Toxicity of Necrostatin-1 in Parkinson’s Disease Models

 , ,
, ,  and
and 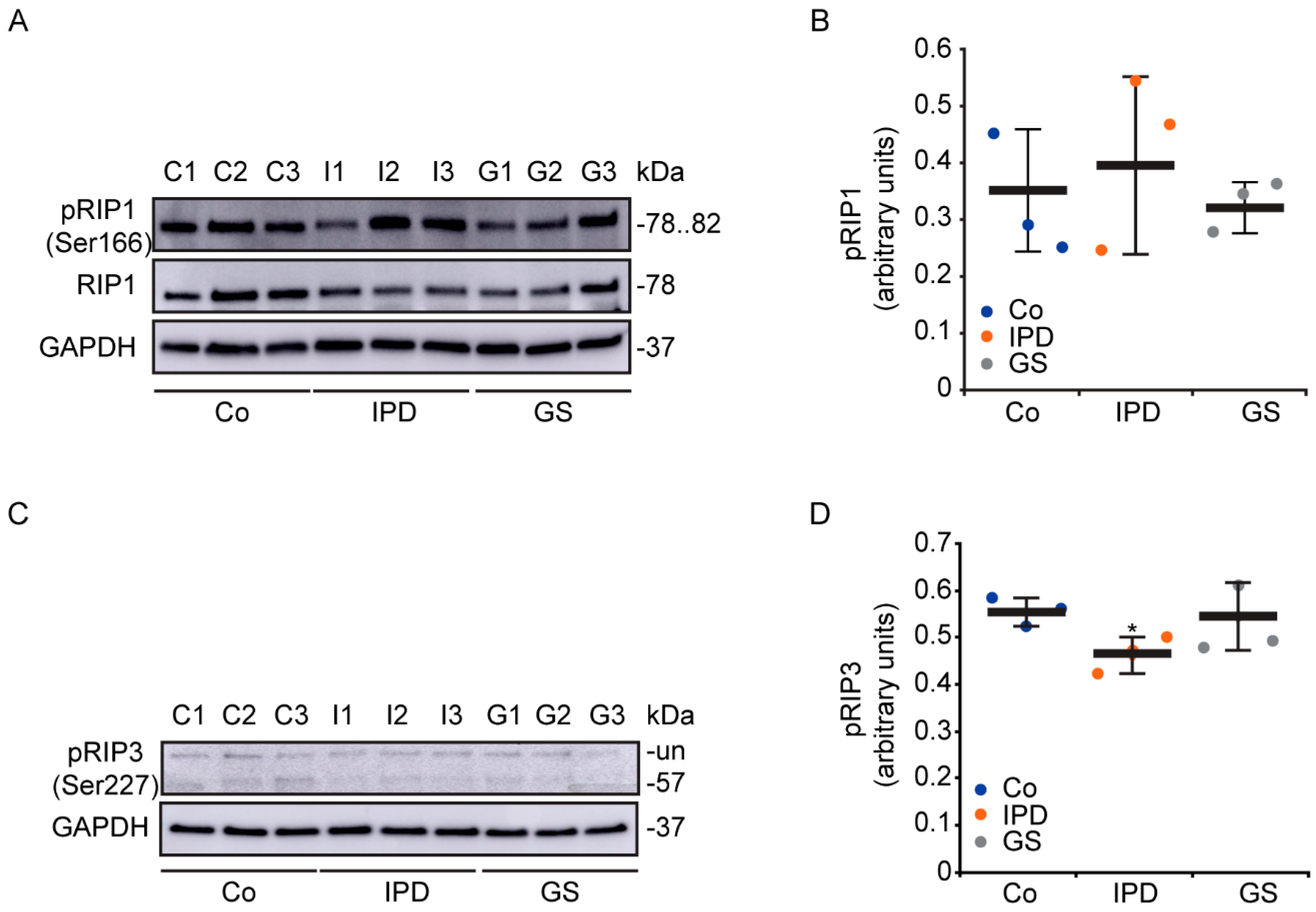{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. PD Models
2.2. Western Blotting Analysis
2.3. Flow Cytometry Assay
2.4. Immunofluorescence Microscopy
2.5. Statistics
3. Results
3.1. Characterization of Necroptosis in PD Models
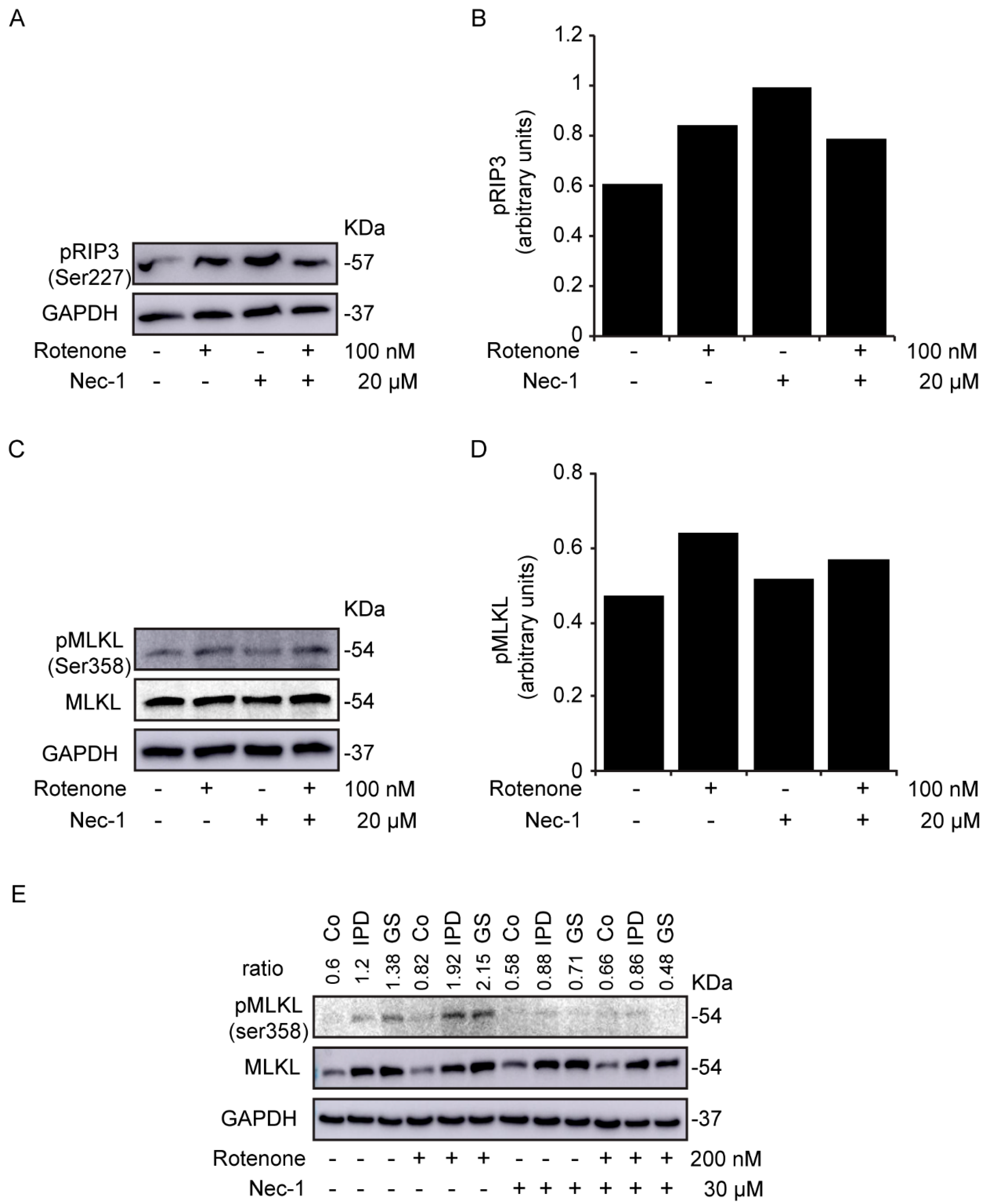
3.2. Study of Necroptosis in Rotenone-Induced Models
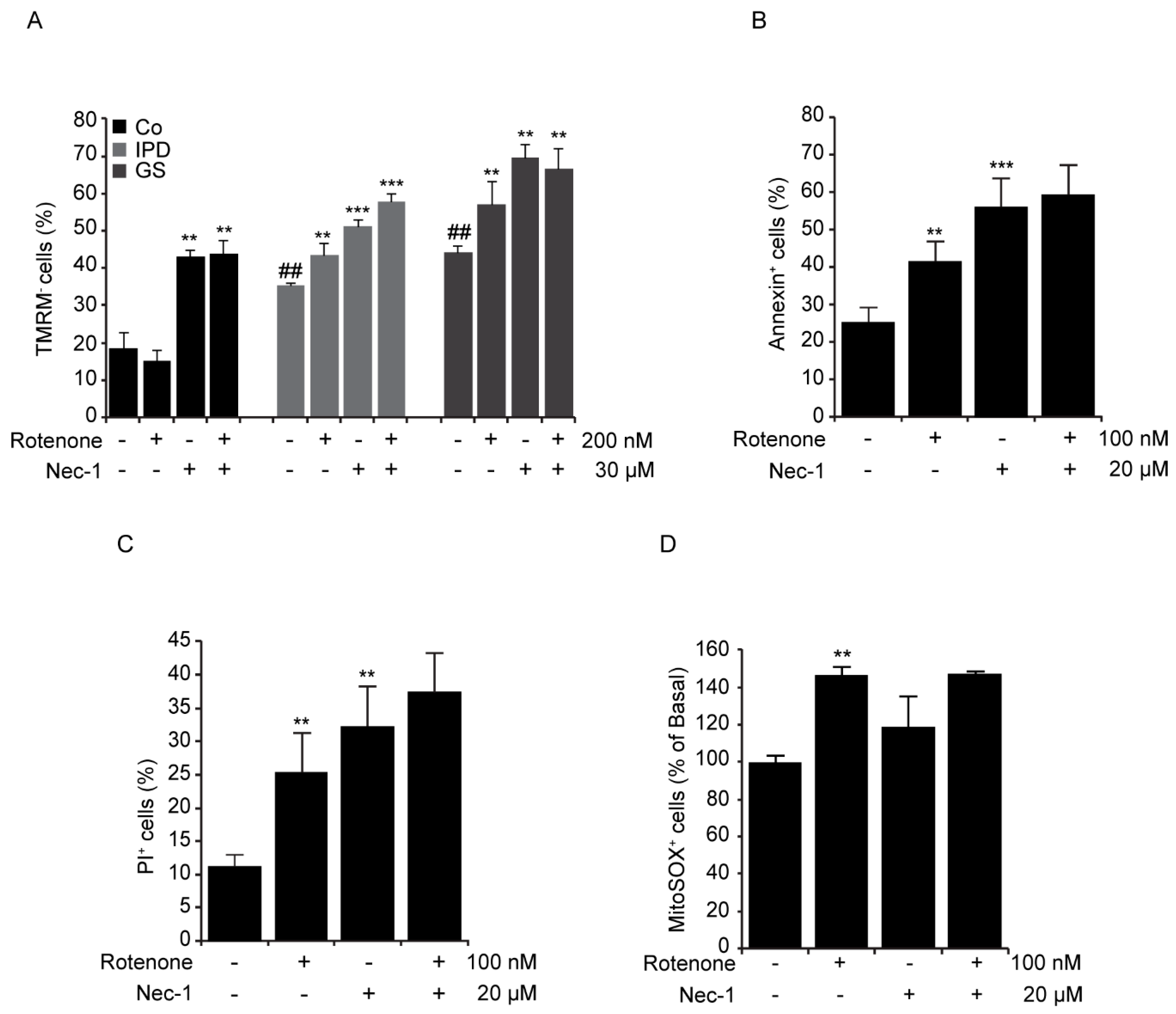
3.3. Cellular Toxicity of Necrostatin-1
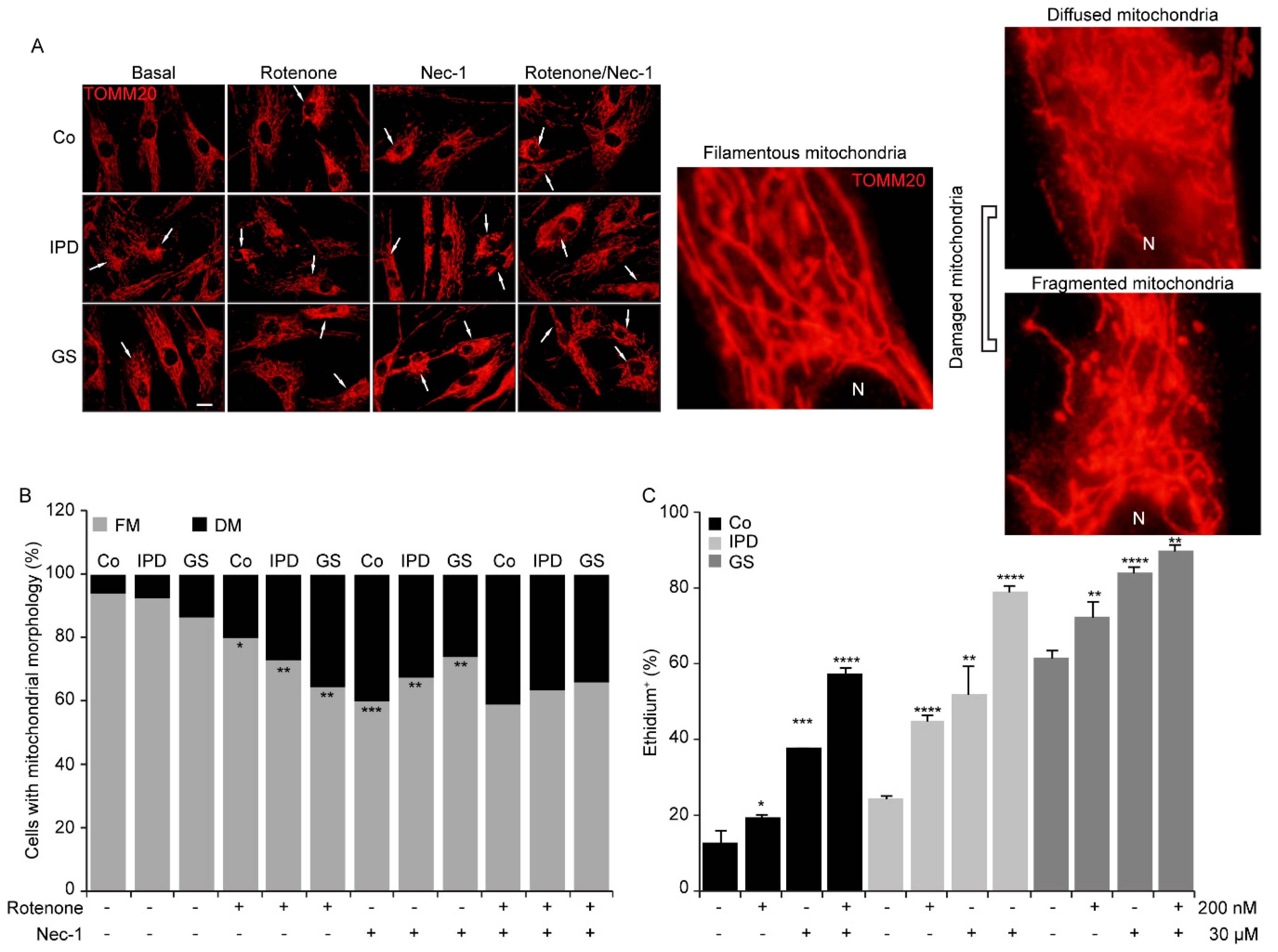
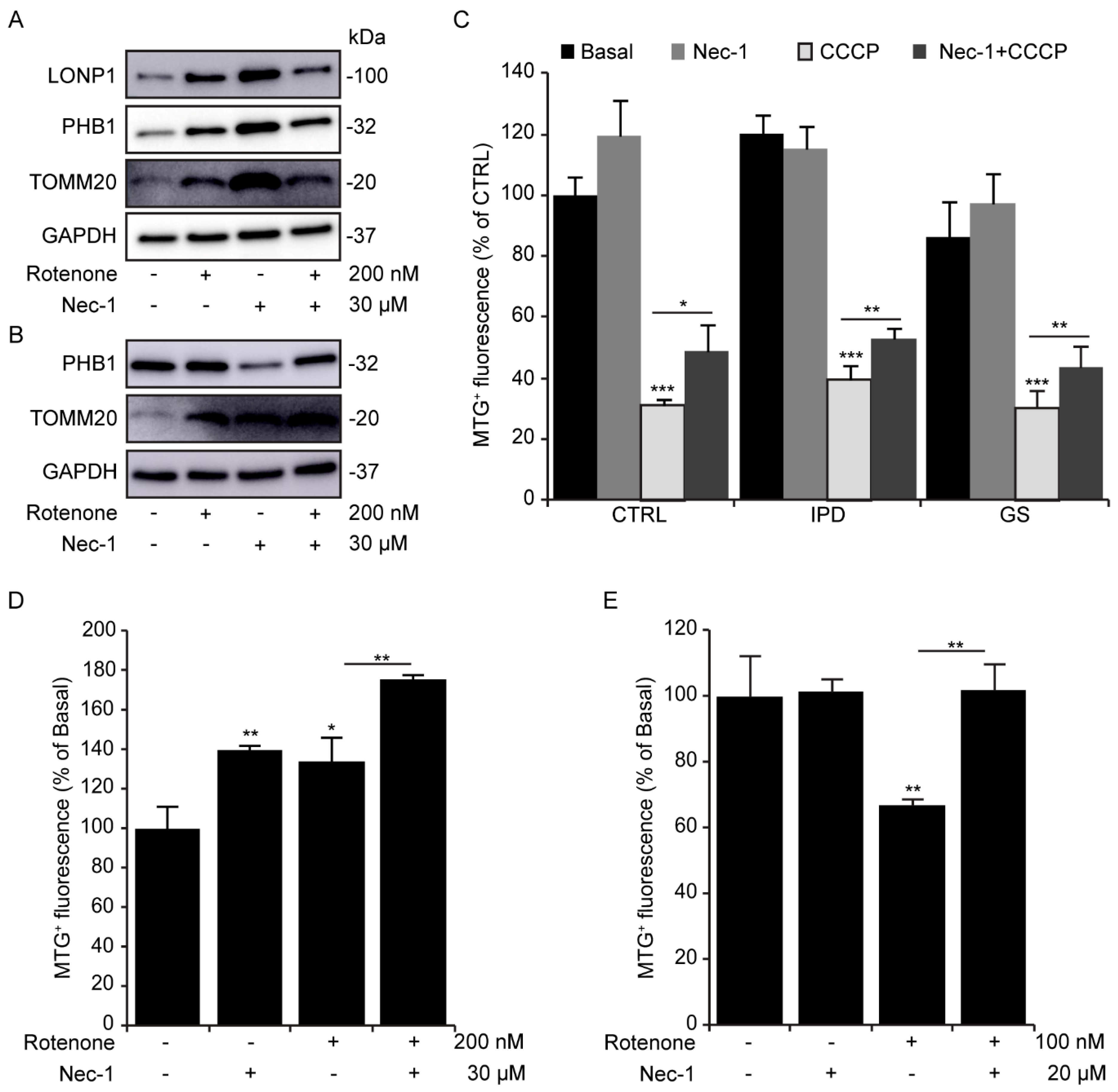
3.4. Mitochondrial Morphological Changes with Necrostatin-1
3.5. Necrostatin-1 Impairs Mitochondrial Clearance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giguere, N.; Burke Nanni, S.; Trudeau, L.E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef]
- González-Polo, R.A.; Rodríguez-Martín, A.; Morán, J.M.; Niso, M.; Soler, G.; Fuentes, J.M. Paraquat-induced apoptotic cell death in cerebellar granule cells. Brain Res. 2004, 1011, 170–176. [Google Scholar] [CrossRef]
