Theoretical Study of the Iron Complexes with Aminoguanidine: Investigating Secondary Antioxidant Activity



Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Complexes of Fe(III) with Protonated AG
3.2. Complexes of Fe(III) with AG
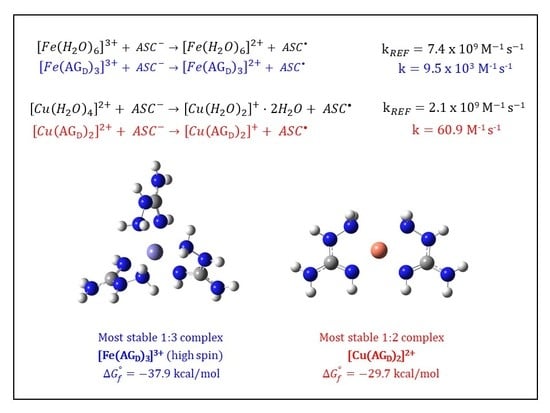
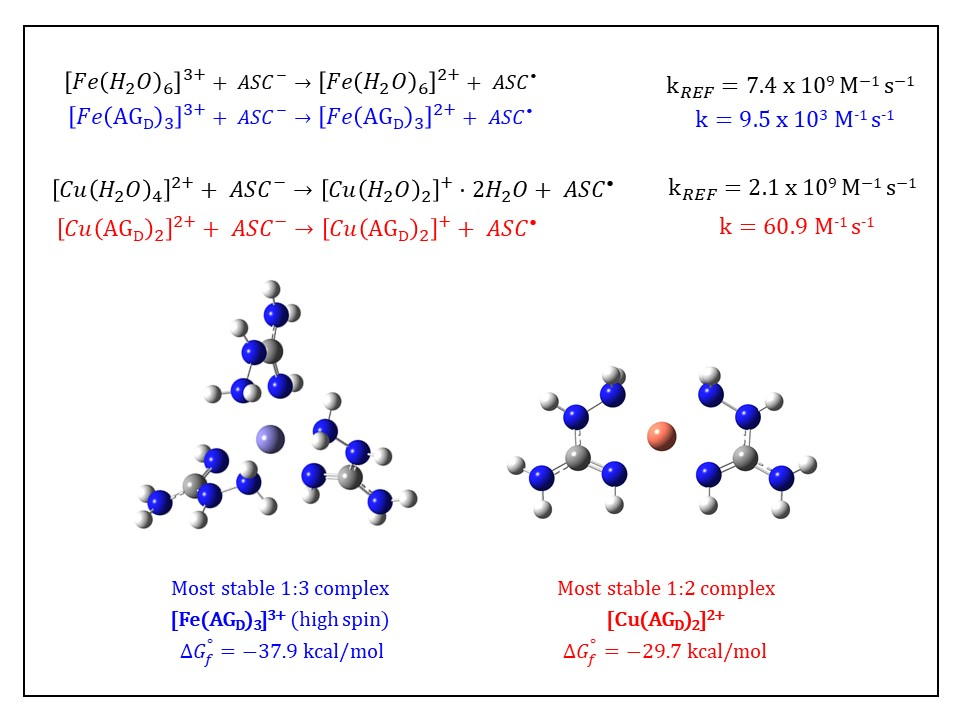
3.3. Kinetic Calculations for the Reduction of Fe(III): Comparison with the Cu(II)/Cu(I) Reduction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thorpe, S.R.; Baynes, J.W. Maillard reaction products in tissue proteins: New products and new perspectives. Amino Acids 2003, 25, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, P.; Cerami, A. Protein glycation, diabetes, and aging. Recent Prog. Horm. Res. 2001, 56, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Münch, G.; Mayer, S.; Michaelis, J.; Hipkiss, A.R.; Riederer, P.; Müller, R.; Neumann, A.; Schinzel, R.; Cunningham, A.M. Influenced of advanced glycation end-products and AGE-inhibitors on nucleation-dependent polymerization of beta-amyloid peptide. Biochim. Biophys. Acta 1997, 1360, 17–29. [Google Scholar] [CrossRef]
- Stitt, A.W. The maillard reaction in eye diseases. Ann. N. Y. Acad. Sci. 2005, 1043, 582–597. [Google Scholar] [CrossRef]
- Colzani, M.; De Maddis, D.; Casali, G.; Carini, M.; Vistoli, G.; Aldini, G. Reactivity, selectivity, and reaction mechanisms of aminoguanidine, hydralazine, pyridoxamine, and carnosine as sequestering agents of reactive carbonyl species: A comparative study. Chem. Med. Chem. 2016, 11, 1778–1789. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. On the catalyst of hydroperoxide. Naturwissenschaften 1932, 20, 948–950. [Google Scholar] [CrossRef]
- Zhao, M.J.; Jung, L. Kinetics of the competitive degradation of deoxyribose and other molecules by hydroxyl radicals produced by the fenton reaction in the presence of ascorbic acid. Free Radic. Res. 1995, 23, 229–243. [Google Scholar] [CrossRef]
- Burkitt, M.J.; Gilbert, B.C. Model studies of the iron-catalysed haber-weiss cycle and the ascorbate-driven fenton reaction. Free Radic. Res. Commun. 1990, 10, 265–280. [Google Scholar] [CrossRef]
- Price, D.L.; Rhett, P.M.; Thorpe, S.R.; Baynes, J.W. Chelating activity of advanced glycation end-products inhibitors. J. Biol. Chem. 2001, 276, 48967–48972. [Google Scholar] [CrossRef]
