The Impact of Acetyl-CoA and Aspartate Shortages on the N-Acetylaspartate Level in Different Models of Cholinergic Neurons

 , , , ,
, , , , 
Abstract
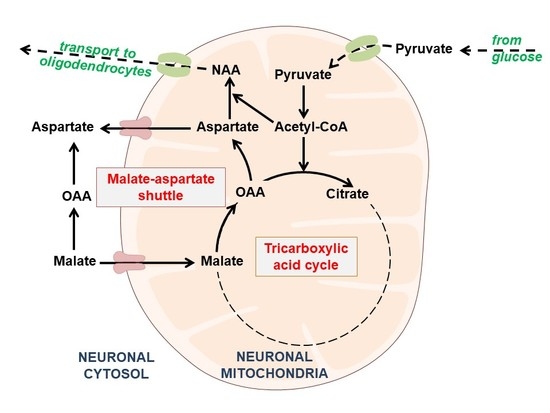
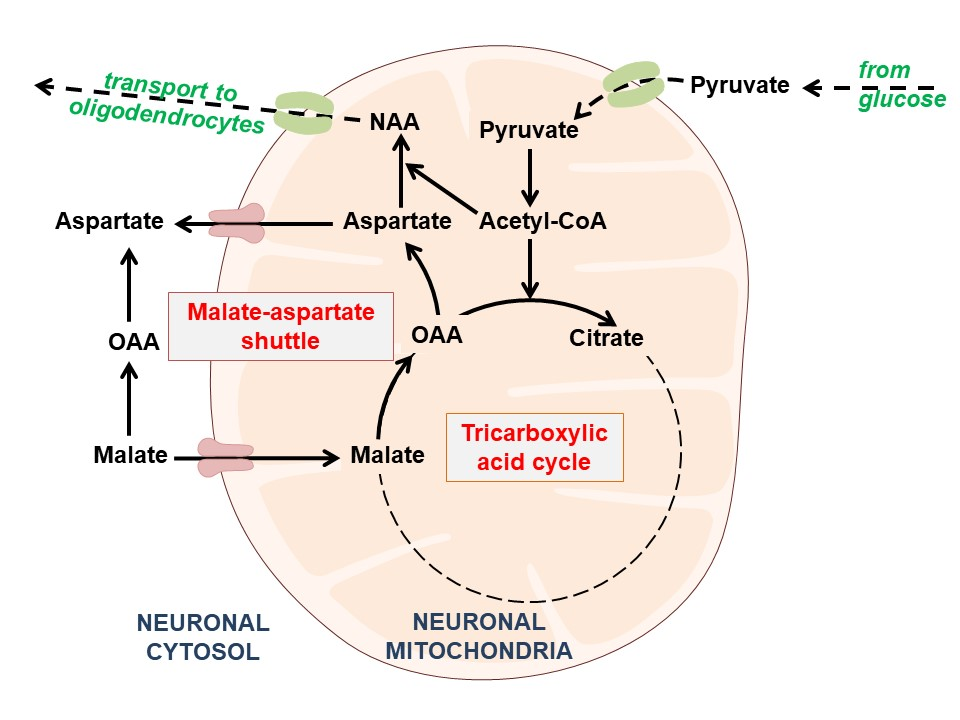
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Cell Culture
2.4. Sample Preparation
2.5. Mitochondria Isolation
2.6. Enzymatic Assays
2.7. Metabolic Assays
2.8. Western Blot Analysis
2.9. Real-Time RT-qPCR Analysis of NAT8L mRNA Levels
2.10. Protein Assay
2.11. Statistics
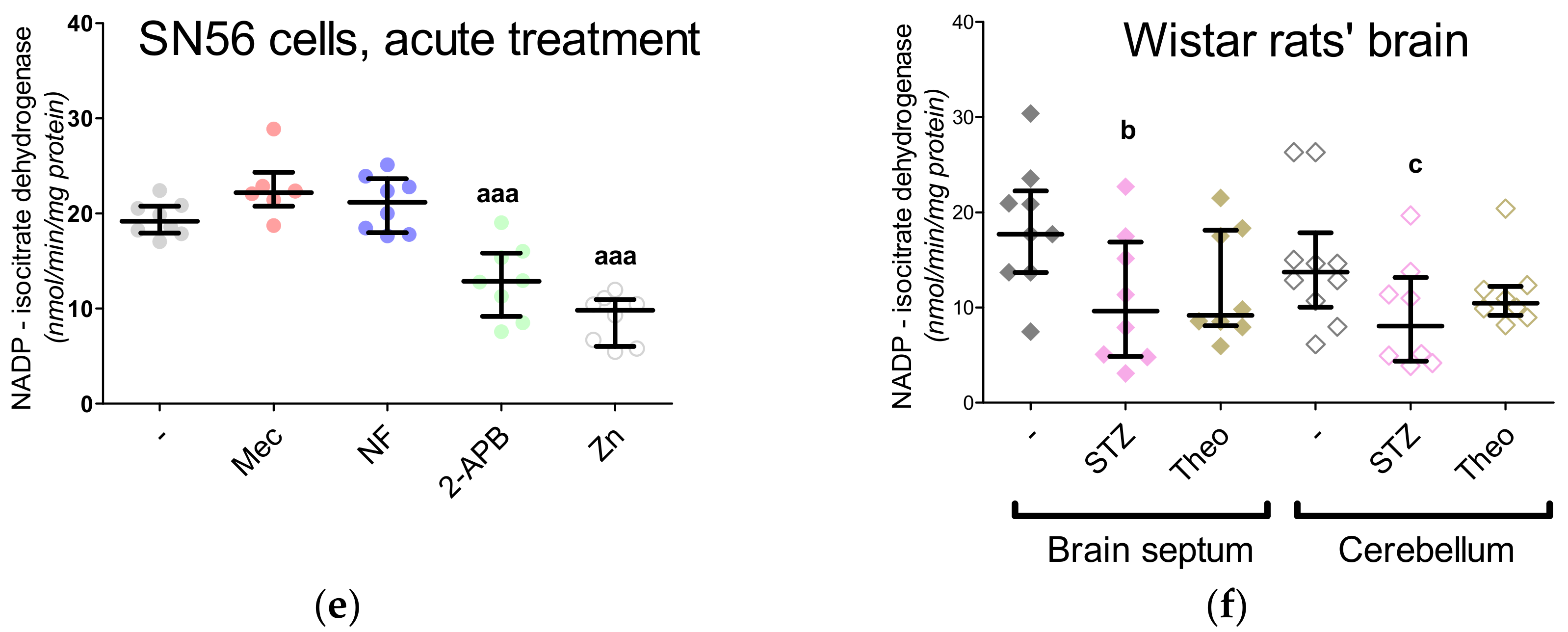
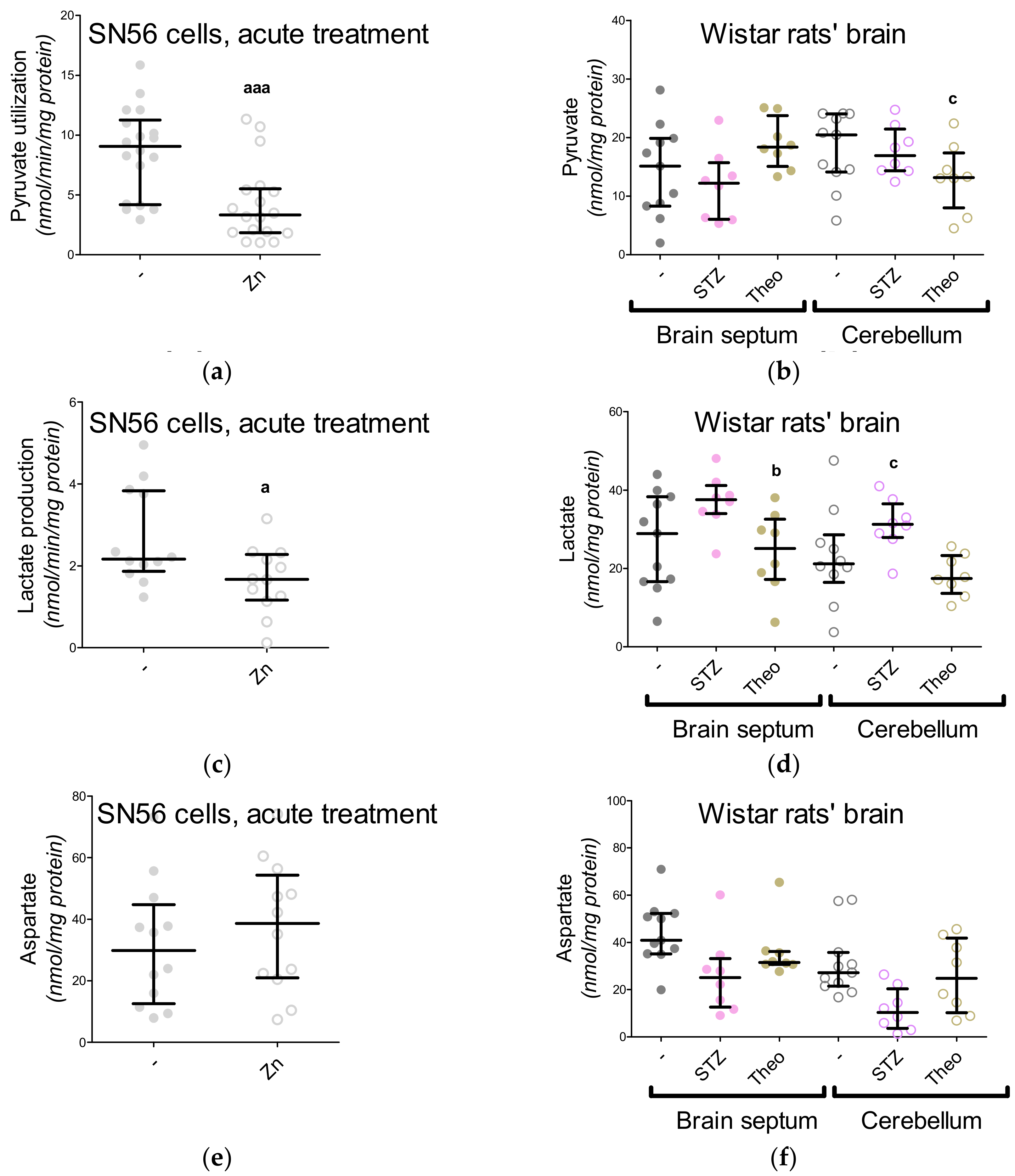
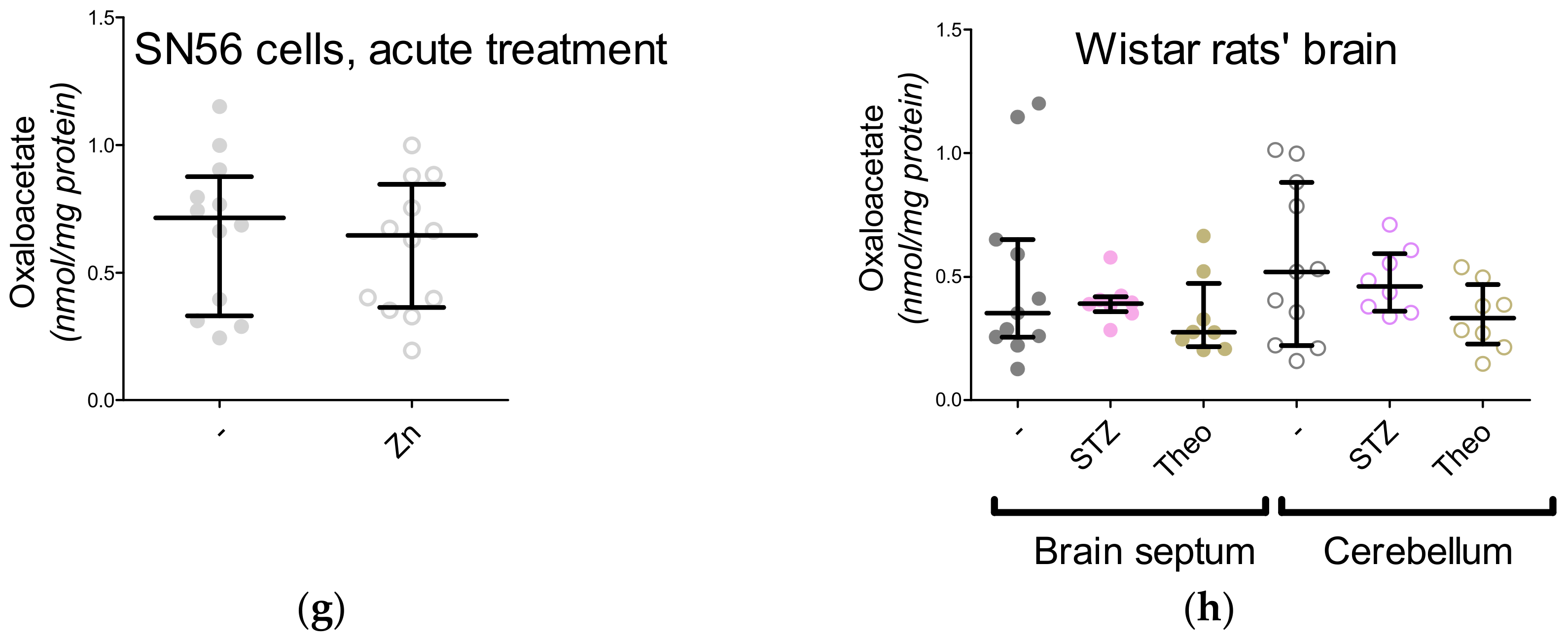
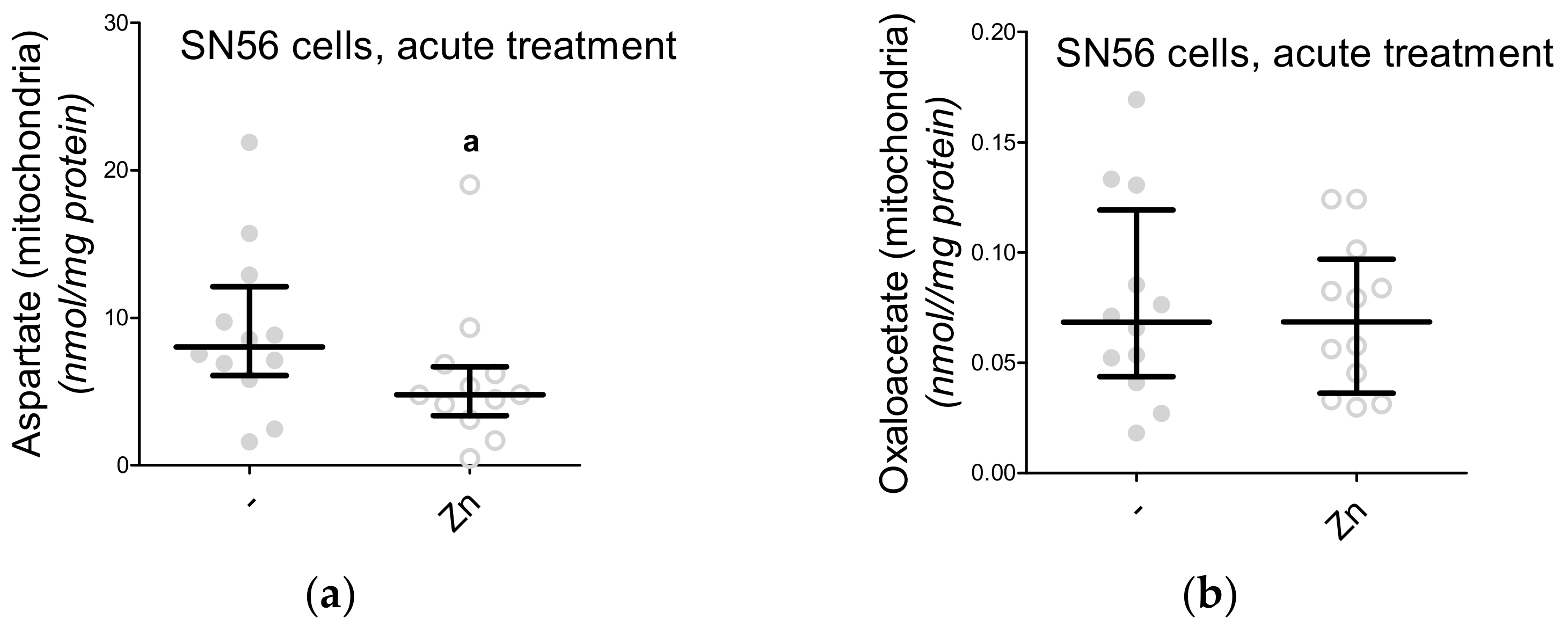
3. Results
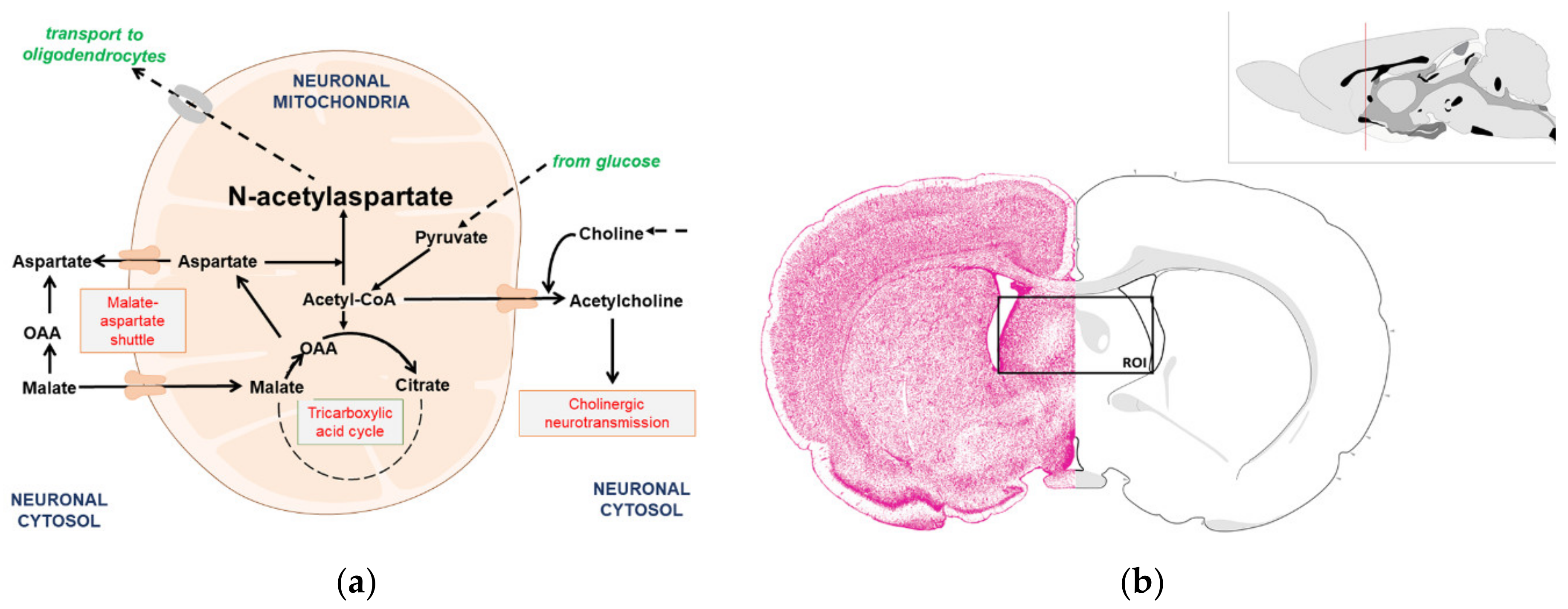
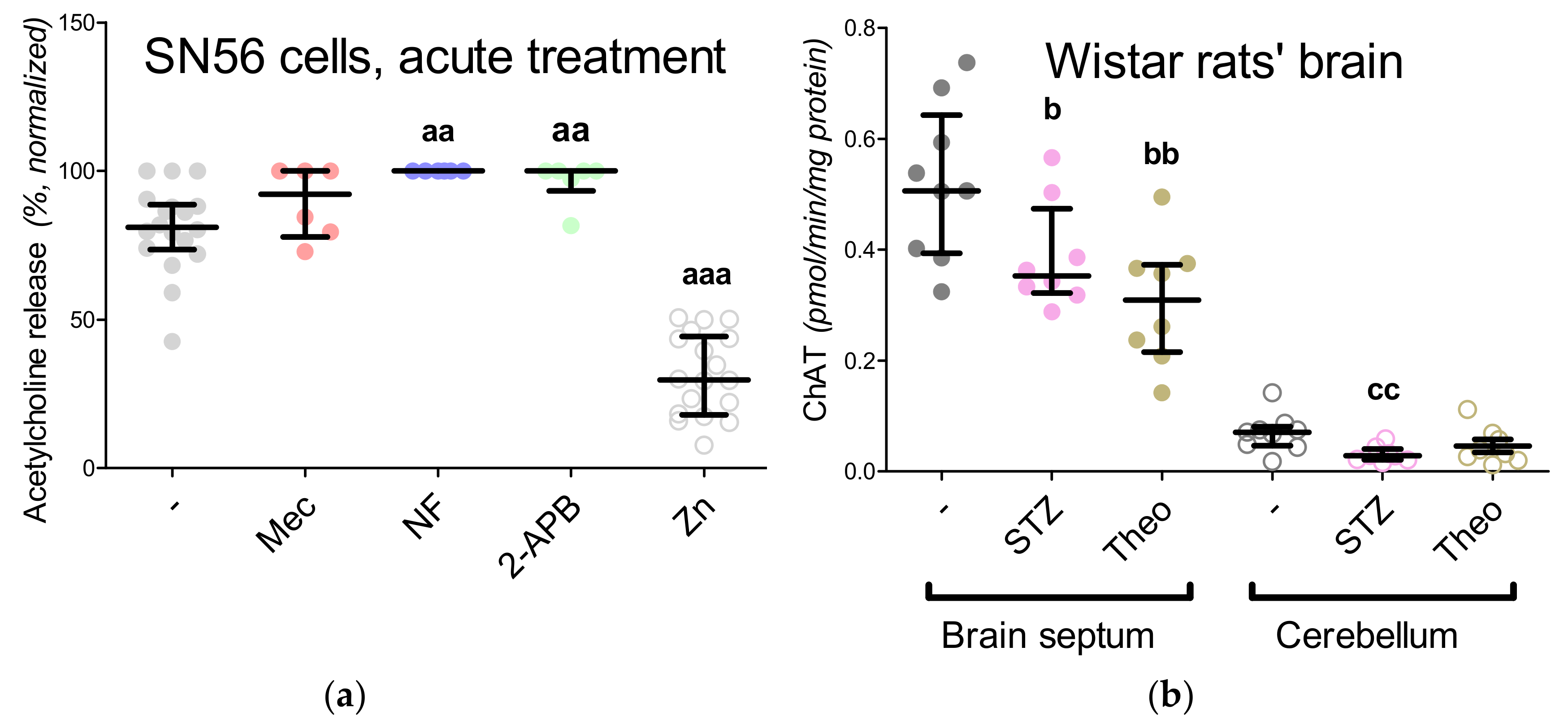
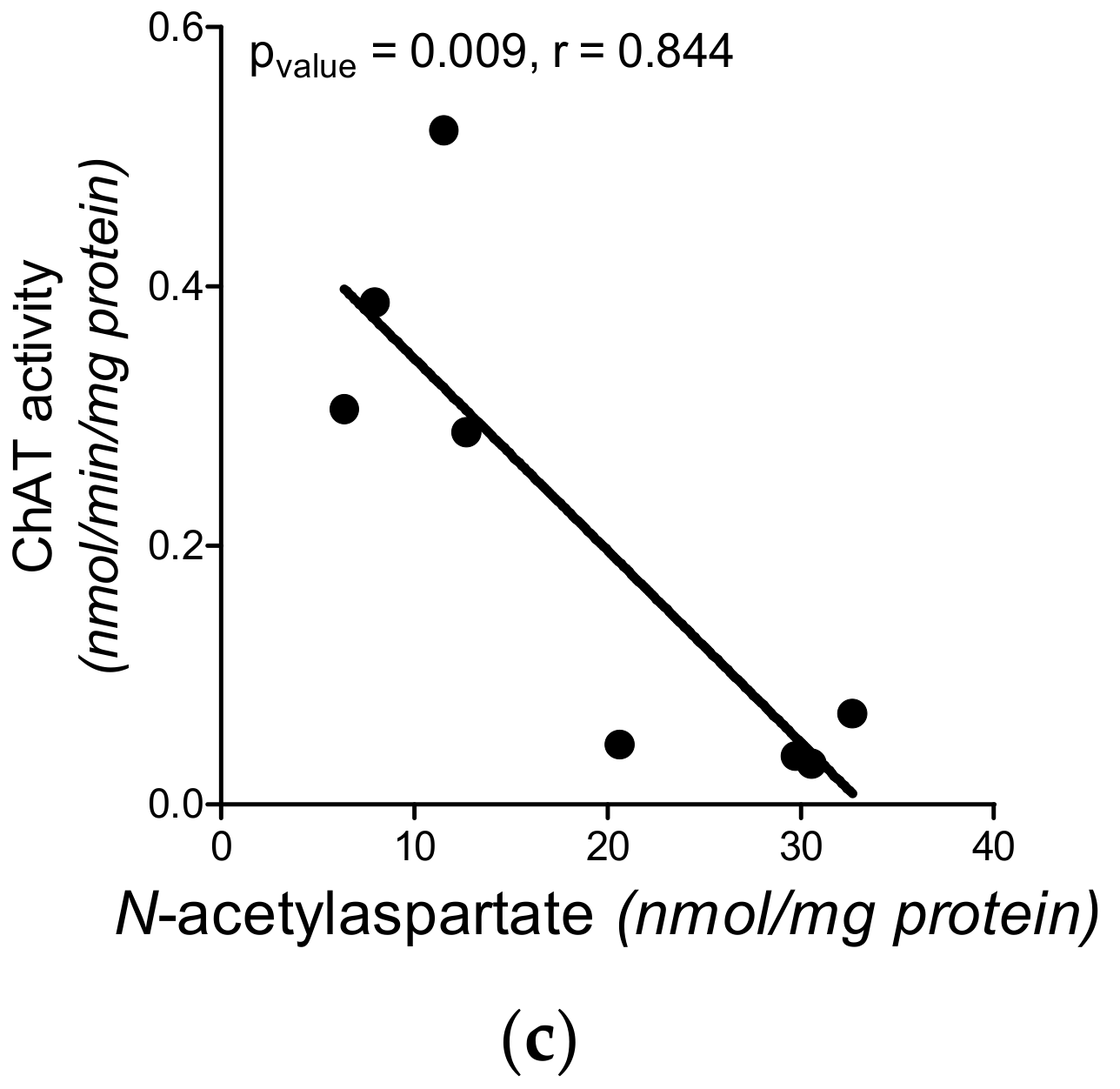
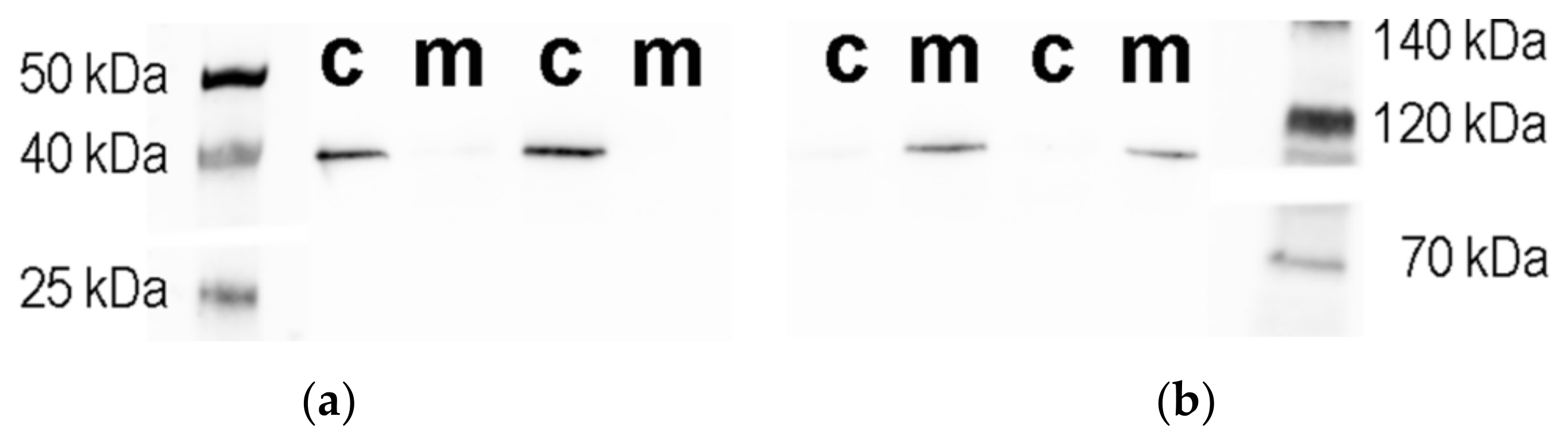
3.1. Cholinergic Phenotype in SN56 Cell Line and Wistar rats’ Brain
3.2. Isolation and Characterization of the Subcellular Fractions of the SN56 Cells
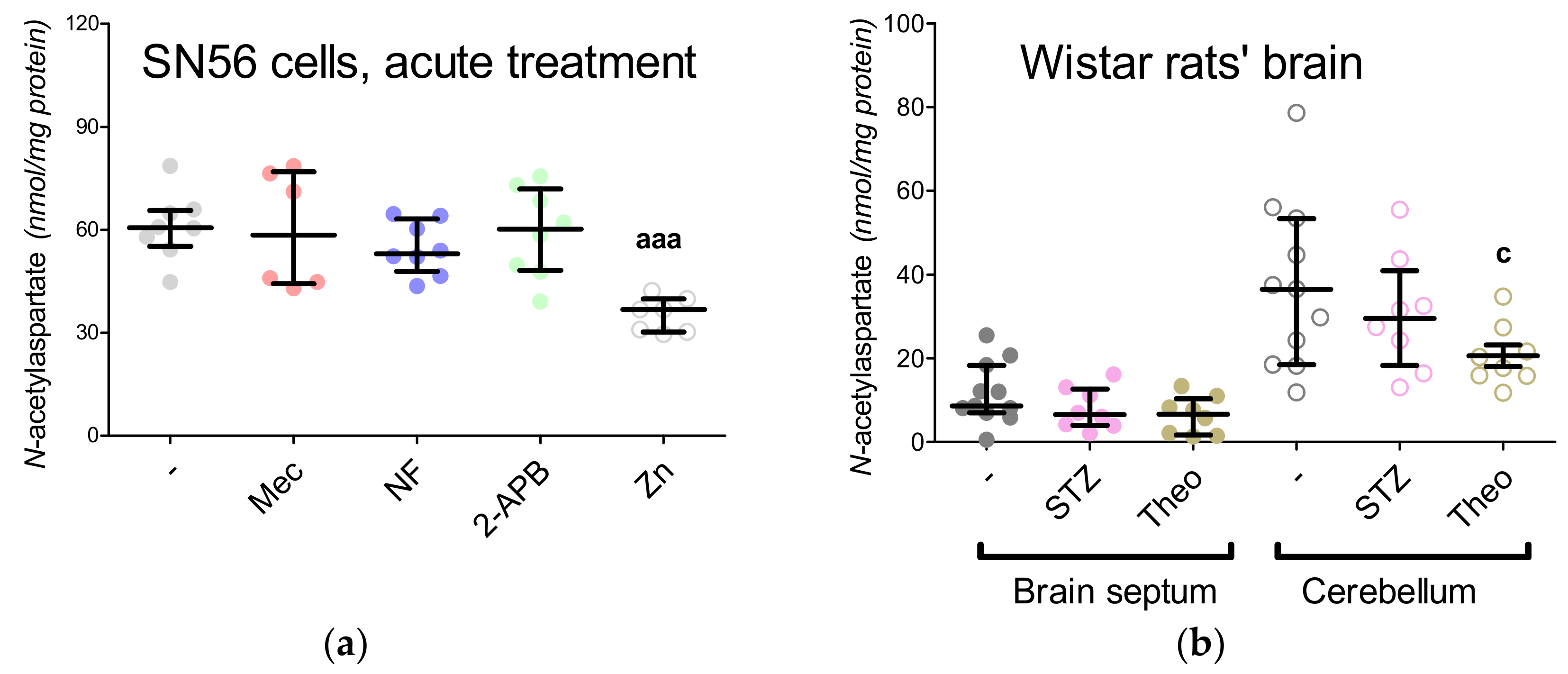
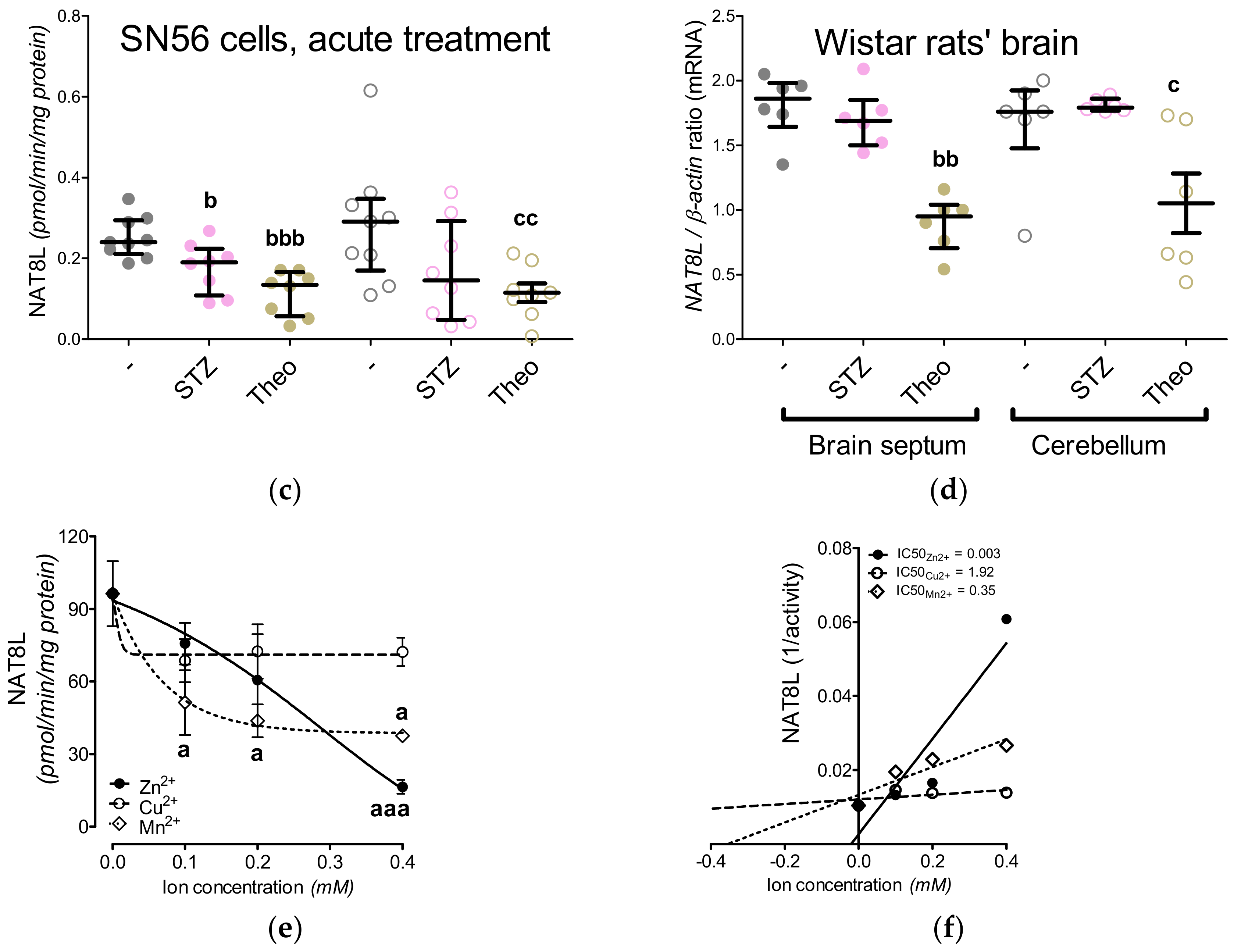
3.3. Chronic Effect of 0.15-mM Zn2+ on the SN56 Cells
3.4. Aspartate N-Acetyltransferase Activity in the SN56 Cells
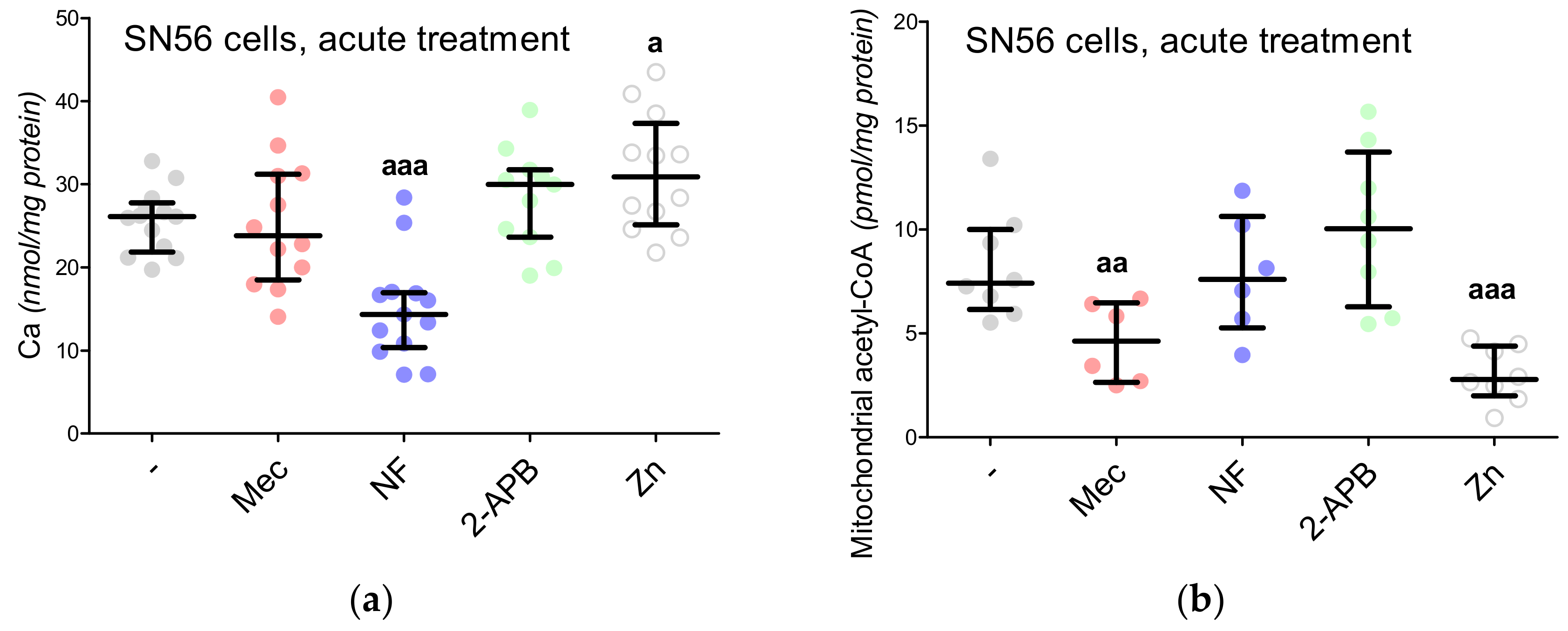
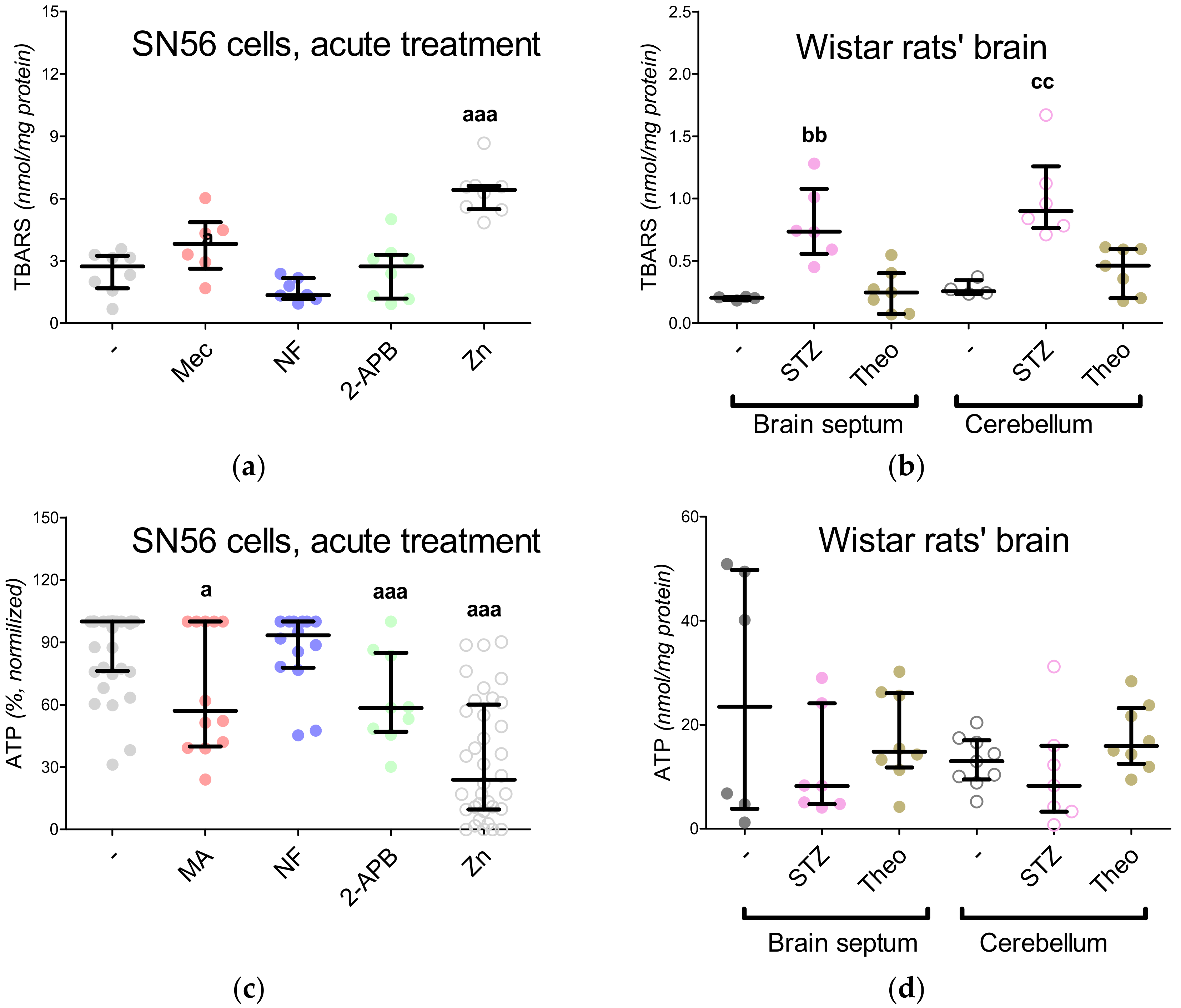
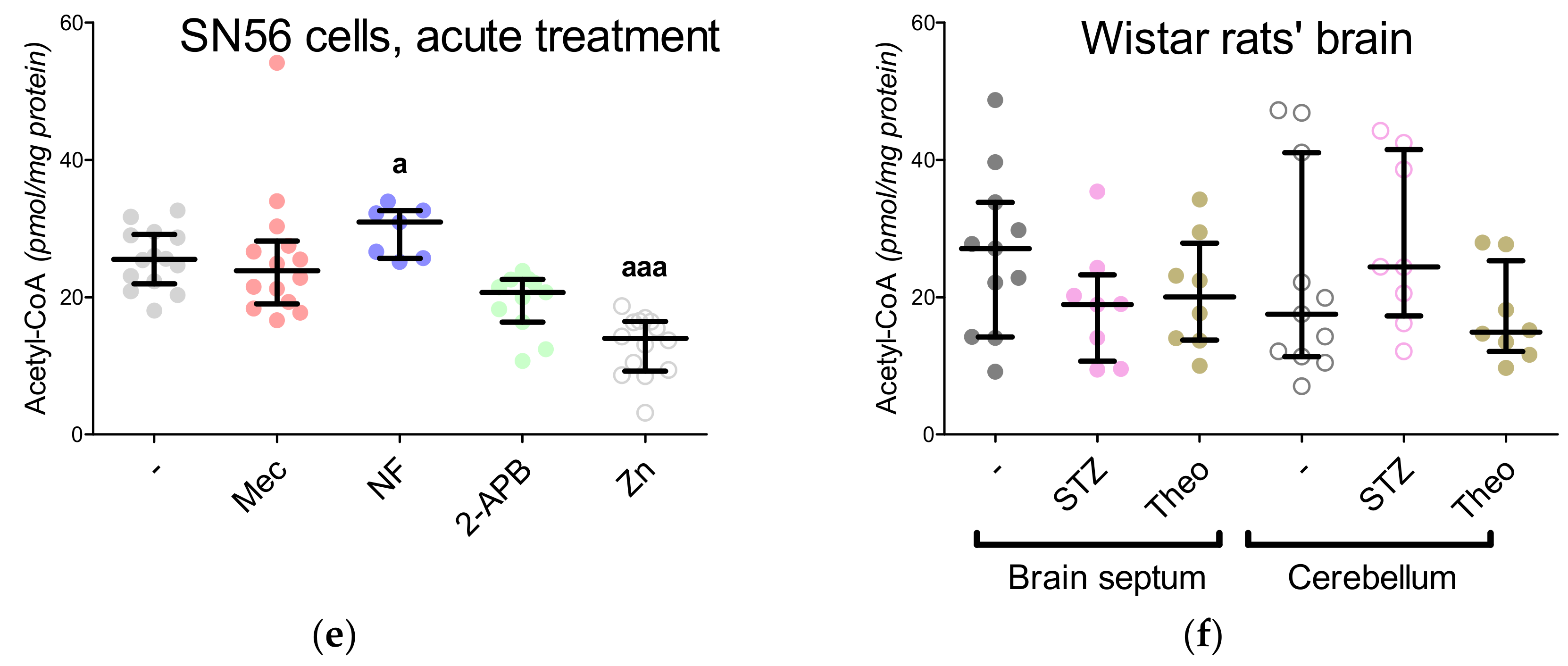
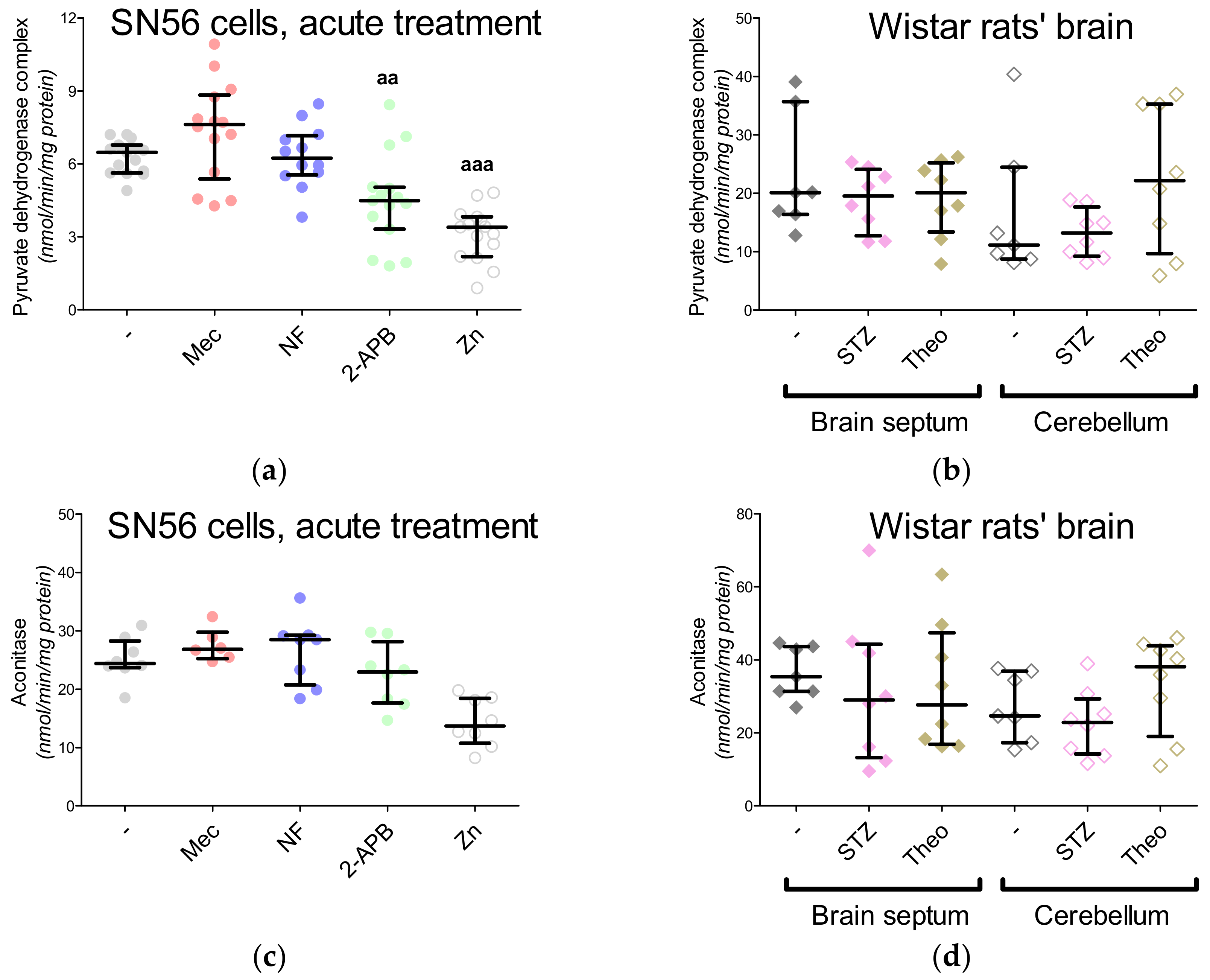
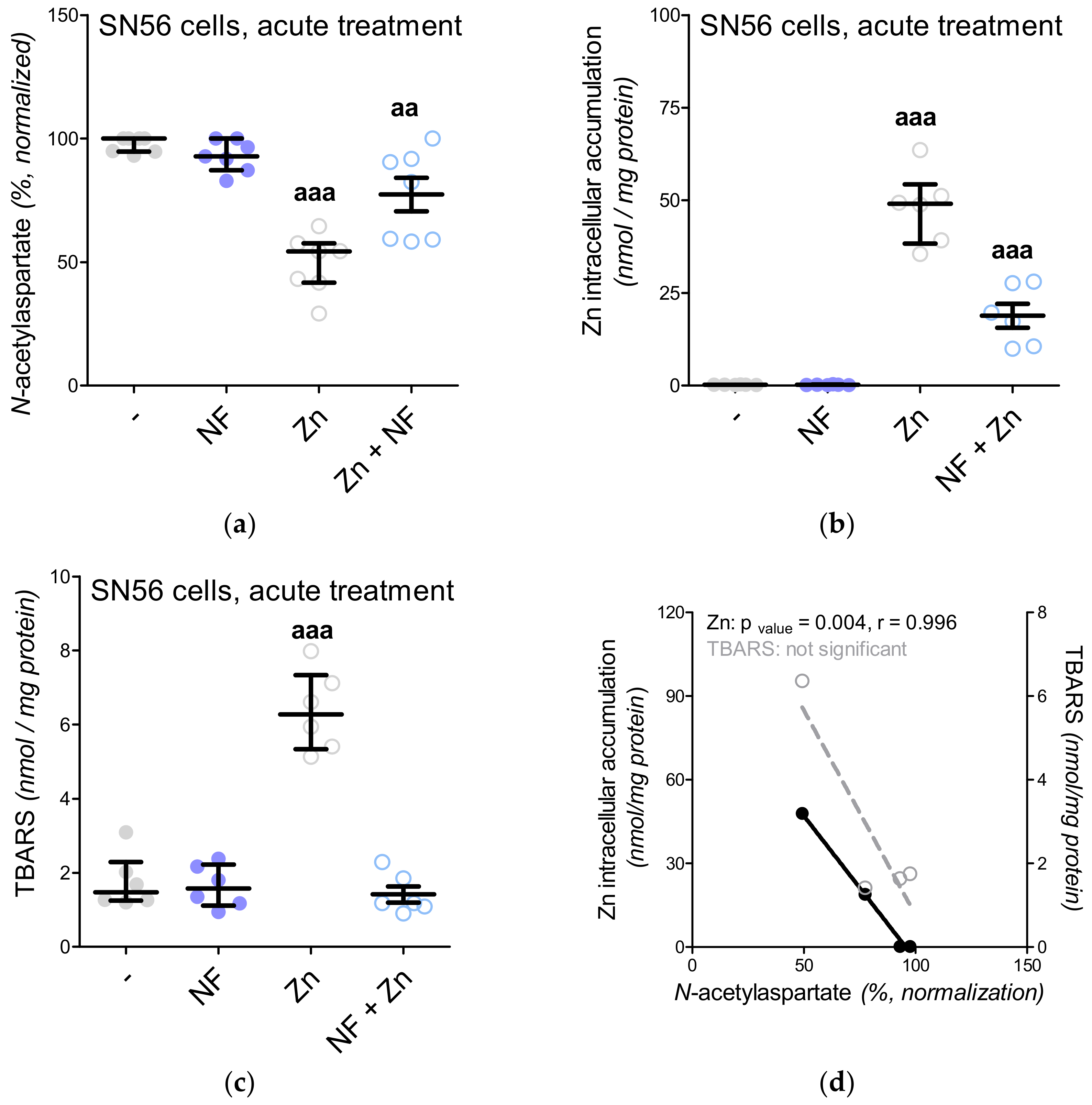
3.5. The Acute Effect of 0.15-mM Zn2+ on SN56 Cells
3.6. The Impact of Hyperglycemia and Theophylline Treatment on Wistar rats’ Bran Septum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miyamoto, Y.; Ishikawa, Y.; Iegaki, N.; Sumi, K.; Fu, K.; Sato, K.; Furukawa-Hibi, Y.; Muramatsu, S.; Nabeshima, T.; Uno, K.; et al. Overexpression of Shati/Nat8l, an N-acetyltransferase, in the nucleus accumbens attenuates the response to methamphetamine via activation of group II mGluRs in mice. Int. J. Neuropsychopharmacol. 