The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity





Abstract
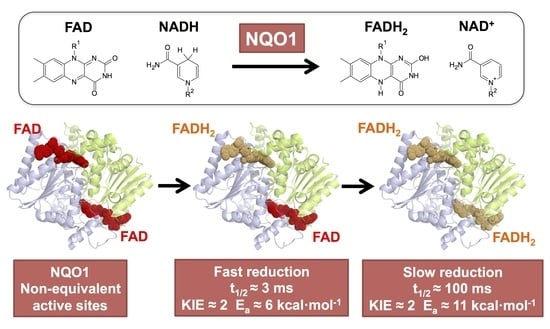
1. Introduction
2. Materials and Methods
2.1. Materials
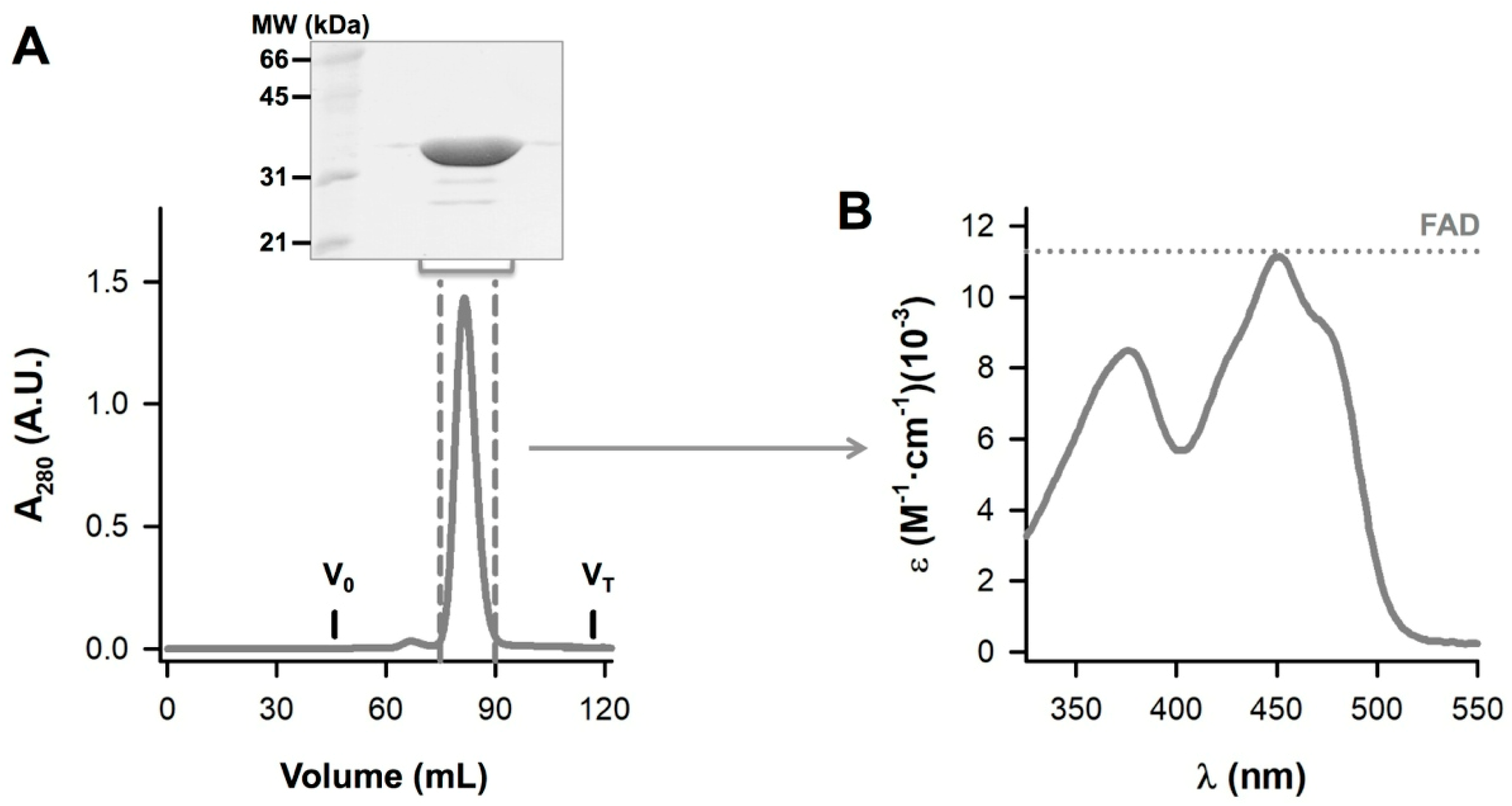
2.2. Protein Expression and Purification
2.3. NQO1 Redox Properties Evaluated by Absorption Spectroscopy
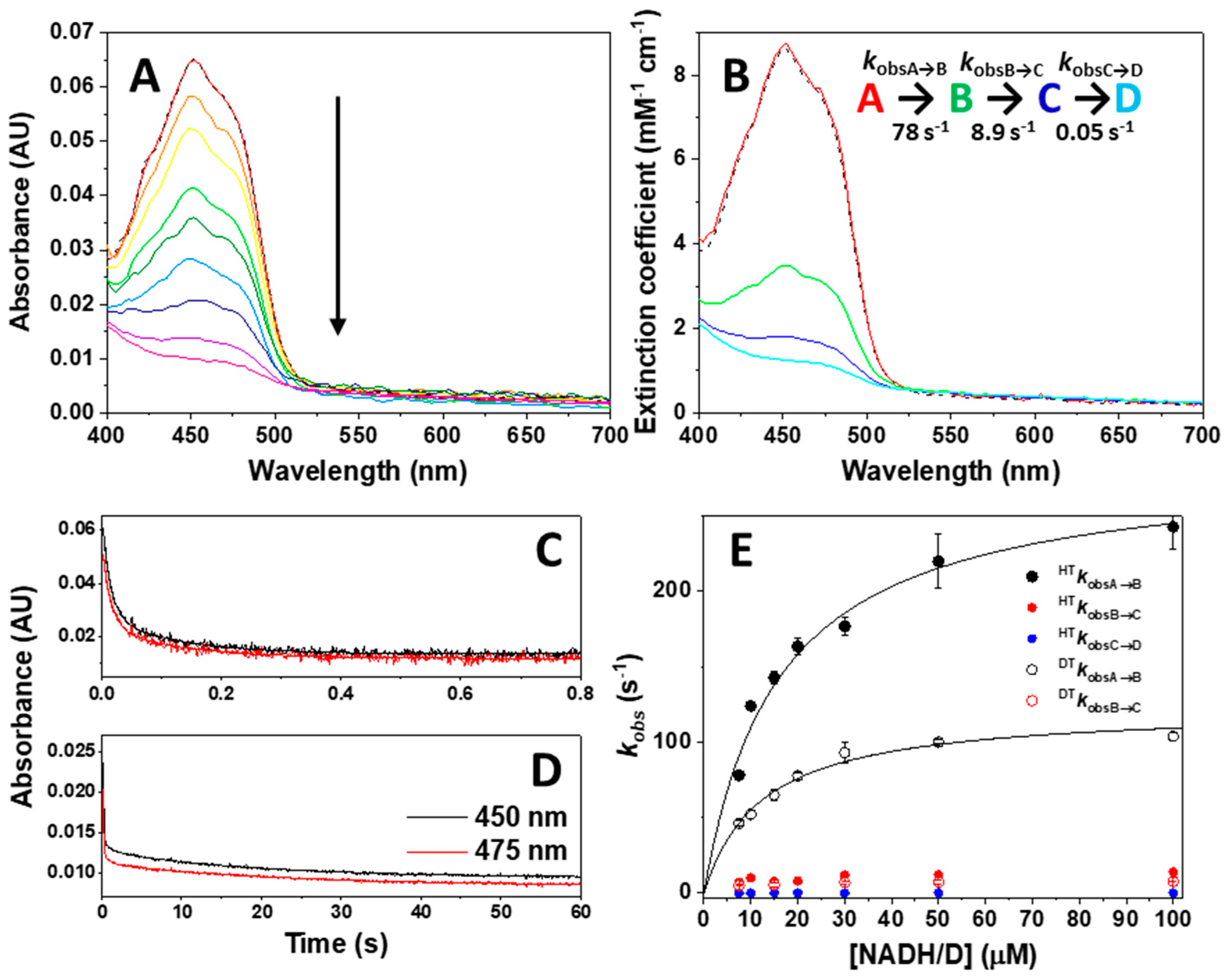
2.4. Stopped-Flow Pre-Steady-State Kinetic Measurements
2.5. Kinetic Isotopic Effects (KIEs)
3. Results and Discussion
3.1. Human NQO1 Does Not Stabilize Intermediate Semiquinone States upon Photoreduction
3.2. The Catalytic Cycle of NQO1
3.2.1. Non-Equivalent Active Sites in the NQO1 Dimer throughout the Reductive Half-Reaction
3.2.2. Non-Equivalent Active Sites in the NQO1 Dimer throughout the Oxidative Half-Reaction
3.3. Dynamics at the NQO1 Active Sites Differentially Contribute to the Two HT Events Representing the Reductive Half-Reaction
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Kinetic Solvent Viscosity Effects (KSVEs)

References
- Ross, D.; Siegel, D. NQO1 in protection against oxidative stress. Curr. Opin. Toxicol. 2018, 7, 67–72. [Google Scholar] [CrossRef]
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Anusevicius, Z.; Sarlauskas, J.; Cenas, N. Two-electron reduction of quinones by rat liver NAD(P)H: Quinone oxidoreductase: Quantitative structure-activity relationships. Arch. Biochem. Biophys. 2002, 404, 254–262. [Google Scholar] [CrossRef]
- Salido, E.; Timson, D.J.; Betancor-Fernández, I.; Palomino-Morales, R.; Pey, A.L. Targeting HIF-1alpha Function in Cancer through the Chaperone Action of NQO1. Preprints 2020, 2020, 030285. [Google Scholar]
- Ingram, B.O.; Turbyfill, J.L.; Bledsoe, P.J.; Jaiswal, A.K.; Stafford, D.W. Assessment of the contribution of NAD(P)H-dependent quinone oxidoreductase 1 (NQO1) to the reduction of vitamin K in wild-type and NQO1-deficient mice. Biochem. J. 2013, 456, 47–54. [Google Scholar] [CrossRef]
- Ernster, L.; Danielson, L.; Ljunggren, M. DT diaphorase. I. Purification from the soluble fraction of rat-liver cytoplasm, and properties. Biochim. Biophys. Acta 1962, 58, 171–188. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef]
- Beyer, R.E.; Segura-Aguilar, J.; Bernardo, S.D.; Cavazzoni, M.; Fato, R.; Fiorentini, D.; Galli, M.C.; Setti, M.; Landi, L.; Lenaz, G. The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc. Natl. Acad. Sci. USA 1996, 93, 2528–2532. [Google Scholar] [CrossRef]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H: Quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharm. 2004, 65, 1238–1247. [Google Scholar] [CrossRef]
