, S.V.D.; Koelewijn, S.-F.; Beckham, G.T.; Sels, B. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Doehring, A.; Fonseca, M.T.H.; Job, A.; Torres, R.R. A Practical, efficient, and atom economic alternative to the Wittig and Horner–Wadsworth–Emmons reactions for the synthesis of (E)-α,β-unsaturated esters from aldehydes. Tetrahedron 2006, 62, 476–482. [Google Scholar] [CrossRef]
- Yadav, V.G.; Chandalia, S.B. An efficient method for synthesis of 2-ethylhexyl 4-methoxycinnamate, a raw material for the cosmetics industry. Indian J. Chem. Technol. 1999, 6, 19–23. [Google Scholar]
- Kumar, V.; Jahan, F.; Kameswaran, K.; Mahajan, R.V.; Saxena, R.K. Eco-friendly methodology for efficient synthesis and scale-up of 2-ethylhexyl-p-methoxycinnamate using Rhizopus oryzae lipase and its biological evaluation. J. Ind. Microbiol. Biotechnol. 2014, 41, 907–912. [Google Scholar] [CrossRef]
- Yvergnaux, F.; Forveille, M.; Callegari, J.P. Enzymatic Synthesis of Esters of Cinnamic Acid and Its Derivatives. France Patent 2736354A1, 10 January 1997. [Google Scholar]
- Monhaphol, T.; Albinsson, B.; Wanichwecharungruang, S. 2-Ethylhexyl-2,4,5-trimethoxycinnamate and di-(2-ethylhexyl)-2,4,5-trimethoxybenzalmalonate as novel UVA filters. J. Pharm. Pharmacol. 2007, 59, 279–288. [Google Scholar] [CrossRef]
- Zeng, Q.-Y. New process for the synthesis of 2-ethylhexyl p-methoxy cinnamate. Huaxue Gongchengshi 2007, 21, 13–15. [Google Scholar]
- Mention, M.M.; Flourat, A.; Peyrot, C.; Allais, F. Biomimetic regioselective and high-yielding Cu(I)-catalyzed dimerization of sinapate esters in green solvent Cyrene™: Towards sustainable antioxidant and anti-UV ingredients. Green Chem. 2020, 22, 2077–2085. [Google Scholar] [CrossRef]
- Horbury, M.D.; Holt, E.L.; Mouterde, L.M.M.; Balaguer, P.; Cebrián, J.; Blasco, L.; Allais, F.; Stavros, V.G. Towards symmetry driven and nature inspired UV filter design. Nat. Commun. 2019, 10, 4748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allais, F.; Ducrot, P.-H.; Martinet, S. Straightforward total synthesis of 2-O-Feruloyl-l-malate, 2-O-Sinapoyl-l-malate and 2-O-5-Hydroxyferuloyl-l-malate. Synthesis 2009, 21, 3571–3578. [Google Scholar] [CrossRef]
- Dean, J.C.; Kusaka, R.; Walsh, P.S.; Allais, F.; Zwier, T. Plant sunscreens in the UV-B: Ultraviolet spectroscopy of jet-cooled sinapoyl malate, sinapic acid, and sinapate ester derivatives. J. Am. Chem. Soc. 2014, 136, 14780–14795. [Google Scholar] [CrossRef]
- Baker, L.A.; Horbury, M.D.; Greenough, S.E.; Allais, F.; Walsh, P.S.; Habershon, S.; Stavros, V.G. Ultrafast photoprotecting sunscreens in natural plants. J. Phys. Chem. Lett. 2015, 7, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Jaufurally, A.S.; Teixeira, A.R.S.; Hollande, L.; Allais, F.; Ducrot, P.H. Optimization of the laccase-catalyzed synthesis of (±)-syringaresinol and study of its thermal and antiradical activities. ChemistrySelect 2016, 1, 5165–5171. [Google Scholar] [CrossRef]