- Niso-Santano, M.; González-Polo, R.A.; Pedro, J.M.B.-S.; Gómez-Sánchez, R.; Lastres-Becker, I.; Ortiz-Ortiz, M.A.; Soler, G.; Moran, J.M.; Cuadrado, A.; Fuentes, J.M. Activation of apoptosis signal-regulating kinase 1 is a key factor in paraquat-induced cell death: Modulation by the Nrf2/Trx axis. Free Radic. Boil. Med. 2010, 48, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Yakhine-Diop, S.M.; Pedro, J.M.B.-S.; Gómez-Sánchez, R.; Pizarro-Estrella, E.; Arribas, M.R.; Climent, V.; Aiastui, A.; De Munain, A.L.; Fuentes, J.M.; González-Polo, R.A. G2019S LRRK2 mutant fibroblasts from Parkinson’s disease patients show increased sensitivity to neurotoxin 1-methyl-4-phenylpyridinium dependent of autophagy. Toxicology 2014, 324, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pedro, J.M.B.-S.; Niso-Santano, M.; Gómez-Sánchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; De Maturana, R.L.; Sanchez-Pernaute, R.; De Munain, A.L.; et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell. Mol. Life Sci. 2012, 70, 121–136. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2011, 21, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sánchez, R.; Yakhine-Diop, S.M.S.; Pedro, J.M.B.-S.; Pizarro-Estrella, E.; Rodríguez-Arribas, M.; Climent, V.; Cano, F.E.M.; González-Soltero, M.E.; Tandon, A.; Fuentes, J.M.; et al. PINK1 deficiency enhances autophagy and mitophagy induction. Mol. Cell. Oncol. 2015, 3, e1046579. [Google Scholar] [CrossRef] [PubMed]