2014, 17, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Nitta, A.; Mizoguchi, H.; Ito, Y.; Noda, Y.; Nagai, T.; Nabeshima, T. A novel molecule shati is involved in methamphetamine-induced hyperlocomotion, sensitization, and conditioned place preference. J. Neurosci. 2007, 27, 7604–7615. [Google Scholar] [CrossRef]
- Sumi, K.; Uno, K.; Noike, H.; Tomohiro, T.; Hatanaka, Y.; Furukawa-Hibi, Y.; Nabeshima, T.; Miyamoto, Y.; Nitta, A. Behavioral impairment in SHATI/NAT8L knockout mice via. Dysfunction of myelination development. Sci. Rep. 2017, 7, 16872. [Google Scholar] [CrossRef] [PubMed]
- Zyśk, M.; Bielarczyk, H.; Gul-Hinc, S.; Dyś, A.; Gapys, B.; Ronowska, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A. Phenotype-Dependent Interactions between N-acetyl-L-Aspartate and Acetyl CoA in Septal SN56 Cholinergic Cells Exposed to an Excess of Zinc. J. Alzheimers Dis. 2017, 56, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Nitta, A.; Noike, H.; Sumi, K.; Miyanishi, H.; Tanaka, T.; Takaoka, K.; Nagakura, M.; Iegaki, N.; Kaji, J.I.; Miyamoto, Y.; et al. Shati/nat8l and N-acetylaspartate (NAA) have important roles in regulating nicotinic acetylcholine receptors in neuronal and psychiatric diseases in animal models and humans. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Akaike, A., Shimoham, S., Misu, Y., Eds.; Springer: Singapore, 2018; Chapter 6; pp. 89–111. ISBN 978-981-10-8488-1. [Google Scholar]
- Szutowicz, A.; Bielarczyk, H.; Jankowska-Kulawy, A.; Pawełczyk, T.; Ronowska, A. Acetyl-CoA the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. Neurochem. Res. 2013, 38, 1523–1542. [Google Scholar] [CrossRef]