- Walton, M.I.; Smith, P.J.; Workman, P. The role of NAD(P)H: Quinone reductase (EC 1.6.99.2, DT-diaphorase) in the reductive bioactivation of the novel indoloquinone antitumor agent EO9. Cancer Commun. 1991, 3, 199–206. [Google Scholar] [CrossRef]
- Brunmark, A.; Cadenas, E.; Lind, C.; Segura-Aguilar, J.; Ernster, L. DT-diaphorase-catalyzed two-electron reduction of quinone epoxides. Free Radic. Biol. Med. 1987, 3, 181–188. [Google Scholar] [CrossRef]
- Siegel, D.; Gibson, N.W.; Preusch, P.C.; Ross, D. Metabolism of mitomycin C by DT-diaphorase: Role in mitomycin C-induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990, 50, 7483–7489. [Google Scholar] [PubMed]
- Siegel, D.; Beall, H.; Senekowitsch, C.; Kasai, M.; Arai, H.; Gibson, N.W.; Ross, D. Bioreductive activation of mitomycin C by DT-diaphorase. Biochemistry 1992, 31, 7879–7885. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Yan, C.; Ross, D. NAD(P)H: Quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharm. 2012, 83, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Kim, J.W.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; Lee, C.H.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef]
- Ross, D.; Kepa, J.K.; Winski, S.L.; Beall, H.D.; Anwar, A.; Siegel, D. NAD(P)H:quinone oxidoreductase 1 (NQO1): Chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact. 2000, 129, 77–97. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar]
- Nioi, P.; Hayes, J.D. Contribution of NAD(P)H: Quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. 2004, 555, 149–171. [Google Scholar] [CrossRef]
- Brauze, D.; Widerak, M.; Cwykiel, J.; Szyfter, K.; Baer-Dubowska, W. The effect of aryl hydrocarbon receptor ligands on the expression of AhR, AhRR, ARNT, Hif1alpha, CYP1A1 and NQO1 genes in rat liver. Toxicol. Lett. 2006, 167, 212–220. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The Mediator Subunit MED16 Transduces NRF2-Activating Signals into Antioxidant Gene Expression. Mol. Cell. Biol. 2016, 36, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Valerio, L.G., Jr.; Kepa, J.K.; Pickwell, G.V.; Quattrochi, L.C. Induction of human NAD(P)H:quinone oxidoreductase (NQO1) gene expression by the flavonol quercetin. Toxicol. Lett. 2001, 119, 49–57. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Lu, X.; Wang, Z.; Wu, J.M. Induction of quinone reductase NQO1 by resveratrol in human K562 cells involves the antioxidant response element ARE and is accompanied by nuclear translocation of transcription factor Nrf2. Med. Chem. 2006, 2, 275–285. [Google Scholar]