- Heeman, B.; Haute, C.V.D.; Aelvoet, S.A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.H.; Willems, P.H.; et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar] [CrossRef]
- Arduíno, D.M.; Raquel Esteves, A.; Cortes, L.; Silva, D.F.; Patel, B.; Grazina, M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum. Mol. Genet. 2012, 21, 4680–4702. [Google Scholar] [CrossRef]
- Ito, K.; Eguchi, Y.; Imagawa, Y.; Akai, S.; Mochizuki, H.; Tsujimoto, Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Zhang, D.; Lin, J.; Han, J. Receptor-interacting protein (RIP) kinase family. Cell. Mol. Immunol. 2010, 7, 243–249. [Google Scholar] [CrossRef]
- Moriwaki, K.; Chan, F.K.L. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013, 27, 1640–1649. [Google Scholar] [CrossRef]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.; Xu, Y.M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [PubMed]
- Negroni, A.; Colantoni, E.; Pierdomenico, M.; Palone, F.; Costanzo, M.; Oliva, S.; Tiberti, A.; Cucchiara, S.; Stronati, L. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Dig. Liver Dis. 2017, 49, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.S.; Chen, P.; Wang, W.X.; Lin, C.C.; Zhou, Y.; Yu, L.H.; Lin, Y.X.; Xu, Y.F.; Kang, D.Z. