Reversible Oxidative Modifications in Myoglobin and Functional Implications

 ,
, 
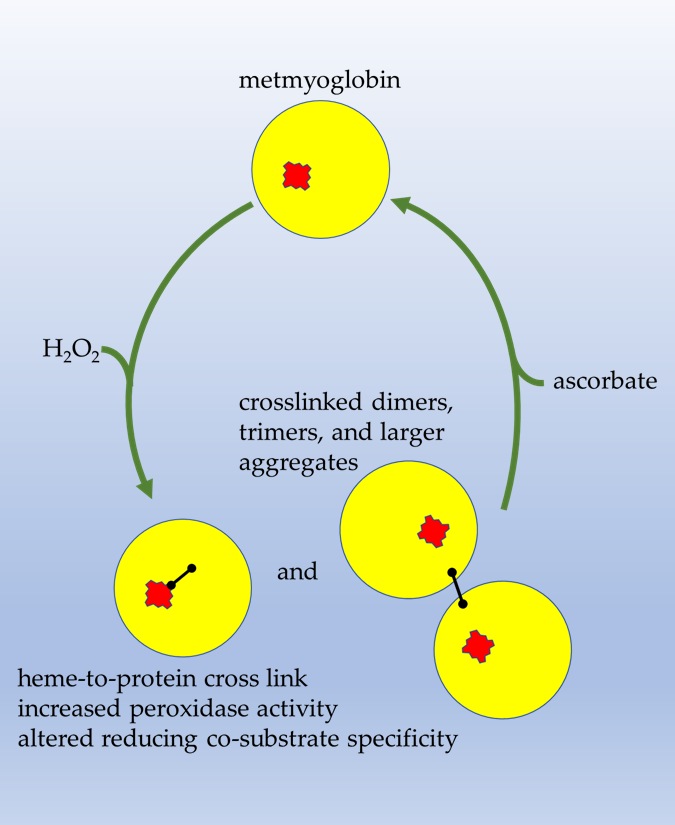
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Metmyoglobin Peroxidase Activity Assays
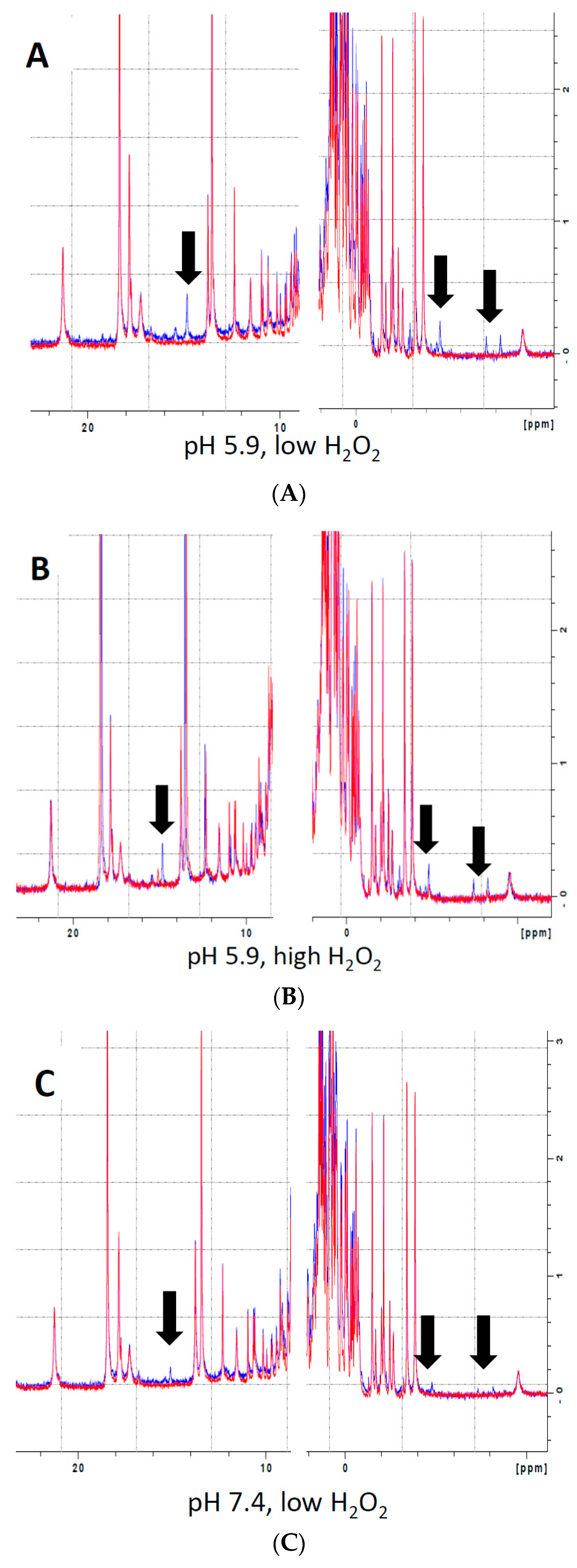
2.3. Nuclear Magnetic Resonance (NMR) Methods
2.4. Heme Stain
2.5. Heme Extraction
2.6. Mb-X Peroxidase Activity Assays
2.7. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS)
2.8. Detection of Dityrosine Using Fluorescence
2.9. Western Blot Using Anti-dityrosine Antibody
2.10. Tyrosine Acetylation Using N-Acetylimidazole
2.11. Statistical Methods
3. Results
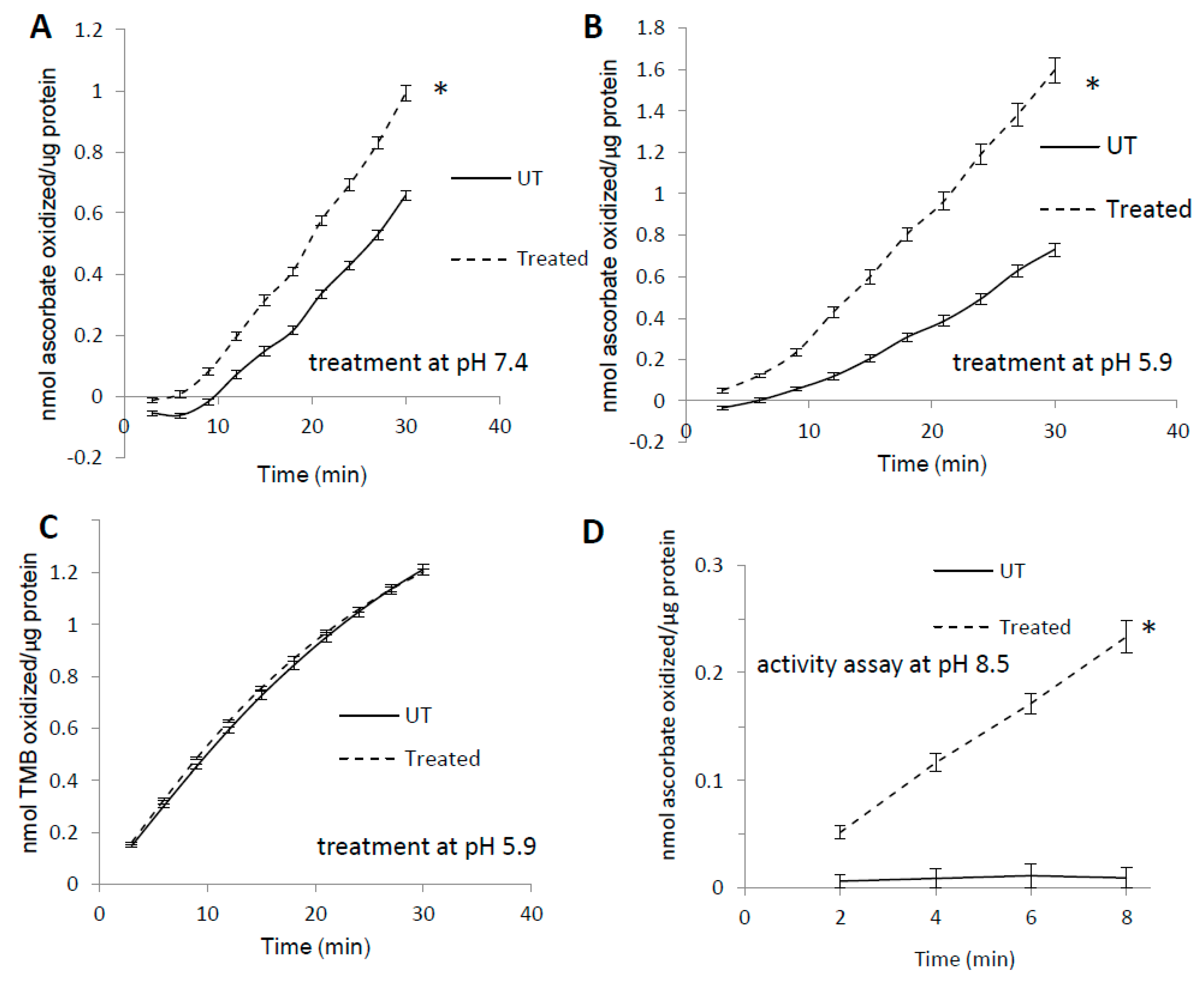
3.1. Pre-treatment with H2O2 Increases Mb Peroxidase Activity
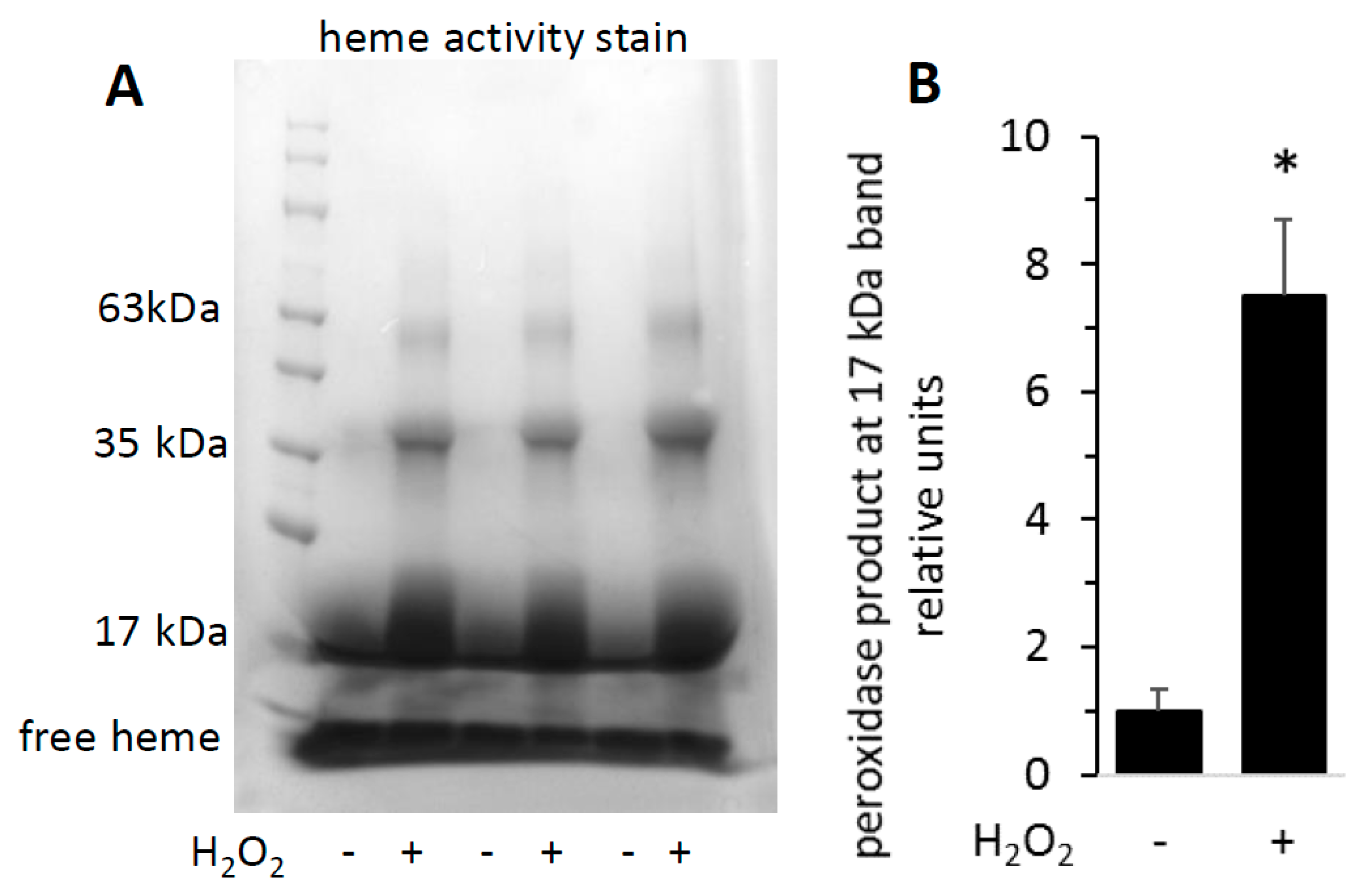
3.2. Heme Activity Stains of H2O2-Reacted metMb
3.3. Unique Activity of Heme-Coupled Mb (Mb-X)
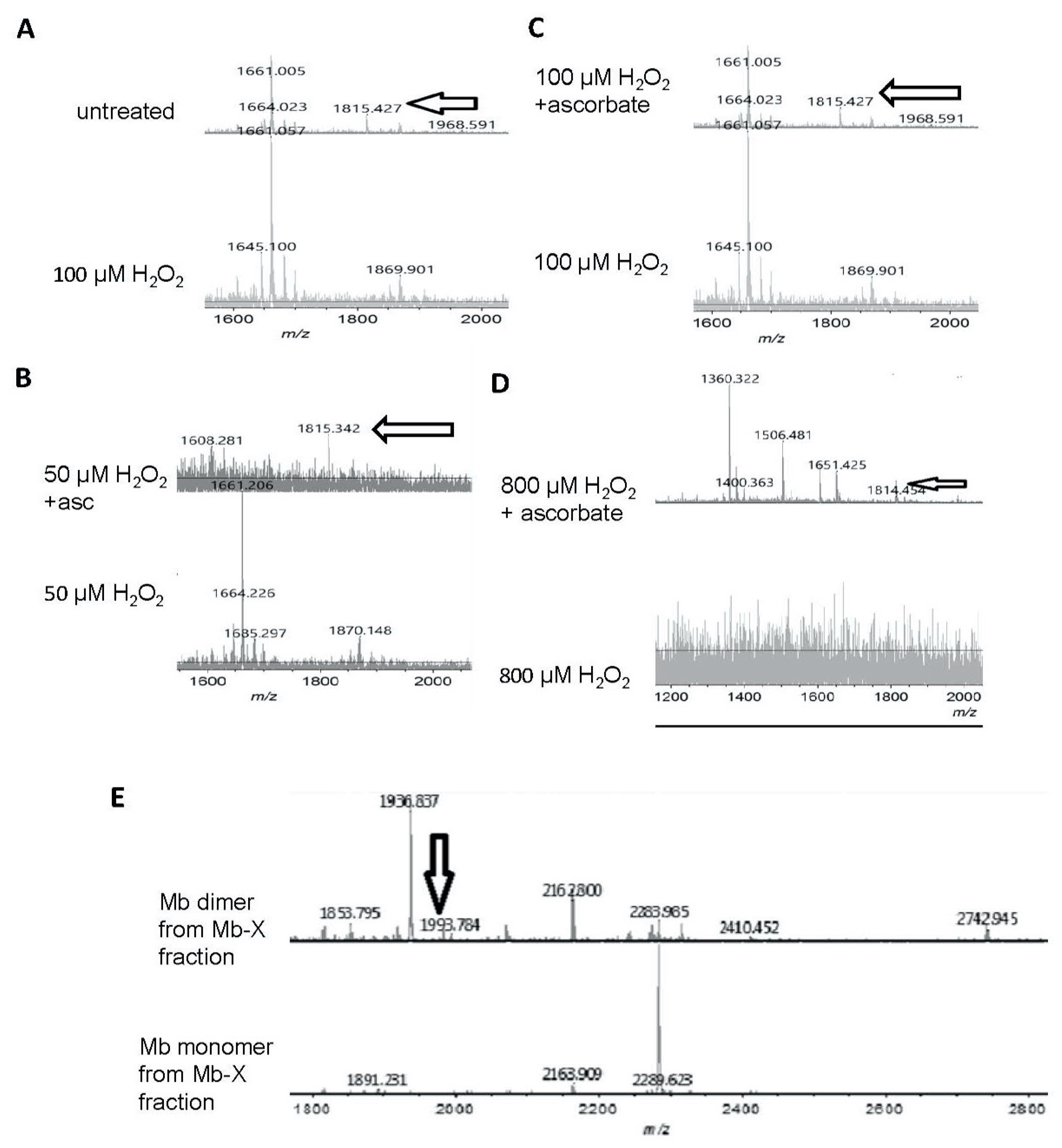
3.4. Reversibility of Mb-X Species
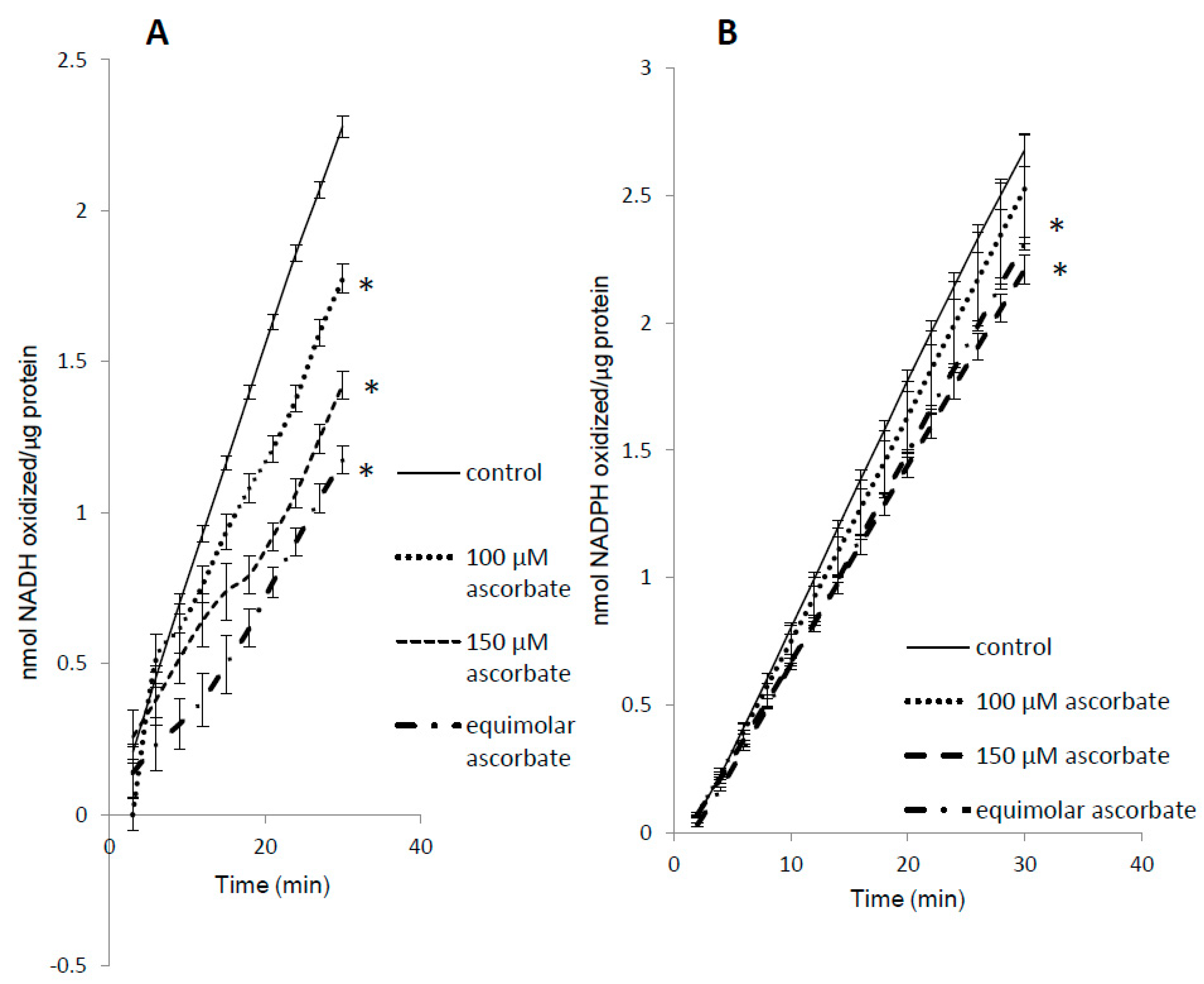
3.5. Complex Substrate Specificity of metMb Peroxidase Activity
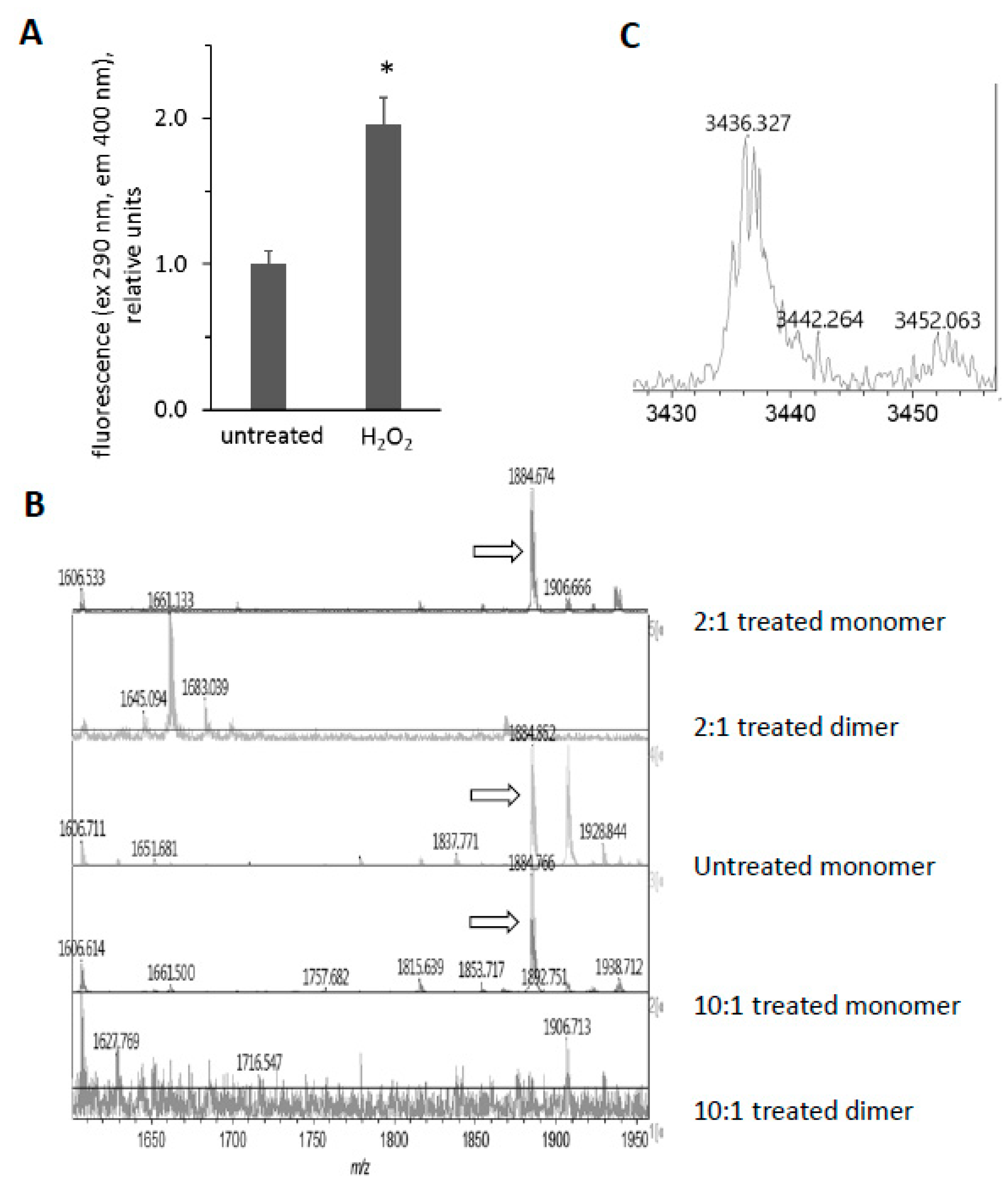
3.6. H2O2-Dependent Dimerization of metMb
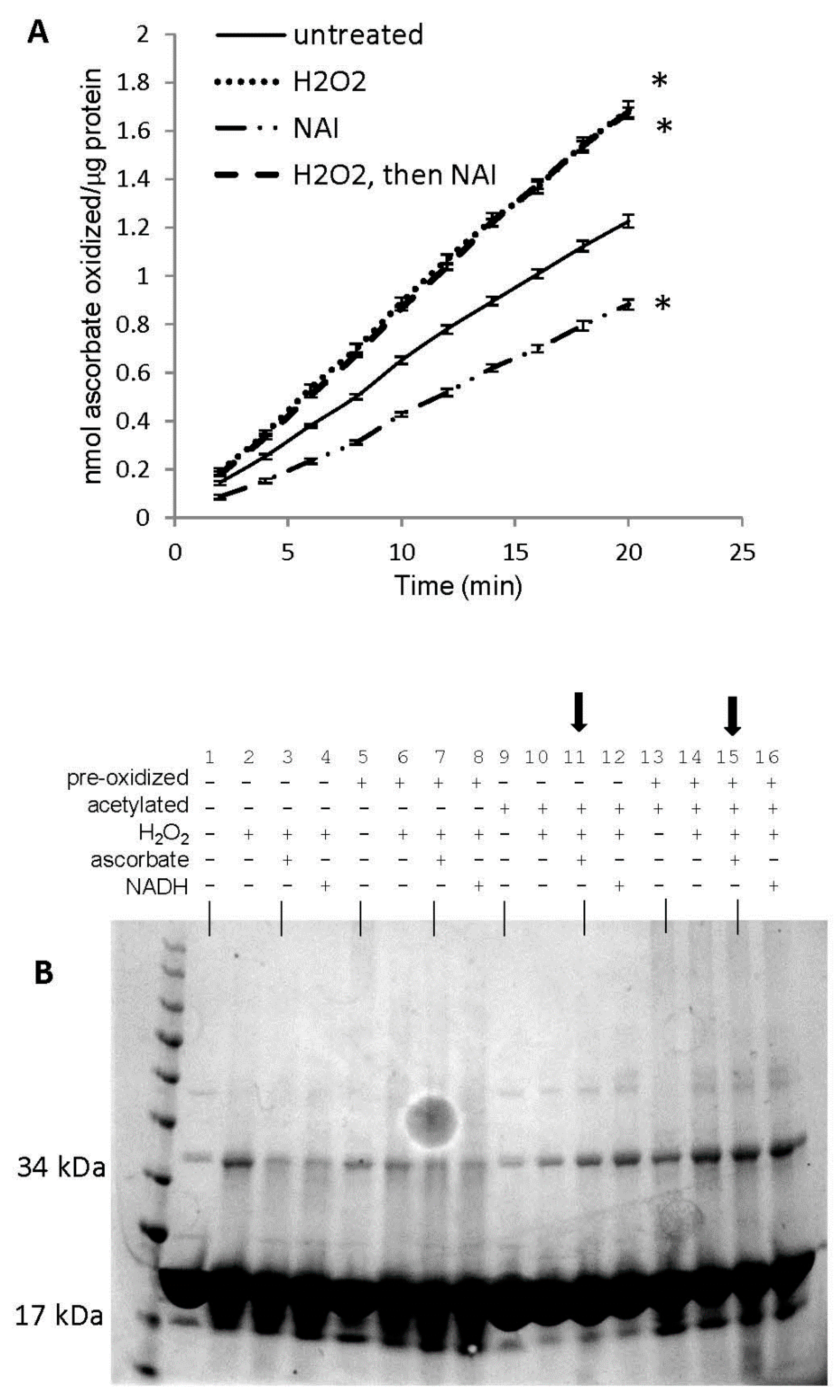
3.7. Reversal of Protein-to-Protein Cross-Links
3.8. Potential Role of Tyrosine Residues in Breaking Mb–Mb Cross-Links
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Boronat, S.; Garcia-Santamarina, S.; Hidalgo, E. Gel-free proteomic methodologies to study reversible cysteine oxidation and irreversible protein carbonyl formation. Free Radic. Res. 2015, 49, 494–510. [Google Scholar] [CrossRef] [PubMed]
- Reeg, S.; Grune, T. Protein oxidation in aging: Does it play a role in aging progression? Antioxid. Redox Signal. 2015, 23, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Jeong, M.S.; Choi, S.Y.; Kang, J.H. Oxidative modification of cytochrome c by hydrogen peroxide. Mol. Cells 2006, 22, 220–227. [Google Scholar] [PubMed]
- Xiang, W.; Weisbach, V.; Sticht, H.; Seebahn, A.; Bussmann, J.; Zimmermann, R.; Becker, C.M. Oxidative stress-induced posttranslational modifications of human hemoglobin in erythrocytes. Arch. Biochem. Biophys. 2013, 529, 34–44. [Google Scholar] [CrossRef]