- Nolan, K.A.; Scott, K.A.; Barnes, J.; Doncaster, J.; Whitehead, R.C.; Stratford, I.J. Pharmacological inhibitors of NAD(P)H quinone oxidoreductase, NQO1: Structure/activity relationships and functional activity in tumour cells. Biochem. Pharm. 2010, 80, 977–981. [Google Scholar] [CrossRef]
- Nolan, K.A.; Zhao, H.; Faulder, P.F.; Frenkel, A.D.; Timson, D.J.; Siegel, D.; Ross, D.; Burke, T.R., Jr.; Stratford, I.J.; Bryce, R.A. Coumarin-based inhibitors of human NAD(P)H: Quinone oxidoreductase-1. Identification, structure-activity, off-target effects and in vitro human pancreatic cancer toxicity. J. Med. Chem. 2007, 50, 6316–6325. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Palomino-Morales, R.J.; Fuchs, J.E.; Padín-Gonzalez, E.; Mesa-Torres, N.; Salido, E.; Timson, D.J.; Pey, A.L. Conformational dynamics is key to understanding loss-of-function of NQO1 cancer-associated polymorphisms and its correction by pharmacological ligands. Sci. Rep. 2016, 6, 20331. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD binding overcomes defects in activity and stability displayed by cancer-associated variants of human NQO1. Biochim. Biophys. Acta 2014, 1842, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.S.; Forrest, G.L.; Akman, S.A.; Hu, L.T. NAD(P)H: Quinone oxidoreductase expression and mitomycin C resistance developed by human colon cancer HCT 116 cells. Cancer Res. 1995, 55, 330–335. [Google Scholar] [PubMed]
- Medina-Carmona, E.; Neira, J.L.; Salido, E.; Fuchs, J.E.; Palomino-Morales, R.; Timson, D.J.; Pey, A.L. Site-to-site interdomain communication may mediate different loss-of-function mechanisms in a cancer-associated NQO1 polymorphism. Sci. Rep. 2017, 7, 44352. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, W.D.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the native structure caused by a single amino acid exchange in human NAD(P)H: Quinone oxidoreductase (1). FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Deng, P.S.; Bailey, J.M.; Swiderek, K.M. A two-domain structure for the two subunits of NAD(P)H: Quinone acceptor oxidoreductase. Protein Sci. 1994, 3, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouse NAD(P)H: Quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The three-dimensional structure of NAD(P)H: Quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef]
- Bianchet, M.A.; Faig, M.; Amzel, L.M. Structure and mechanism of NAD[P]H:quinone acceptor oxidoreductases (NQO). Methods Enzym. 2004, 382, 144–174. [Google Scholar]
- Hosoda, S.; Nakamura, W.; Hayashi, K. Properties and reaction mechanism of DT diaphorase from rat liver. J. Biol. Chem. 1974, 249, 6416–6423. [Google Scholar]