- Pohanka, M. Alpha7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int. J. Mol. Sci. 2012, 13, 2219–2238. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Iegaki, N.; Fu, K.; Ishikawa, Y.; Sumi, K.; Azuma, S.; Uno, K.; Muramatsu, S.I.; Nitta, A. Striatal n-acetylaspartate synthetase Shati/Nat8l regulates depression-like behaviors via mGluR3-mediated serotonergic suppression in mice. Int. J. Neuropsychopharm. 2017, 20, 1027–1035. [Google Scholar] [CrossRef]
- Wang, H.; Tan, L.; Wang, H.F.; Liu, Y.; Yin, R.H.; Wang, W.Y.; Chang, X.L.; Jiang, T.; Yu, J.T. Magnetic Resonance Spectroscopy in Alzheimer’s Disease: Systematic Review and Meta Analysis. J. Alzheimers Dis. 2015, 46, 1049–1070. [Google Scholar] [CrossRef]
- Ronowska, A.; Gul-Hinc, S.; Bielarczyk, H.; Pawełczyk, T.; Szutowicz, A. Effects of zinc on SN56 cholinergic neuroblastoma cells. J. Neurochem. 2007, 103, 972–983. [Google Scholar] [CrossRef]
- Ronowska, A.; Dyś, A.; Jankowska-Kulawy, A.; Klimaszewska-Łata, J.; Bielarczyk, H.; Romianowski, P.; Pawełczyk, T.; Szutowicz, A. Short-term effects of zinc on acetylcholine Metabolism and viability of SN56 cholinergic neuroblastoma cells. Neurochem. Int. 2010, 56, 143–151. [Google Scholar] [CrossRef]
- Zyśk, M.; Gapys, B.; Ronowska, A.; Gul-Hinc, S.; Erlandsson, A.; Iwanicki, A.; Sakowicz Burkiewicz, M.; Szutowicz, A.; Bielarczyk, H. Protective effects of voltage-gated calcium channel antagonists against zinc toxicity in SN56 neuroblastoma cholinergic cells. PLoS ONE 2018, 13, e0209363. [Google Scholar] [CrossRef]
- Hammond, D.N.; Lee, H.J.; Tonsgard, J.H.; Wainer, B.H. Development and characterization of clonal cell lines derived from septal cholinergic neurons. Brain Res. 1990, 512, 190–200. [Google Scholar] [CrossRef]