- Rahbar, S.; Natarajan, R.; Yerneni, K.K.; Scott, S.; Gonzales, N.; Nadler, J.L. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin. Chim. Acta 2000, 301, 65–77. [Google Scholar] [CrossRef]
- Ortega-Castro, J.; Adrover, M.; Frau, J.; Donoso, J.; Muñoz, F. Chelating power of LR-74, a new AGE-inhibitor. Chem. Phys. Lett. 2008, 465, 120–125. [Google Scholar] [CrossRef]
- Casasnovas, R.; Ortega-Castro, J.; Donoso, J.; Frau, F.; Muñoz, F. Theoretical calculations of stability constants and pKa values of metal complexes in solution: Application to pyridoxamine-copper(II) complexes and their biological implications in AGE inhibition. Phys. Chem. Chem. Phys. 2013, 15, 16303–16313. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, E.; Bolton, W.K. Pimagedine: A novel therapy for diabetic nephropathy. Expert Opin. Investig. Drugs 2002, 11, 565–574. [Google Scholar]
- Li, Y.M.; Steffes, M.; Donnelly, T.; Liu, C.; Fuh, H.; Basgen, J.; Bucala, R.; Vlassara, H. Prevention of cardiovascular and renal pathology of aging by the advanced glycation inhibitor aminoguanidine. Proc. Natl. Acad. Sci. USA 1996, 93, 3902–3907. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Castro, J.; Frau, J.; Casasnovas, R.; Fernández, D.; Donoso, J.; Muñoz, F. High- and low-spin Fe(III) complexes of various AGE inhibitors. J. Phys. Chem. A 2012, 116, 2961–2971. [Google Scholar] [CrossRef]
- Ortega-Castro, J.; Adrover, M.; Frau, J.; Donoso, J.; Muñoz, F. Cu2+ complexes of some AGEs inhibitors. Chem. Phys. Lett. 2009, 475, 277–284. [Google Scholar] [CrossRef]
- Ortega-Castro, J.; Adrover, M.; Frau, J.; Salvà, A.; Donoso, J.; Muñoz, F. DFT studies on schiff base formation of vitamin B6 analogues. Reaction between a pyridoxamine-analogue and carbonyl compounds. J. Phys. Chem. A 2010, 114, 4634–4640. [Google Scholar] [CrossRef]
- Adrover, M.; Vilanova, B.; Muñoz, F.; Donoso, J. Pyridxamine, a scavenger agent of carbohydrates. Int. J. Chem. Kinet. 2007, 39, 154–167. [Google Scholar] [CrossRef]
- Adrover, M.; Vilanova, B.; Muñoz, F.; Donoso, J. Inhibition of glycosylation processes: The reaction between pyridoxamine and glucose. Chem. Biodivers. 2005, 2, 964–975. [Google Scholar] [CrossRef]
- Solís-Calero, C.; Ortega-Castro, J.; Frau, J.; Muñoz, F. Scavenger mechanism of methylglyoxal by metformin. A DFT study. Theor. Chem. Acc. 2015, 134, 48–62. [Google Scholar] [CrossRef]