- Bianchet, M.A.; Erdemli, S.B.; Amzel, L.M. Structure, function, and mechanism of cytosolic quinone reductases. Vitam. Horm. 2008, 78, 63–84. [Google Scholar]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. NAD(P)H quinone oxidoreductase (NQO1): An enzyme which needs just enough mobility, in just the right places. Biosci. Rep. 2019, 39, BSR20180459. [Google Scholar] [CrossRef] [PubMed]
- Vankova, P.; Salido, E.; Timson, D.J.; Man, P.; Pey, A.L. A dynamic core in human NQO1 controls the functional and stability effects of ligand binding and their communication across the enzyme dimer. Biomolecules 2019, 9, 728. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, G.; Chen, S.; Massey, V. Active site studies of DT-diaphorase employing artificial flavins. J. Biol. Chem. 1995, 270, 2512–2516. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, G.; Chen, S.; Massey, V. DT-diaphorase. Redox potential, steady-state, and rapid reaction studies. J. Biol. Chem. 1995, 270, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Cavelier, G.; Amzel, L.M. Role of Fluctuations in Quinone Reductase Hydride Transfer: A Combined Quantum Mechanics and Molecular Dynamics Study. In Proceedings of the AIP Conference, Zaragoza, Spain, 8–11 February 2006; Volume 851. Meeting: FROM PHYSICS TO BIOLOGY: The Interface between Experiment and Computation - BIFI 2006 II International. [Google Scholar]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.J. Dicoumarol: A Drug which Hits at Least Two Very Different Targets in Vitamin K Metabolism. Curr. Drug Targets 2017, 18, 500–510. [Google Scholar] [CrossRef]
- Megarity, C.F.; Abdel-Bettley, H.; Caraher, M.C.; Scott, K.A.; Whitehead, R.C.; Jowitt, T.A.; Gutierrez, A.; Bryce, R.A.; Nolan, K.A.; Stratford, I.J.; et al. Negative cooperativity in NADP(H) quinone oxidoreductase 1 (NQO1). ChemBioChem 2019, 20, 2841–2849. [Google Scholar] [CrossRef]
- Megarity, C.F.; Timson, D.J. Cancer-associated variants of human NQO1: Impacts on inhibitor binding and cooperativity. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Fuchs, J.E.; Gavira, J.A.; Mesa-Torres, N.; Neira, J.L.; Salido, E.; Palomino-Morales, R.; Burgos, M.; Timson, D.J.; Pey, A.L. Enhanced vulnerability of human proteins towards disease-associated inactivation through divergent evolution. Hum. Mol. Genet. 2017, 26, 3531–3544. [Google Scholar] [CrossRef]
- Munoz, I.G.; Morel, B.; Medina-Carmona, E.; Pey, A.L. A mechanism for cancer-associated inactivation of NQO1 due to P187S and its reactivation by the consensus mutation H80R. FEBS Lett. 2017, 591, 2826–2835. [Google Scholar] [CrossRef]