- Ray, R.S.; Rai, S.; Katyal, A. Cholinergic receptor blockade by scopolamine and mecamylamine exacerbates global cerebral ischemia induced memory dysfunction in C57BL/6J mice. Nitric Oxide 2014, 43, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Di Mise, A.; Ranieri, M.; Centrone, M.; Venneri, M.; Tamma, G.; Valenti, D.; Valenti, G. Activation of the calcium-sensing receptor corrects the impaired mitochondrial energy status observed in renal polycystin-1 knockdown cells modeling autosomal dominant polycystic kidney disease. Front. Mol. Biosci. 2018, 5, 77. [Google Scholar] [CrossRef]
- Swanson, L.W. Brain maps 4.0—Structure of the rat brain: An open access atlas with global nervous system nomenclature ontology and flatmaps. J. Comp. Neurol. 2018, 526, 93. [Google Scholar] [CrossRef] [PubMed]
- Kreft, E.; Kowalski, R.; Jankowski, M.; Szczepańska-Konkel, M. Renal vasculature reactivity to agonist of P2X7 receptor is increased in streptozotocin-induced diabetes. Pharmacol. Rep. 2016, 68, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Chaudhary, M.J.; Tiwari, P.C.; Babu, S.; Pant, K.K. Protective role of theophylline and their interaction with nitric oxide (NO) in adjuvant-induced rheumatoid arthritis in rats. Int. Immunopharmacol. 2015, 29, 854–862. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H. Elimination of CoASH interference from acetyl-CoA cycling assay by maleic anhydride. Anal. Biochem. 1987, 164, 292–296. [Google Scholar] [CrossRef]
- De Villafranca, G.W.; Haines, V.E. Paramyosin from arthropod cross-striated muscle. Comp. Biochem. Physiol. B 1974, 47, 9–26. [Google Scholar] [CrossRef]
- Itoh, H.; Srere, P.A. An assay and screening procedure for serum glutamic oxaloacetic transaminase. Clin. Chem. 1971, 17, 86–88. [Google Scholar] [CrossRef]
- Wiame, E.; Tyteca, D.; Pierrot, N.; Collard, F.; Amyere, M.; Noel, G.; Desmedt, J.; Nassogne, M.C.; Vikkula, M.; Octave, J.N.; et al. Molecular identification of aspartate N-acetyltransferase and its mutation in hypoacetylaspartia. Biochem. J. 2010, 425, 127–136. [Google Scholar] [CrossRef]