- Rahbar, S.; Figarola, J.L. Novel inhibitors of advanced glycation endproducts. Arch. Biochem. Biophys. 2003, 419, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Ramis, R.; Casasnovas, R.; Mariño, L.; Frau, J.; Adrover, M.; Vilanova, B.; Mora-Diez, N.; Ortega-Castro, J. A density functional theory study of the free-radical scavenging activity of aminoguanidine. Comparison with its reactive carbonyl compound and metal scavenging activities. Int. J. Quantum Chem. 2019, 119, e25911. [Google Scholar] [CrossRef]
- García-Díez, G.; Ramis, R.; Mora-Diez, N. Theoretical study of the copper complexes with aminoguanidine: Investigating secondary antioxidant activity. ACS Omega 2020, 5, 14502–14512. [Google Scholar] [CrossRef] [PubMed]
- Monreal-Corona, R.; Ippolito, A.A.; Biddlecombe, J.R.; Mora-Diez, N. Theoretical study of the iron complexes with lipoic and dihydrolipoic acids: Exploring secondary antioxidant activity. Antioxidants 2020, 9, 674. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Exchange-correlation functional with broad accuracy for metallic and non-metallic compounds, kinetics, and noncovalent interactions. J. Chem. Phys. 2005, 123, 161103–161106. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functional for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition metals: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Marcus, R.A. Electrons transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Marcus, R.A. Transfer reactions in chemistry. Theory and experiment. Pure Appl. Chem. 1997, 69, 13–30. [Google Scholar] [CrossRef]
- Collins, F.C.; Kimball, G.E. Diffusion-controlled reaction rates. J. Colloid. Sci. 1949, 4, 425–437. [Google Scholar] [CrossRef]
- Smoluchowskim, M.Z. Versuch einer mathematischen theorie der koagulationskinetik kolloider Lösungen. J. Phys. Chem. 1917, 92, 129–168. [Google Scholar]
- Einstein, A. Über die von der molekularkinetischen theorie der wärme geforderte bewegung von in ruhenden flüssigkeiten suspendierten teilchen. Ann. Phys. 1905, 322, 549–560. [Google Scholar] [CrossRef]
- Stokes, G.G. Mathematical and Physical Papers; Cambridge University Press: Cambridge, UK, 1903; Volume 3. [Google Scholar]
- Castañeda-Arriaga, R.; Alvarez-Idaboy, J.R.; Mora-Diez, N. Theoretical study of copper complexes with lipoic and dihydrolipoic acids. RSC Adv. 2016, 6, 107924–107932. [Google Scholar] [CrossRef]