- Archakov, A.I.; Zgoda, V.G.; Karuzina, I.I. Oxidative modification of cytochrome P450 and other macromolecules during its turnover. Vopr. Med. Khim. 1998, 44, 3–27. [Google Scholar]
- Giulivi, C.; Cadenas, E. Ferrylmyoglobin: Formation and chemical reactivity toward electron-donating compounds. Methods Enzym. 1994, 233, 189–202. [Google Scholar]
- Reeder, B.J.; Cutruzzola, F.; Bigotti, M.G.; Watmough, N.J.; Wilson, M.T. Histidine and not tyrosine is required for the peroxide-induced formation of haem to protein cross-linked myoglobin. IUBMB Life 2007, 59, 477–489. [Google Scholar] [CrossRef]
- Svistunenko, D.A.; Reeder, B.J.; Wankasi, M.M.; Silaghi-Dumitrescu, R.L.; Cooper, C.E.; Rinaldo, S.; Cutruzzola, F.; Wilson, M.T. Reaction of Aplysia limacina metmyoglobin with hydrogen peroxide. Dalton Trans. 2007, 840–850. [Google Scholar] [CrossRef]
- Catalano, C.E.; Choe, Y.S.; Ortiz de Montellano, P.R. Reactions of the protein radical in peroxide-treated myoglobin. Formation of a heme-protein cross-link. J. Biol. Chem. 1989, 264, 10534–10541. [Google Scholar]
- Fox, J.B., Jr.; Nicholas, R.A.; Ackerman, S.A.; Swift, C.E. A multiple wavelength analysis of the reaction between hydrogen peroxide and metmyoglobin. Biochemistry 1974, 13, 5178–5186. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Korzekwa, K. Oxidative modification by low levels of HOOH can transform myoglobin to an oxidase. Proc. Natl. Acad. Sci. USA 1991, 88, 7081–7085. [Google Scholar] [CrossRef] [PubMed]
- Vuletich, J.L.; Osawa, Y.; Aviram, M. Enhanced lipid oxidation by oxidatively modified myoglobin: Role of protein-bound heme. Biochem. Biophys. Res. Commun. 2000, 269, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Williams, M.S. Covalent crosslinking of the heme prosthetic group to myoglobin by H2O2: Toxicological implications. Free Radic. Biol. Med. 1996, 21, 35–41. [Google Scholar] [CrossRef]
- Boutaud, O.; Moore, K.P.; Reeder, B.J.; Harry, D.; Howie, A.J.; Wang, S.; Carney, C.K.; Masterson, T.S.; Amin, T.; Wright, D.W.; et al. Acetaminophen inhibits hemoprotein-catalyzed lipid peroxidation and attenuates rhabdomyolysis-induced renal failure. Proc. Natl. Acad. Sci. USA 2010, 107, 2699–2704. [Google Scholar] [CrossRef]