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. A J. Tech. Methods Pathol. 2019, 100, 503–551. [Google Scholar] [CrossRef] [PubMed]
- Oñate, M.; Catenaccio, A.; Salvadores, N.; Saquel, C.; Martinez, A.; Moreno-Gonzalez, I.; Gamez, N.; Soto, P.; Soto, C.; Hetz, C.; et al. The necroptosis machinery mediates axonal degeneration in a model of Parkinson disease. Cell Death Differ. 2019, 27, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.B.; Wu, J.R.; Wang, J.; Zhou, S.K.; Yang, L.; Yin, J.L.; Cao, J.P. Necrostatin-1 protection of dopaminergic neurons. Neural Regen. Res. 2015, 10, 1120–1124. [Google Scholar] [CrossRef]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Yakhine-Diop, S.M.S.; Niso-Santano, M.; Rodríguez-Arribas, M.; Gómez-Sánchez, R.; Chacón, G.M.; Uribe-Carretero, E.; Navarro-García, J.A.; Ruiz-Hurtado, G.; Aiastui, A.; Cooper, J.M.; et al. Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients. Mol. Neurobiol. 2018, 56, 2466–2481. [Google Scholar] [CrossRef]
- Callizot, N.; Combes, M.; Henriques, A.; Poindron, P. Necrosis, apoptosis, necroptosis, three modes of action of dopaminergic neuron neurotoxins. PLoS ONE 2019, 14, e0215277. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, P.J. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. J. Boil. Chem. 2002, 278, 8516–8525. [Google Scholar] [CrossRef]