- Martinez-Limon, A.; Alriquet, M.; Lang, W.H.; Calloni, G.; Wittig, I.; Vabulas, R.M. Recognition of enzymes lacking bound cofactor by protein quality control. Proc. Natl. Acad. Sci. USA 2016, 113, 12156–12161. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Anwar, A.; Winski, S.L.; Kepa, J.K.; Zolman, K.L.; Ross, D. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H: Quinone oxidoreductase 1. Mol. Pharm. 2001, 59, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Claveria-Gimeno, R.; Velazquez-Campoy, A.; Pey, A.L. Thermodynamics of cooperative binding of FAD to human NQO1: Implications to understanding cofactor-dependent function and stability of the flavoproteome. Arch. Biochem. Biophys. 2017, 636, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L. Biophysical and functional perturbation analyses at cancer-associated P187 and K240 sites of the multifunctional NADP(H): Quinone oxidoreductase 1. Int. J. Biol. Macromol. 2018, 118, 1912–1923. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Betancor-Fernández, I.; Santos, J.; Mesa-Torres, N.; Grottelli, S.; Batlle, C.; Naganathan, A.N.; Oppici, O.; Cellini, B.; Ventura, S.; et al. Insight into the specificity and severity of pathogenic mechanisms associated with missense mutations through experimental and structural perturbation analyses. Hum. Mol. Genet. 2019, 28, 1–15. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Rizzuti, B.; Martin-Escolano, R.; Pacheco-Garcia, J.L.; Mesa-Torres, N.; Neira, J.L.; Guzzi, R.; Pey, A.L. Phosphorylation compromises FAD binding and intracellular stability of wild-type and cancer-associated NQO1: Insights into flavo-proteome stability. Int. J. Biol. Macromol. 2019, 125, 1275–1288. [Google Scholar] [CrossRef]
- Reigan, P.; Colucci, M.A.; Siegel, D.; Chilloux, A.; Moody, C.J.; Ross, D. Development of indolequinone mechanism-based inhibitors of NAD(P)H: Quinone oxidoreductase 1 (NQO1): NQO1 inhibition and growth inhibitory activity in human pancreatic MIA PaCa-2 cancer cells. Biochemistry 2007, 46, 5941–5950. [Google Scholar] [CrossRef]
- Colucci, M.A.; Reigan, P.; Siegel, D.; Chilloux, A.; Ross, D.; Moody, C.J. Synthesis and evaluation of 3-aryloxymethyl-1,2-dimethylindole-4,7-diones as mechanism-based inhibitors of NAD(P)H: Quinone oxidoreductase 1 (NQO1) activity. J. Med. Chem. 2007, 50, 5780–5789. [Google Scholar] [CrossRef]
- Dehn, D.L.; Siegel, D.; Swann, E.; Moody, C.J.; Ross, D. Biochemical, cytotoxic, and genotoxic effects of ES936, a mechanism-based inhibitor of NAD(P)H:quinone oxidoreductase 1, in cellular systems. Mol. Pharm. 2003, 64, 714–720. [Google Scholar] [CrossRef]