- Holt, S.; Reeder, B.; Wilson, M.; Harvey, S.; Morrow, J.D.; Roberts, L.J., 2nd; Moore, K. Increased lipid peroxidation in patients with rhabdomyolysis. Lancet 1999, 353, 1241. [Google Scholar] [CrossRef]
- Malencik, D.A.; Anderson, S.R. Dityrosine as a product of oxidative stress and fluorescent probe. Amino Acids 2003, 25, 233–247. [Google Scholar] [CrossRef]
- Cheng, G.; Li, H.; Cao, Z.; Qiu, X.; McCormick, S.; Thannickal, V.J.; Nauseef, W.M. Vascular peroxidase-1 is rapidly secreted, circulates in plasma, and supports dityrosine cross-linking reactions. Free Radic. Biol. Med. 2011, 51, 1445–1453. [Google Scholar] [CrossRef]
- DiMarco, T.; Giulivi, C. Current analytical methods for the detection of dityrosine, a biomarker of oxidative stress, in biological samples. Mass Spectrom. Rev. 2007, 26, 108–120. [Google Scholar] [CrossRef]
- Fukuchi, Y.; Miura, Y.; Nabeno, Y.; Kato, Y.; Osawa, T.; Naito, M. Immunohistochemical detection of oxidative stress biomarkers, dityrosine and N(epsilon)-(hexanoyl)lysine, and C-reactive protein in rabbit atherosclerotic lesions. J. Atheroscler. Thromb. 2008, 15, 185–192. [Google Scholar] [CrossRef]
- Colombo, G.; Reggiani, F.; Cucchiari, D.; Portinaro, N.M.; Giustarini, D.; Rossi, R.; Garavaglia, M.L.; Saino, N.; Milzani, A.; Badalamenti, S.; et al. Plasma protein-bound di-tyrosines as biomarkers of oxidative stress in end stage renal disease patients on maintenance haemodialysis. BBA Clin. 2017, 7, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Il’yasova, D.; Scarbrough, P.; Spasojevic, I. Urinary biomarkers of oxidative status. Clin. Chim. Acta 2012, 413, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.G.; Morris, K.L.; Sada, A.A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of Amyloid-beta in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Biasetti, L.; Blakeman, B.J.; Pollack, S.J.; Zibaee, S.; Abdul-Sada, A.; Thorpe, J.R.; Xue, W.F.; Serpell, L.C. The involvement of dityrosine crosslinking in alpha-synuclein assembly and deposition in Lewy Bodies in Parkinson’s disease. Sci. Rep. 2016, 6, 39171. [Google Scholar] [CrossRef] [PubMed]