- Rodríguez-Padrón, D.; Zhao, D.; Ortega, R.N.G.; Len, C.; Balu, A.M.; Garcia, A.; Luque, R. Characterization and antioxidant activity of microwave-extracted phenolic compounds from biomass residues. ACS Sustain. Chem. Eng. 2020, 8, 1513–1519. [Google Scholar] [CrossRef]
- Mishra, K.; Ojha, H.; Chaudhury, N.K. Estimation of antiradical properties of antioxidants using DPPH assay: A critical review and results. Food Chem. 2012, 130, 1036–1043. [Google Scholar] [CrossRef]
- Tararov, V.I.; Korostylev, A.; König, G.; Börner, A. Facile preparation and purification of mono tert-butyl malonate. Synth. Commun. 2006, 36, 187–191. [Google Scholar] [CrossRef]
- Menezes, J.C.J.M.D.S.; Kamat, S.P.; Cavaleiro, J.A.; Gaspar, A.; Garrido, E.M.J.; Borges, F. Synthesis and antioxidant activity of long chain alkyl hydroxycinnamates. Eur. J. Med. Chem. 2011, 46, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoevenagel, E. Ueber eine Darstellungsweise der Glutarsäure. Eur. J. Inorg. Chem. 1894, 27, 2345–2346. [Google Scholar] [CrossRef] [Green Version]
- Peyrot, C.; Peru, A.A.M.; Mouterde, L.M.M.; Allais, F. Proline-mediated Knoevenagel–Doebner condensation in ethanol: A sustainable access to p-hydroxycinnamic acids. ACS Sustain. Chem. Eng. 2019, 7, 9422–9427. [Google Scholar] [CrossRef]
- Mouterde, L.M.M.; Allais, F. Microwave-assisted Knoevenagel-Doebner reaction: An efficient method for naturally occurring phenolic acids synthesis. Front. Chem. 2018, 6, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Ngwa, C.; Zheng, Q. A simple and efficient procedure for Knoevenagel reaction promoted by imidazolium-based ionic liquids. Curr. Org. Synth. 2015, 13, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Padwal, J.; Lewis, W.; Moody, C.J. Synthesis of the reported structure of crassiflorone, a naturally occurring quinone isolated from the African ebony Diospyros crassiflora, and regioisomeric pentacyclic furocoumarin naphthoquinones. Org. Biomol. Chem. 2011, 9, 3484–3493. [Google Scholar] [CrossRef]
- Padilha, G.; Birmann, P.T.; Domingues, M.; Kaufman, T.S.; Savegnago, L.; Silveira, C.C. Convenient Michael addition/β-elimination approach to the synthesis of 4-benzyl- and 4-aryl-selenyl coumarins using diselenides as selenium sources. Tetrahedron Lett. 2017, 58, 985–990. [Google Scholar] [CrossRef]
- Johansen, M.; Larsen, C. A comparison of the chemical stability and the enzymatic hydrolysis of a series of aliphatic and aromatic ester derivatives of metronidazole. Int. J. Pharm. 1985, 26, 227–241. [Google Scholar] [CrossRef]
- Sánchez-Moreno, C.; Larrauri, J.A.; Saura-Calixto, F. A procedure to measure the antiradical efficiency of polyphenols. J. Sci. Food Agric. 1998, 76, 270–276. [Google Scholar] [CrossRef]
- Nimse, S.B.; Pal, D.; Mazumder, A.; Mazumder, R. Synthesis of cinnamanilide derivatives and their antioxidant and antimicrobial activity. J. Chem. 2015, 208920. [Google Scholar] [CrossRef] [Green Version]