- Dehn, D.L.; Siegel, D.; Zafar, K.S.; Reigan, P.; Swann, E.; Moody, C.J.; Ross, D. 5-Methoxy-1,2-dimethyl-3-[(4-nitrophenoxy)methyl] indole-4,7-dione, a mechanism-based inhibitor of NAD(P)H: Quinone oxidoreductase 1, exhibits activity against human pancreatic cancer in vitro and in vivo. Mol. Cancer 2006, 5, 1702–1709. [Google Scholar] [CrossRef][Green Version]
- Xu, S.; Yao, H.; Pei, L.; Hu, M.; Li, D.; Qiu, Y.; Wang, G.; Wu, L.; Yao, H.; Zhu, Z.; et al. Design, synthesis, and biological evaluation of NAD(P)H: Quinone oxidoreductase (NQO1)-targeted oridonin prodrugs possessing indolequinone moiety for hypoxia-selective activation. Eur. J. Med. Chem. 2017, 132, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bian, J.; Li, X.; Wu, X.; Dong, Y.; You, Q. 2-Substituted 3,7,8-trimethylnaphtho [1–b]furan-4,5-diones as specific L-shaped NQO1-mediated redox modulators for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2017, 138, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Klinman, J.P.; Offenbacher, A.R. Understanding Biological Hydrogen Transfer Through the Lens of Temperature Dependent Kinetic Isotope Effects. Acc. Chem. Res. 2018, 51, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Pollock, V.V.; Barber, M.J. Kinetic and mechanistic properties of biotin sulfoxide reductase. Biochemistry 2001, 40, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Whitby, L.G. A new method for preparing flavin-adenine dinucleotide. Biochem. J. 1953, 54, 437–442. [Google Scholar] [CrossRef]
- Frago, S.; Goni, G.; Herguedas, B.; Peregrina, J.R.; Serrano, A.; Perez-Dorado, I.; Molina, R.; Gomez-Moreno, C.; Hermoso, J.A.; Martinez-Julvez, M.; et al. Tuning of the FMN binding and oxido-reduction properties by neighboring side chains in Anabaena flavodoxin. Arch. Biochem. Biophys. 2007, 467, 206–217. [Google Scholar] [CrossRef]
- Sanchez-Azqueta, A.; Catalano-Dupuy, D.L.; Lopez-Rivero, A.; Tondo, M.L.; Orellano, E.G.; Ceccarelli, E.A.; Medina, M. Dynamics of the active site architecture in plant-type ferredoxin-NADP(+) reductases catalytic complexes. Biochim. Biophys. Acta 2014, 1837, 1730–1738. [Google Scholar] [CrossRef]
- Perez-Amigot, D.; Taleb, V.; Boneta, S.; Anoz-Carbonell, E.; Sebastian, M.; Velazquez-Campoy, A.; Polo, V.; Martinez-Julvez, M.; Medina, M. Towards the competent conformation for catalysis in the ferredoxin-NADP(+) reductase from the Brucella ovis pathogen. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 148058. [Google Scholar] [CrossRef]
- Tejero, J.; Peregrina, J.R.; Martinez-Julvez, M.; Gutierrez, A.; Gomez-Moreno, C.; Scrutton, N.S.; Medina, M. Catalytic mechanism of hydride transfer between NADP+/H and ferredoxin-NADP+ reductase from Anabaena PCC 7119. Arch. Biochem. Biophys. 2007, 459, 79–90. [Google Scholar] [CrossRef]