- Mannino, M.H.; Patel, R.S.; Eccardt, A.M.; Perez Magnelli, R.A.; Robinson, C.L.C.; Janowiak, B.E.; Warren, D.E.; Fisher, J.S. Myoglobin as a versatile peroxidase: Implications for a more important role for vertebrate striated muscle in antioxidant defense. Comp. Biochem. Physiol. Part B 2019, 234, 9–17. [Google Scholar] [CrossRef] [PubMed]
- La Mar, G.; Satterlee, J.; De Ropp, J. Nuclear magnetic resonance of hemoproteins. In The Porphrylin Handbook; Kadish, K., Smith, K., Guilard, R., Eds.; Academic Press: New York, NY, USA, 2000; pp. 185–298. [Google Scholar]
- Yamamoto, Y. NMR study of active sites in paramagnetic haemoproteins. Annu. Rep. NMR Spectrosc. 1998, 36, 1–77. [Google Scholar] [CrossRef]
- Luthje, S.; Meisrimler, C.N.; Hopff, D.; Schutze, T.; Koppe, J.; Heino, K. Class III peroxidases. Methods Mol. Biol. 2014, 1072, 687–706. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Basu, S.; Kirley, T.L. Identification of a tyrosine residue responsible for N-acetylimidazole-induced increase of activity of ecto-nucleoside triphosphate diphosphohydrolase 3. Purinergic Signal. 2005, 1, 271–280. [Google Scholar] [CrossRef][Green Version]
- Lin, Y.W. The broad diversity of heme-protein cross-links: An overview. Biochim. Biophys. Acta 2015, 1854, 844–859. [Google Scholar] [CrossRef]
- Silaghi-Dumitrescu, R.; Reeder, B.J.; Nicholls, P.; Cooper, C.E.; Wilson, M.T. Ferryl haem protonation gates peroxidatic reactivity in globins. Biochem. J. 2007, 403, 391–395. [Google Scholar] [CrossRef] [PubMed]
- DeGray, J.A.; Gunther, M.R.; Tschirret-Guth, R.; Ortiz de Montellano, P.R.; Mason, R.P. Peroxidation of a specific tryptophan of metmyoglobin by hydrogen peroxide. J. Biol. Chem. 1997, 272, 2359–2362. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kroger-Ohlsen, M.V.; Ostdal, H.; Andersen, M.L. The effect of pH on the oxidation of bovine serum albumin by hypervalent myoglobin species. Arch. Biochem. Biophys. 2003, 416, 202–208. [Google Scholar] [CrossRef]
- Mitsos, S.E.; Kim, D.; Lucchesi, B.R.; Fantone, J.C. Modulation of myoglobin-H2O2-mediated peroxidation reactions by sulfhydryl compounds. Lab. Investig. 1988, 59, 824–830. [Google Scholar] [PubMed]