- Han, C.H.; Guan, Z.B.; Zhang, P.X.; Fang, H.L.; Li, L.; Zhang, H.M.; Zhou, F.J.; Mao, Y.F.; Liu, W.W. Oxidative stress induced necroptosis activation is involved in the pathogenesis of hyperoxic acute lung injury. Biochem. Biophys. Res. Commun. 2018, 495, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Gräber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.E.; Andrews, L.A.; Burman, J.L.; Lin, W.Y.; Pallanck, L.J. PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix. PLoS Genet. 2014, 10, e1004279. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, O.; Tyurin, V.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef]
- Venderova, K.; Park, D.S. Programmed Cell Death in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009365. [Google Scholar] [CrossRef]
- Hussain, M.; Zimmermann, V.; Van Wijk, S.J.L.; Fulda, S. Mouse lung fibroblasts are highly susceptible to necroptosis in a reactive oxygen species-dependent manner. Biochem. Pharmacol. 2018, 153, 242–247. [Google Scholar] [CrossRef]
- Wu, J.; Huang, Z.; Ren, J.; Zhang, Z.; He, P.; Li, Y.; Ma, J.; Chen, W.; Zhang, Y.; Zhou, X.; et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013, 23, 994–1006. [Google Scholar] [CrossRef]
- Jie, H.; He, Y.; Huang, X.; Zhou, Q.; Han, Y.; Li, X.; Bai, Y.; Sun, E. Necrostatin-1 enhances the resolution of inflammation by specifically inducing neutrophil apoptosis. Oncotarget 2016, 7, 19367–19381. [Google Scholar] [CrossRef]
- Hong, Y.; Nie, H.; Wu, D.; Wei, X.; Ding, X.; Ying, W. NAD+ treatment prevents rotenone-induced apoptosis and necrosis of differentiated PC12 cells. Neurosci. Lett. 2014, 560, 46–50. [Google Scholar] [CrossRef]
- Yonekawa, T.; Gamez, G.; Kim, J.; Tan, A.C.; Thorburn, J.; Gump, J.; Thorburn, A.; Morgan, M.J. RIP 1 negatively regulates basal autophagic flux through TFEB to control sensitivity to apoptosis. EMBO Rep. 2015, 16, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Dionísio, P.A.; Oliveira, S.R.; Gaspar, M.M.; Gama, M.J.; Castro-Caldas, M.; Amaral, J.D.; Rodrigues, C.M.P. Ablation of RIP3 protects from dopaminergic neurodegeneration in experimental Parkinson’s disease. Cell Death Dis. 2019, 10, 840. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alegre-Cortés, E.; Muriel-González, A.; Canales-Cortés, S.; Uribe-Carretero, E.; Martínez-Chacón, G.; Aiastui, A.; López de Munain, A.; Niso-Santano, M.; Gonzalez-Polo, R.A.; Fuentes, J.M.; et al. Toxicity of Necrostatin-1 in Parkinson’s Disease Models. Antioxidants 2020, 9, 524. https://doi.org/10.3390/antiox9060524
Alegre-Cortés E, Muriel-González A, Canales-Cortés S, Uribe-Carretero E, Martínez-Chacón G, Aiastui A, López de Munain A, Niso-Santano M, Gonzalez-Polo RA, Fuentes JM, et al. Toxicity of Necrostatin-1 in Parkinson’s Disease Models. Antioxidants. 2020; 9(6):524. https://doi.org/10.3390/antiox9060524
Chicago/Turabian StyleAlegre-Cortés, Eva, Alicia Muriel-González, Saray Canales-Cortés, Elisabet Uribe-Carretero, Guadalupe Martínez-Chacón, Ana Aiastui, Adolfo López de Munain, Mireia Niso-Santano, Rosa A. Gonzalez-Polo, José M. Fuentes, and et al. 2020. "Toxicity of Necrostatin-1 in Parkinson’s Disease Models" Antioxidants 9, no. 6: 524. https://doi.org/10.3390/antiox9060524
APA StyleAlegre-Cortés, E., Muriel-González, A., Canales-Cortés, S., Uribe-Carretero, E., Martínez-Chacón, G., Aiastui, A., López de Munain, A., Niso-Santano, M., Gonzalez-Polo, R. A., Fuentes, J. M., & Yakhine-Diop, S. M. S. (2020). Toxicity of Necrostatin-1 in Parkinson’s Disease Models. Antioxidants, 9(6), 524. https://doi.org/10.3390/antiox9060524