- Massey, V.; Muller, F.; Feldberg, R.; Schuman, M.; Sullivan, P.A.; Howell, L.G.; Mayhew, S.G.; Matthews, R.G.; Foust, G.P. The reactivity of flavoproteins with sulfite. Possible relevance to the problem of oxygen reactivity. J. Biol. Chem. 1969, 244, 3999–4006. [Google Scholar]
- Clark, W.M. Oxidation-Reduction Potentials of Organic Systems; The Williams & Wilkins Company: Baltimore, MD, USA, 1960. [Google Scholar]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H: Quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Burton, K. The enthalpy change for the reduction of nicotinamide—Adenine dinucleotide. Biochem. J. 1974, 143, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Lind, C.; Cadenas, E.; Hochstein, P.; Ernster, L. DT-diaphorase: Purification, properties, and function. Methods Enzymol. 1990, 186, 287–301. [Google Scholar] [PubMed]
- Merker, M.P.; Audi, S.H.; Bongard, R.D.; Lindemer, B.J.; Krenz, G.S. Influence of pulmonary arterial endothelial cells on quinone redox status: Effect of hyperoxia-induced NAD(P)H: Quinone oxidoreductase 1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L607–L619. [Google Scholar] [CrossRef] [PubMed]
- Bongard, R.D.; Krenz, G.S.; Gastonguay, A.J.; Williams, C.L.; Lindemer, B.J.; Merker, M.P. Characterization of the threshold for NAD(P)H: Quinone oxidoreductase activity in intact sulforaphane-treated pulmonary arterial endothelial cells. Free Radic. Biol. Med. 2011, 50, 953–962. [Google Scholar] [CrossRef]
- Klinman, J.P. Moving Through Barriers in Science and Life. Annu. Rev. Biochem. 2019, 88, 1–24. [Google Scholar] [CrossRef]
- Nelson, S.D.; Trager, W.F. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metab. Dispos. 2003, 31, 1481–1498. [Google Scholar] [CrossRef]
- Yoshimoto, F.K.; Zhou, Y.; Peng, H.M.; Stidd, D.; Yoshimoto, J.A.; Sharma, K.K.; Matthew, S.; Auchus, R.J. Minor activities and transition state properties of the human steroid hydroxylases cytochromes P450c17 and P450c21, from reactions observed with deuterium-labeled substrates. Biochemistry 2012, 51, 7064–7077. [Google Scholar] [CrossRef][Green Version]
- Nagel, Z.D.; Klinman, J.P. Update 1 of: Tunneling and dynamics in enzymatic hydride transfer. Chem. Rev. 2010, 110, PR41–PR67. [Google Scholar] [CrossRef]
- Nagel, Z.D.; Klinman, J.P. A 21st century revisionist’s view at a turning point in enzymology. Nat. Chem. Biol. 2009, 5, 543–550. [Google Scholar] [CrossRef]
- Mesa-Torres, N.; Betancor-Fernández, I.; Oppici, E.; Cellini, B.; Salido, E.; Pey, A.L. Evolutionary Divergent Suppressor Mutations in Conformational Diseases. Genes 2018, 9, 352. [Google Scholar] [CrossRef] [PubMed]