- Reeder, B.J.; Svistunenko, D.A.; Sharpe, M.A.; Wilson, M.T. Characteristics and mechanism of formation of peroxide-induced heme to protein cross-linking in myoglobin. Biochemistry 2002, 41, 367–375. [Google Scholar] [CrossRef]
- Detweiler, C.D.; Lardinois, O.M.; Deterding, L.J.; de Montellano, P.R.; Tomer, K.B.; Mason, R.P. Identification of the myoglobin tyrosyl radical by immuno-spin trapping and its dimerization. Free Radic. Biol. Med. 2005, 38, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Svistunenko, D.A.; Dunne, J.; Fryer, M.; Nicholls, P.; Reeder, B.J.; Wilson, M.T.; Bigotti, M.G.; Cutruzzola, F.; Cooper, C.E. Comparative study of tyrosine radicals in hemoglobin and myoglobins treated with hydrogen peroxide. Biophys. J. 2002, 83, 2845–2855. [Google Scholar] [CrossRef]
- Richardson, R.S.; Noyszewski, E.A.; Kendrick, K.F.; Leigh, J.S.; Wagner, P.D. Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J. Clin. Investig. 1995, 96, 1916–1926. [Google Scholar] [CrossRef]
- Richardson, R.S.; Newcomer, S.C.; Noyszewski, E.A. Skeletal muscle intracellular PO(2) assessed by myoglobin desaturation: Response to graded exercise. J. Appl. Physiol. 2001, 91, 2679–2685. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef]
- Rainey, W.T.J.; McDufie, H.F.; Hess, D.N.; Yeatts, L.B., Jr. Kinetics of the Thermal Decomposition of Biphenyl; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1964. [Google Scholar]
- Paviani, V.; Queiroz, R.F.; Marques, E.F.; Di Mascio, P.; Augusto, O. Production of lysozyme and lysozyme-superoxide dismutase dimers bound by a ditryptophan cross-link in carbonate radical-treated lysozyme. Free Radic. Biol. Med. 2015, 89, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Leinisch, F.; Mariotti, M.; Rykaer, M.; Lopez-Alarcon, C.; Hagglund, P.; Davies, M.J. Peroxyl radical- and photo-oxidation of glucose 6-phosphate dehydrogenase generates cross-links and functional changes via oxidation of tyrosine and tryptophan residues. Free Radic. Biol. Med. 2017, 112, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Leo, G.; Altucci, C.; Bourgoin-Voillard, S.; Gravagnuolo, A.M.; Esposito, R.; Marino, G.; Costello, C.E.; Velotta, R.; Birolo, L. Ultraviolet laser-induced cross-linking in peptides. Rapid Commun. Mass Spectrom. 