- Fonnum, F. Radiochemical micro assays for the determination of choline acetyltransferase and acetylcholinesterase activities. Biochem. J. 1969, 115, 465–472. [Google Scholar] [CrossRef]
- Wright, J.A.; Maeba, P.; Sanwal, B.D. Allosteric regulation of the activity of citrate synthetase of Escherichia coli by alpha-ketoglutarate. Biochem. Biophys. Res. Commun. 1967, 29, 34–38. [Google Scholar] [CrossRef]
- Botman, D.; Tigchelaar, W.; Van Noorden, C.J. Determination of glutamate dehydrogenase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry). J. Histochem. Cytochem. 2014, 62, 802–812. [Google Scholar] [CrossRef]
- Slein, M.W.; Cori, G.T.; Cori, C.F. A comparative study of hexokinase from yeast and animal tissues. J. Biol. Chem. 1950, 186, 763–780. [Google Scholar] [PubMed]
- Plaut, G.W.; Aogaichi, T. Purification and properties of diphosphopyridine nuleotide-linked isocitrate dehydrogenase of mammalian liver. J. Biol. Chem. 1968, 243, 5572–5583. [Google Scholar]
- Koh, J.Y.; Choi, D.W. Effect of anticonvulsant drugs on glutamate neurotoxicity in cortical cell culture. Neurology 1987, 37, 319–322. [Google Scholar] [CrossRef]
- Szutowicz, A.; Stepien, M.; Piec, G. Determination of pyruvate dehydrogenase and acetyl-CoA synthetase activities using citrate synthase. Anal. Biochem. 1981, 115, 81–87. [Google Scholar] [CrossRef]
- Bergmeyer, H.U.; Rozalskis, G. The Km of malate dehydrogenase from pig heart with oxaloacetate as substrate. Z. Klin. Chem. Klin. Biochem. 1975, 13, 509. [Google Scholar] [PubMed]