- Castañeda-Arriaga, R.; Mora-Diez, N.; Alvarez-Idaboy, J.R. Modelling the chemical repair of protein carbon-centered radicals formed via oxidative damage with dihydrolipoic acid. RCS Adv. 2015, 5, 96714–96719. [Google Scholar] [CrossRef]
- Castañeda-Arriaga, R.; Domínguez-Casto, A.; Lee, J.; Alvarez-Idaboy, J.R.; Mora-Diez, N. Chemical repair of protein-centred radicals: Long-distance dynamic factors. Can. J. Chem. 2016, 94, 1119–1126. [Google Scholar] [CrossRef]
- Ramis, R.; Casasnovas, R.; Ortega-Castro, J.; Frau, J.; Alvarez-Idaboy, J.R.; Mora-Diez, N. Modelling the repair of carbon-centered protein radicals by the antioxidants glutathione and Trolox. New J. Chem. 2019, 43, 2085–2097. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. A computational methodology for accurate predictions of rate constants in solution: Application to the assessment of primary antioxidant activity. J. Comput. Chem. 2013, 34, 2430–2445. [Google Scholar] [CrossRef]
- Romeo, I.; Parise, A.; Galano, A.; Russo, N.; Alvarez-Idaboy, J.R.; Marino, T. The antioxidant capability of higenamine: Insights from theory. Antioxidants 2020, 9, 358. [Google Scholar] [CrossRef]
- Goel, S.; Masunov, A.E. Pairwise spin-contamination correction method and dft study of mnh and h2 dissociation curves. In Computational Science—ICCS 2009; Allen, G., Nabrzyski, J., Seidel, E., van Albada, G.D., Dongarra, J., Sloot, P.M.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 5545. [Google Scholar]
- Koskinen, M.; Mutikainen, I.; Tilus, P.; Pelttari, E.; Korvela, M.; Elo, H. Structure of aminoguanidine hemioxalate. Implications for the synthesis of amidinohydrazones. Mon. Chem. 1997, 128, 767–775. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Butterworth-Heinemann: Leeds, UK, 1997. [Google Scholar]
- Edler, E.; Stein, M. Spin-state-dependent properties of an Iron(III) hydrogenase mimic. Eur. J. Inorg. Chem. 2014, 22, 3587–3599. [Google Scholar] [CrossRef]
- Boldyrev, V.V.; Tukhtaev, R.K.; Gavrilov, A.I.; Larionov, S.V.; Savel’eva, Z.A.; Lavrenova, L.G. Combustion of nickel and copper nitrate complexes of hydrazine derivatives as a method for manufacturing fine-grained and porous metals. Russ. J. Inorg. Chem. 1998, 43, 302–305. [Google Scholar]