- Wyman, J.; Gill, S.J. Binding and Linkage. Functional Chemistry of Biological Macromolecules; University Science Books: Mill Valley, CA, USA, 1990. [Google Scholar]
- Gadda, G.; Sobrado, P. Kinetic Solvent Viscosity Effects as Probes for Studying the Mechanisms of Enzyme Action. Biochemistry 2018, 57, 3445–3453. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample in Tonometer 1 | Sample in Tonometer 2 | kobsA→B (s−1) | kobsB→C (s−1) |
|---|---|---|---|
| NQO1 (1) | NADH (1) | 78 ± 1 | 8.9 ± 0.9 |
| NQO1 (1) | 4R-NADD (1) | 44 ± 2 | 6.3 ± 0.2 |
| NQO1 (1) | NADPH (1) | 261 ± 13 | 7.8 ± 0.3 |
| NQO1 (1) + Dic (1) | NADH (1) | 0.034 ± 0.003 | 0.0065 ± 0.0005 |
| NQO1 (1) + Dic (1) | NADH (6.6) | 0.036 ± 0.002 | 0.010 ± 0.001 |
| NQO1 (1) + Dic (4) | NADH (1) | 0.018 ± 0.003 | 0.0015 ± 0.0001 |
| NQO1 (1) + Dic (1) | NADPH (1) | 0.036 ± 0.006 | 0.0070 ± 0.0008 |
| NQO1 (1) + Dic (4) | NADPH (1) | 0.017 ± 0.001 | 0.0020 ± 0.0001 |
| NQO1 (1) + NADH (1) | DCPIP (1) | >500 | 160 ± 14 |
| NQO1 (1) + NADH (1) | DCPIP (1) + Dic (1) | 38 ± 3 | 6.3 ± 1.2 |
| NQO1 (1) + NADH (1) | DCPIP (1) + Dic (4) | 7.7 ± 0.2 | 1.3 ± 0.1 |
| NQO1 (1) + NADH (1) | Ferricyanide | 219 ± 12 | 29 ± 4 |
| HT | DT | KIE | ΔEa EaDT − EaHT (kcal·mol−1) | AH/AD | |||||
|---|---|---|---|---|---|---|---|---|---|
| HTkobsa (s−1) | EaHT (kcal·mol−1) | AH (s−1) | DTkobsa (s−1) | EaDT (kcal·mol−1) | AD (s−1) | ||||
| A→B | 78 ± 1 | 6.1 ± 0.2 | (5.3 ± 1.2)·106 | 44 ± 2 | 6.3 ± 0.4 | (4.1 ± 1.1)·106 | 1.8 ± 0.1 | 0.2 ± 0.4 | 1.3 ± 0.6 |
| B→C | 8.9 ± 0.9 | 10.9 ± 0.5 | (3.4 ± 0.9)·109 | 5.3 ± 0.2 | 9.8 ± 0.5 | (2.6 ± 0.6)·108 | 1.8 ± 0.3 | −1.1 ± 0.7 | 13 ± 6 |
| Activation Parameters | HT | DT | ||
|---|---|---|---|---|
| A→B | B→C | A→B | B→C | |
| ΔH‡ (kcal·mol−1) | 5.3 ± 0.3 | 11 ± 1 | 5.7 ± 0.6 | 9.8 ± 0.8 |
| ΔS‡ (cal·mol−1·K−1) | −31 ± 1 | −16 ± 2 | −30 ± 2 | −20 ± 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anoz-Carbonell, E.; Timson, D.J.; Pey, A.L.; Medina, M. The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity. Antioxidants 2020, 9, 772. https://doi.org/10.3390/antiox9090772
Anoz-Carbonell E, Timson DJ, Pey AL, Medina M. The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity. Antioxidants. 2020; 9(9):772. https://doi.org/10.3390/antiox9090772
Chicago/Turabian StyleAnoz-Carbonell, Ernesto, David J. Timson, Angel L. Pey, and Milagros Medina. 2020. "The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity" Antioxidants 9, no. 9: 772. https://doi.org/10.3390/antiox9090772
APA StyleAnoz-Carbonell, E., Timson, D. J., Pey, A. L., & Medina, M. (2020). The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity. Antioxidants, 9(9), 772. https://doi.org/10.3390/antiox9090772