- Szutowicz, A.; Tomaszewicz, M.; Jankowska, A.; Kisielevski, Y. Acetylcholine synthesis in nerve terminals of diabetic rats. Neuroreport 1994, 5, 2421–2424. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R.; Corkey, B.E. Assays of intermediates of the citric acid cycle and related compounds by fluorometric enzyme methods. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1969; Volume 13, pp. 434–513. [Google Scholar] [CrossRef]
- Di Pierro, D.; Tavazzi, B.; Perno, C.F.; Bartolini, M.; Balestra, E.; Caliò, R.; Giardina, B.; Lazzarino, G. An ion-pairing high-performance liquid chromatographic method for the direct simultaneous determination of nucleotides, deoxynucleotides, nicotinic coenzymes, oxypurines, nucleosides, and bases in perchloric acid cell extracts. Anal. Biochem. 1995, 231, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Panse, M.; Block, H.U.; Förster, W.; Mest, H.J. An improved malondialdehyde assay for estimation of thromboxane synthase activity in washed human blood platelets. Prostaglandins 1985, 30, 1031–1040. [Google Scholar] [CrossRef]
- Sakowicz-Burkiewicz, M.; Kuczkowski, J.; Przybyla, T.; Grden, M.; Starzynska, A.; Pawelczyk, T. Gene expression profile of collagen types, osteopontin in the tympanic membrane of patients with tympanosclerosis. Adv. Clin. Exp. Med. 2017, 26, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Yau, W.M.; Dorsett, J.A.; Youther, M.L. Stimulation of acetylcholine release from myenteric neurons of guinea pig small intestine by forskolin and cyclic AMP. J. Pharmacol. Exp. Ther. 1987, 243, 506–510. [Google Scholar]
- Kamat, P.K. Streptozotocin induced Alzheimer’s disease like changes and the underlying neural degeneration and regeneration mechanism. Neural Regen. Res. 2015, 10, 1050–1052. [Google Scholar] [CrossRef]
- Bathina, S.; Srinivas, N.; Das, U.N. Streptozotocin produces oxidative stress, inflammation and decreases BDNF concentrations to induce apoptosis of RIN5F cells and type 2 diabetes mellitus in Wistar rats. Biochem. Biophys. Res. Commun. 2017, 486, 406–413. [Google Scholar] [CrossRef]