2013, 27, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Osapay, K.; Tran, D.; Ladokhin, A.S.; White, S.H.; Henschen, A.H.; Selsted, M.E. Formation and characterization of a single Trp-Trp cross-link in indolicidin that confers protease stability without altering antimicrobial activity. J. Biol. Chem. 2000, 275, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Paviani, V.; Galdino, G.T.; dos Prazeres, J.N.; Queiroz, R.F.; Augusto, O. Ditryptophan cross-links as novel products of protein oxidation. J. Braz. Chem. Soc. 2018. [Google Scholar] [CrossRef]
- Giulivi, C.; Romero, F.J.; Cadenas, E. The interaction of Trolox C, a water-soluble vitamin E analog, with ferrylmyoglobin: Reduction of the oxoferryl moiety. Arch. Biochem. Biophys. 1992, 299, 302–312. [Google Scholar] [CrossRef]
- Aguirre, E.; Rodriguez-Juarez, F.; Bellelli, A.; Gnaiger, E.; Cadenas, S. Kinetic model of the inhibition of respiration by endogenous nitric oxide in intact cells. Biochim. Biophys. Acta 2010, 1797, 557–565. [Google Scholar] [CrossRef]
- Madej, T.; Lanczycki, C.J.; Zhang, D.; Thiessen, P.A.; Geer, R.C.; Marchler-Bauer, A.; Bryant, S.H. MMDB and VAST+: Tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014, 42, D297–D303. [Google Scholar] [CrossRef]
- Wang, C.S.; Pan, H.; Weerasekare, G.M.; Stewart, R.J. Peroxidase-catalysed interfacial adhesion of aquatic caddisworm silk. J. R. Soc. Interface 2015, 12. [Google Scholar] [CrossRef]
- Raven, D.J.; Earland, C.; Little, M. Occurrence of dityrosine in Tussah silk fibroin and keratin. Biochim. Biophys. Acta 1971, 251, 96–99. [Google Scholar] [CrossRef]
- LaBella, F.; Keeley, F.; Vivian, S.; Thornhill, D. Evidence for dityrosine in elastin. Biochem. Biophys. Res. Commun. 1967, 26, 748–753. [Google Scholar] [CrossRef]
- Foerder, C.A.; Shapiro, B.M. Release of ovoperoxidase from sea urchin eggs hardens the fertilization membrane with tyrosine crosslinks. Proc. Natl. Acad. Sci. USA 1977, 74, 4214–4218. [Google Scholar] [CrossRef]
- Kato, Y.; Maruyama, W.; Naoi, M.; Hashizume, Y.; Osawa, T. Immunohistochemical detection of dityrosine in lipofuscin pigments in the aged human brain. FEBS Lett. 1998, 439, 231–234. [Google Scholar] [CrossRef]