- Buettner, G.G. Ascorbate autoxidation in the presence of iron and copper chelates. Free Rad. Res. Commun. 1986, 1, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Francisco-Marquez, M.; Aguilar-Fernánde, M.; Galano, A. Anthranilic acid as a secondary antioxidant: Implications to the inhibition of OH production and the associated oxidative stress. Comp. Theor. Chem. 2016, 1077, 18–24. [Google Scholar] [CrossRef]
- Martínez, A.; Vargas, R.; Galano, A. Citric acid: A promising copper scavenger. Comp. Theor. Chem. 2018, 1133, 47–50. [Google Scholar] [CrossRef]
- Castañeda-Arriaga, R.; Pérez-González, A.; Reina, M.; Alvarez-Idaboy, J.R.; Galano, A. Comprehensive investigation of the antioxidant and pro-oxidant effects of phenolic compounds: A double-edged sword in the context of oxidative stress? J. Phys. Chem. B 2018, 122, 6198–6214. [Google Scholar] [CrossRef] [PubMed]
- Mravljak, J.; Jakopin, Z. Iron-binding and anti-fenton properties of novel amino acid-derived cyclic imide dioximes. Antioxidants 2019, 8, 473. [Google Scholar] [CrossRef]
- Kubicova, L.; Hadacek, F.; Bachmann, G.; Weckwerth, W.; Chobot, V. Coordination complex formation and redox properties of kynurenic and xanthurenic acid can affect brain tissue homeodynamics. Antioxidants 2019, 8, 476. [Google Scholar] [CrossRef]
- Brovč, E.V.; Pajk, S.; Šink, R.; Mravljak, J. Protein formulations containing polysorbates: Are metal chelators needed at all? Antioxidants 2020, 9, 441. [Google Scholar] [CrossRef]
- Wang, L.; Santos, E.; Schenk, D.; Rabago-Smith, M. Kinetics and mechanistic studies on the reaction between cytochrome c and tea catechins. Antioxidants 2014, 3, 559–568. [Google Scholar] [CrossRef]
- Guerreiro, J.F.; Gomes, M.A.G.B.; Pagliari, F.; Jansen, J.; Marafioti, M.G.; Nistico, C.; Hanley, R.; Costa, R.O.; Ferreira, S.S.; Mendes, F.; et al. Iron and copper complexes with antioxidant activity as inhibitors of the metastatic potential of glioma cells. RSC Adv. 2020, 10, 12699–12710. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity of HO2/O−2 radicals in aqueous solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | |||
|---|---|---|---|
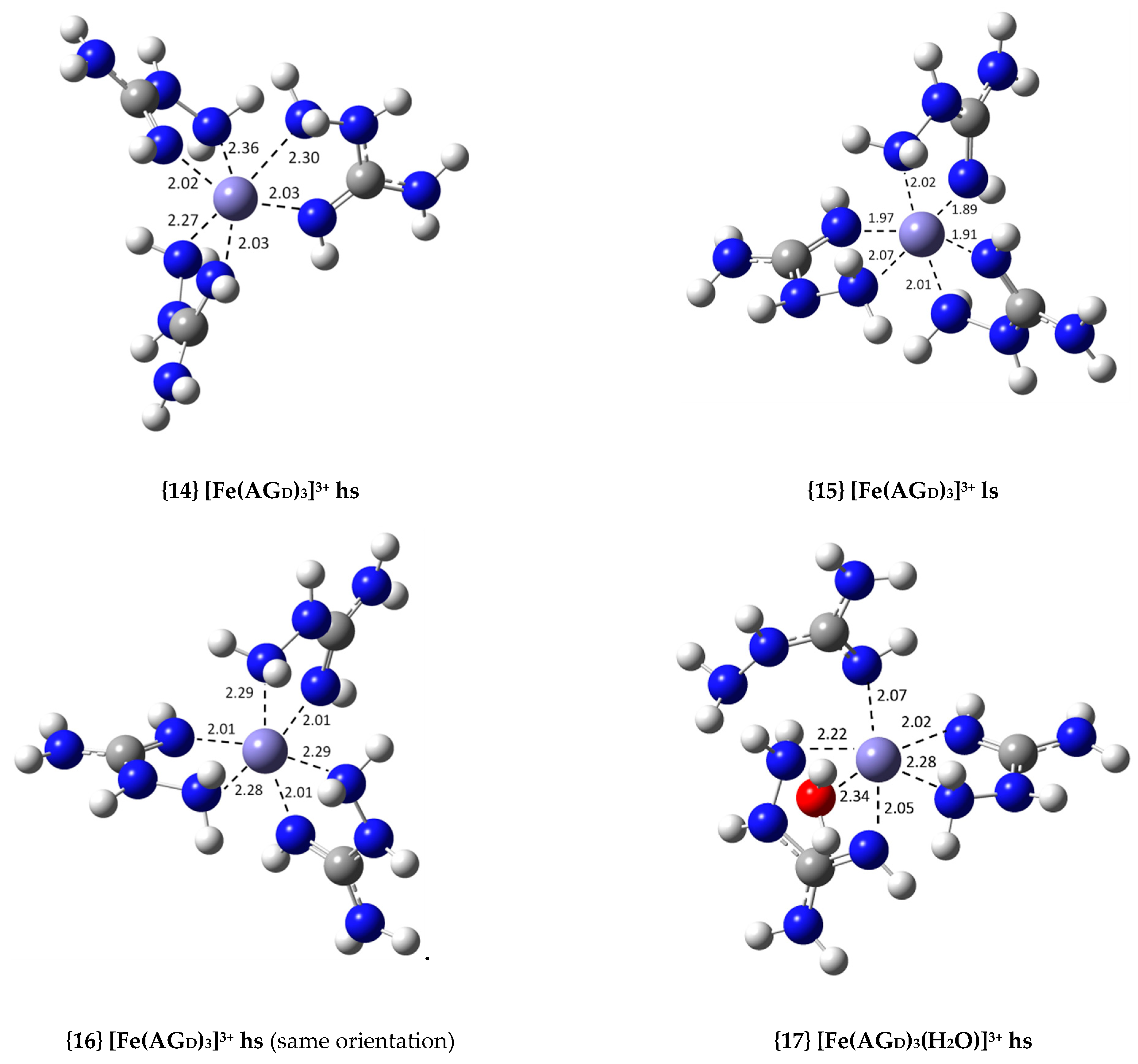
| {14} [Fe(AGD)3]3+ hs (N2, N4) | −36.8 | 9.03 × 1026 | 26.96 |
| {15} [Fe(AGD)3]3+ ls (N2, N4) | −6.7 | 8.22 × 104 | 4.91 |
| {16} [Fe(AGD)3]3+ hs (same orientation, N2, N4) | −37.9 | 5.65 × 1027 | 27.75 |
| {17} [Fe(AGD)3(H2O)]3+ hs (N2, N4, N2′, N4′, N2′) | −24.1 | 4.96 × 1017 | 17.70 |
| {18} [Fe(AGD)3(H2O)2]3+ hs (N2, N4, N2′, N2′) | −19.1 | 1.03 × 1014 | 14.01 |