- Son, S.; Lewis, B.A. Free radical scavenging and antioxidative activity of caffeic acid amide and ester analogues: Structure−activity relationship. J. Agric. Food Chem. 2002, 50, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Harvey, B.G.; Palmese, G.R.; Stanzione, J.F.; Ng, H.W.; Sakkiah, S.D.; Tong, W.; Sadler, J.M. Experimental data extraction and in silico prediction of the estrogenic activity of renewable replacements for bisphenol A. Int. J. Environ. Res. Public Health 2016, 13, 705. [Google Scholar] [CrossRef] [PubMed]
- Janvier, M.; Hollande, L.; Jaufurally, A.S.; Pernes, M.; Ménard, R.; Grimaldi, M.; Beaugrand, J.; Ducrot, P.-H.; Allais, F.; Balaguer, P. Syringaresinol: A renewable and safer alternative to Bisphenol A for epoxy-amine resins. ChemSusChem 2017, 10, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Peyrot, C.; Mention, M.M.; Brunissen, F.; Balaguer, P.; Allais, F. Innovative bio-based organic UV-A and blue light filters from Meldrum’s acid. Molecules 2020, 25, 2178. [Google Scholar] [CrossRef] [PubMed]
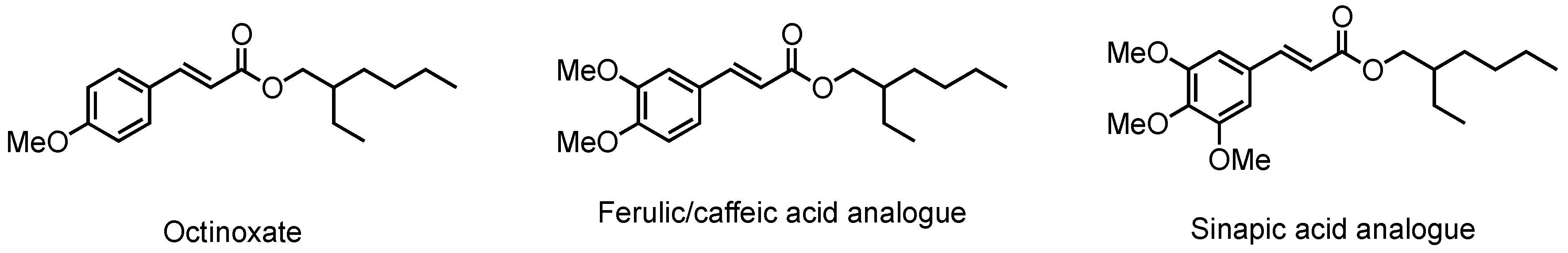

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC50 (nmol) a | TEC50 (s) a | Kinetic |
|---|---|---|---|
| BHA | 6.6 ± 0.2 | >4800 | Slow |
| BHT | 11.8 ± 1.3 | 360 ± 13 | Intermediate |
| Octinoxate | - b | - b | - b |
| 1 | 22.2 ± 1.1 | 180 ± 10 | Rapid |
| 2 | - b | - b | - b |
| 3 | - b | - b | - b |
| 4 | 18.0 ± 0.9 | 360 ± 18 | Intermediate |
| 5 | 17.0 ± 0.7 | 360 ± 15 | Intermediate |
| 6 | 19.9 ± 1.0 | 180 ± 9 | Rapid |
| 7 | 14.4 ± 0.4 | 180 ± 9 | Rapid |
| 8 | 19.8 ± 1.0 | 180 ± 11 | Rapid |
| 9 | 20.9 ± 1.1 | 420 ± 32 | Intermediate |
| 10 | 21.1 ± 1.1 | 180 ± 8 | Rapid |
| 11 | 17.1 ± 0.8 | 360 ± 18 | Intermediate |
| 12 | 15.3 ± 0.5 | 240 ± 12 | Rapid |
| 13 | 13.7 ± 0.6 | 480 ± 24 | Intermediate |
| 14 | 19.4 ± 0.9 | <60 | Rapid |
| 15 | 8.9 ± 0.3 | 600 ± 40 | Intermediate |
| 16 | 15.9 ± 0.7 | 240 ± 12 | Rapid |
| 17 | 9.4 ± 0.4 | 180 ± 10 | Rapid |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peyrot, C.; Mention, M.M.; Brunissen, F.; Allais, F. Sinapic Acid Esters: Octinoxate Substitutes Combining Suitable UV Protection and Antioxidant Activity. Antioxidants 2020, 9, 782. https://doi.org/10.3390/antiox9090782
Peyrot C, Mention MM, Brunissen F, Allais F. Sinapic Acid Esters: Octinoxate Substitutes Combining Suitable UV Protection and Antioxidant Activity. Antioxidants. 2020; 9(9):782. https://doi.org/10.3390/antiox9090782
Chicago/Turabian StylePeyrot, Cédric, Matthieu M. Mention, Fanny Brunissen, and Florent Allais. 2020. "Sinapic Acid Esters: Octinoxate Substitutes Combining Suitable UV Protection and Antioxidant Activity" Antioxidants 9, no. 9: 782. https://doi.org/10.3390/antiox9090782
APA StylePeyrot, C., Mention, M. M., Brunissen, F., & Allais, F. (2020). Sinapic Acid Esters: Octinoxate Substitutes Combining Suitable UV Protection and Antioxidant Activity. Antioxidants, 9(9), 782. https://doi.org/10.3390/antiox9090782