- Mayer, F.; Falk, M.; Huhn, R.; Behmenburg, F.; Ritz-Timme, S. Dityrosine as a marker of acute myocardial infarction? Experiments with the isolated Langendorff heart. Int. J. Leg. Med. 2016, 130, 1053–1060. [Google Scholar] [CrossRef]
- Atwood, C.S.; Perry, G.; Zeng, H.; Kato, Y.; Jones, W.D.; Ling, K.Q.; Huang, X.; Moir, R.D.; Wang, D.; Sayre, L.M.; et al. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry 2004, 43, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Mao, X.O.; Banwait, S.; Jin, K.; Greenberg, D.A. Neuroglobin attenuates beta-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19114–19119. [Google Scholar] [CrossRef]
- Szymanski, M.; Wang, R.; Fallin, M.D.; Bassett, S.S.; Avramopoulos, D. Neuroglobin and Alzheimer’s dementia: Genetic association and gene expression changes. Neurobiol. Aging 2010, 31, 1835–1842. [Google Scholar] [CrossRef]
- Reeder, B.J.; Cutruzzola, F.; Bigotti, M.G.; Hider, R.C.; Wilson, M.T. Tyrosine as a redox-active center in electron transfer to ferryl heme in globins. Free Radic. Biol. Med. 2008, 44, 274–283. [Google Scholar] [CrossRef]
- Reeder, B.J.; Wilson, M.T. The effects of pH on the mechanism of hydrogen peroxide and lipid hydroperoxide consumption by myoglobin: A role for the protonated ferryl species. Free Radic. Biol. Med. 2001, 30, 1311–1318. [Google Scholar] [CrossRef]
- Weller, P.A.; Price, M.; Isenberg, H.; Edwards, Y.H.; Jeffreys, A.J. Myoglobin expression: Early induction and subsequent modulation of myoglobin and myoglobin mRNA during myogenesis. Mol. Cell. Biol. 1986, 6, 4539–4547. [Google Scholar] [CrossRef]
- Moller, P.; Sylven, C. Myoglobin in human skeletal muscle. Scand. J. Clin. Lab. Investig. 1981, 41, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Kreutzer, U.; Jue, T. Role of myoglobin as a scavenger of cellular NO in myocardium. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H985–H991. [Google Scholar] [CrossRef] [PubMed]
- Gussakovsky, E.; Yang, Y.; Rendell, J.; Jilkina, O.; Kupriyanov, V. Mapping the myoglobin concentration, oxygenation, and optical pathlength in heart ex vivo using near-infrared imaging. Anal. Biochem. 2010, 407, 120–127. [Google Scholar] [CrossRef][Green Version]