| {32} [Fe(AGD)2(H2O)2]3+ hs (trans, N2, N4) | −20.1 | 5.35 × 1014 | 14.73 |
| {34} [Fe(AGD)2(H2O)2]3+ hs (mirror image, N2, N4) | −19.0 | 7.91 × 1013 | 13.90 |
| {38} [Fe(AGD)2(H2O)2]3+ hs (cis, N2, N4) | −22.9 | 5.86 × 1016 | 16.77 |
| {43} [Fe(AGD)(H2O)4]3+ hs (N2, N4) | −10.3 | 3.48 × 107 | 7.54 |
| {49} [Fe(AGA)2(H2O)2]3+ hs (4-coord., N2) | −19.6 | 2.20 × 1014 | 14.34 |
| {51} [Fe(AGB)2(H2O)2]3+ hs (4-coord., N2) | −21.6 | 6.35 × 1015 | 15.80 |
| {54} [Fe(AGC)2(H2O)2]3+ hs (4-coord., N2) | −20.5 | 1.09 × 1015 | 15.04 |
| {57} [Fe(AGD)(H2O)3]3+ hs (5-coord., sq. pyr., N2, N4) | −15.3 | 1.63 × 1011 | 11.21 |
| {59} [Fe(AGD)2]3+ hs (4-coord., N2, N4) | −20.5 | 1.02 × 1015 | 15.01 |
| {60} [Fe(AGD)2(H2O)]3+ hs (5-coord., N2, N4) | −23.5 | 1.78 × 1017 | 17.25 |
| Reaction | kapp | Ratio | kapp | Ratio |
|---|---|---|---|---|
| 7.28 × 109 | 7.43 × 109 | |||
| {15} {61} + (ls) | 7.76 × 109 | 0.94 | 4.48 | 1.66 × 109 |
| {16} {62} + | 8.09 × 109 | 0.90 | 9.47 × 103 | 7.85 × 105 |
| {14} {63} + | 8.29 × 109 | 0.88 | 1.96 × 105 | 3.79 × 104 |
| {17} {64} + | 8.30 × 109 | 0.88 | 1.53 × 108 | 48.6 |
| {38} {65} + | 8.08 × 109 | 0.90 | 6.57 × 108 | 11.3 |
| 7.71 × 109 | 2.10 × 109 | |||
| 2.80 × 109 | 2.75 | 60.9 | 3.45 × 107 | |
| 3.49 × 109 | 2.21 | 66.6 | 3.15 × 107 | |
| 7.02 × 109 | 1.10 | 3.56 × 104 | 5.90 × 104 | |
| 7.40 × 109 | 1.04 | 8.01 × 104 | 2.62 × 104 | |
| 7.47 × 109 | 1.03 | 4.83 × 105 | 4.35 × 103 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Díez, G.; Mora-Diez, N. Theoretical Study of the Iron Complexes with Aminoguanidine: Investigating Secondary Antioxidant Activity. Antioxidants 2020, 9, 756. https://doi.org/10.3390/antiox9080756
García-Díez G, Mora-Diez N. Theoretical Study of the Iron Complexes with Aminoguanidine: Investigating Secondary Antioxidant Activity. Antioxidants. 2020; 9(8):756. https://doi.org/10.3390/antiox9080756
Chicago/Turabian StyleGarcía-Díez, Guillermo, and Nelaine Mora-Diez. 2020. "Theoretical Study of the Iron Complexes with Aminoguanidine: Investigating Secondary Antioxidant Activity" Antioxidants 9, no. 8: 756. https://doi.org/10.3390/antiox9080756
APA StyleGarcía-Díez, G., & Mora-Diez, N. (2020). Theoretical Study of the Iron Complexes with Aminoguanidine: Investigating Secondary Antioxidant Activity. Antioxidants, 9(8), 756. https://doi.org/10.3390/antiox9080756