- Pessentheiner, A.R.; Pelzmann, H.J.; Walenta, E.; Schweiger, M.; Groschner, L.N.; Graier, W.F.; Kolb, D.; Uno, K.; Miyazaki, T.; Nitta, A.; et al. NAT8L (N-acetyltransferase 8-like) accelerates lipid turnover and increases energy expenditure in brown adipocytes. J. Biol. Chem. 2013, 288, 36040–36051. [Google Scholar] [CrossRef]
- Arun, P.; Moffett, J.R.; Namboodiri, A.M. Riluzole decreases synthesis of N-acetylaspartate and N-acetylaspartylglutamate in SH-SY5Y human neuroblastoma cells. Brain Res. 2010, 1334, 25–30. [Google Scholar] [CrossRef]
- Lu, Z.H.; Chakraborty, G.; Ledeen, R.W.; Yahya, D.; Wu, G. N-Acetylaspartate synthase is bimodally expressed in microsomes and mitochondria of brain. Brain Res. Mol. Brain Res. 2004, 122, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Jalil, M.A.; Begum, L.; Contreras, L.; Pardo, B.; Iijima, M.; Li, M.X.; Ramos, M.; Marmol, P.; Horiuchi, M.; Shimotsu, K.; et al. Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J. Biol. Chem. 2005, 280, 31333–31339. [Google Scholar] [CrossRef] [PubMed]
- Bielarczyk, H.; Gul, S.; Ronowska, A.; Bizon-Zygmanska, D.; Pawelczyk, T.; Szutowicz, A. RS alpha-lipoic acid protects cholinergic cells against sodium nitroprusside and amyloid-beta neurotoxicity through restoration of acetyl-CoA level. J. Neurochem. 2006, 98, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.M.; Kim, B.C.; Jeong, G.W. Effects of donepezil on brain morphometric and metabolic changes in patients with Alzheimer’s disease: A DARTEL-based VBM and1H-MRS. Magn. Reson. Imaging 2016, 34, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Blamire, A.M.; Watson, R.; He, J.; Hayes, L.; O’Brien, J.T. Whole-brain patterns of 1H magnetic resonance spectroscopy imaging in Alzheimer’s disease and dementia with Lewy bodies. Transl. Psychiatry 2016, 6, e877. [Google Scholar] [CrossRef]