- Swartling, J.; Pålsson, S.; Platonov, P.; Olsson, S.B.; Andersson-Engels, S. Changes in tissue optical properties due to radio-frequency ablation of myocardium. Med. Biol. Eng. Comput. 2003, 41, 403–409. [Google Scholar] [CrossRef]
- Mason, S.A.; Baptista, R.; Della Gatta, P.A.; Yousif, A.; Russell, A.P.; Wadley, G.D. High-dose vitamin C supplementation increases skeletal muscle vitamin C concentration and SVCT2 transporter expression but does not alter redox status in healthy males. Free Radic. Biol. Med. 2014, 77, 130–138. [Google Scholar] [CrossRef]
- Jackson, M.J. Redox regulation of adaptive responses in skeletal muscle to contractile activity. Free Radic. Biol. Med. 2009, 47, 1267–1275. [Google Scholar] [CrossRef]
- Palomero, J.; Pye, D.; Kabayo, T.; Spiller, D.G.; Jackson, M.J. In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid. Redox Signal. 2008, 10, 1463–1474. [Google Scholar] [CrossRef]
- Akl, M.G.; Fawzy, E.; Deif, M.; Farouk, A.; Elshorbagy, A.K. Perturbed adipose tissue hydrogen peroxide metabolism in centrally obese men: Association with insulin resistance. PLoS ONE 2017, 12, e0177268. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mannino, M.H.; Patel, R.S.; Eccardt, A.M.; Janowiak, B.E.; Wood, D.C.; He, F.; Fisher, J.S. Reversible Oxidative Modifications in Myoglobin and Functional Implications. Antioxidants 2020, 9, 549. https://doi.org/10.3390/antiox9060549
Mannino MH, Patel RS, Eccardt AM, Janowiak BE, Wood DC, He F, Fisher JS. Reversible Oxidative Modifications in Myoglobin and Functional Implications. Antioxidants. 2020; 9(6):549. https://doi.org/10.3390/antiox9060549
Chicago/Turabian StyleMannino, Mark H., Rishi S. Patel, Amanda M. Eccardt, Blythe E. Janowiak, David C. Wood, Fahu He, and Jonathan S. Fisher. 2020. "Reversible Oxidative Modifications in Myoglobin and Functional Implications" Antioxidants 9, no. 6: 549. https://doi.org/10.3390/antiox9060549
APA StyleMannino, M. H., Patel, R. S., Eccardt, A. M., Janowiak, B. E., Wood, D. C., He, F., & Fisher, J. S. (2020). Reversible Oxidative Modifications in Myoglobin and Functional Implications. Antioxidants, 9(6), 549. https://doi.org/10.3390/antiox9060549