- Waragai, M.; Moriya, M.; Nojo, T. Decreased N-Acetyl Aspartate/Myo-Inositol Ratio in the Posterior Cingulate Cortex Shown by Magnetic Resonance Spectroscopy May Be One of the Risk Markers of Preclinical Alzheimer’s Disease: A 7-Year Follow-Up Study. J. Alzheimers Dis. 2017, 60, 1411–1427. [Google Scholar] [CrossRef] [PubMed]
- Bagga, P.; Patel, A.B. Regional cerebral metabolism in mouse under chronic manganese exposure: Implications for Manganism. Neurochem. Int. 2012, 60, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Tarnacka, B.; Szeszkowski, W.; Gołȩbiowski, M.; Członkowska, A. Brain proton magnetic spectroscopy in long-term treatment of Wilson’s disease patients. Metab. Brain Dis. 2010, 25, 325–329. [Google Scholar] [CrossRef]
- Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef]
- Sarasamma, S.; Audira, G.; Juniardi, S.; Sampurna, B.P.; Liang, S.T.; Hao, E.; Lai, Y.H.; Hsiao, C.D. Zinc chloride exposure inhibits brain acetylcholine levels, produces neurotoxic signatures, and diminishes memory and motor activities in adult zebrafish. Int. J. Mol. Sci. 2018, 19, 3195. [Google Scholar] [CrossRef]
- Satrústegui, J.; Contreras, L.; Ramos, M.; Marmol, P.; Del Arco, A.; Saheki, T.; Pardo, B. Role of aralar, the mitochondrial transporter of aspartate-glutamate, in brain N-acetylaspartate formation and Ca2+signaling in neuronal mitochondria. J. Neurosci. Res. 2007, 85, 3359–3366. [Google Scholar] [CrossRef]
- Profilo, E.; Peña-Altamira, L.E.; Corricelli, M.; Castegna, A.; Danese, A.; Agrimi, G.; Petralla, S.; Giannuzzi, G.; Porcelli, V.; Sbano, L.; et al. Down-regulation of the mitochondrial aspartate glutamate carrier isoform 1 AGC1 inhibits proliferation and N-acetylaspartate synthesis in Neuro2A cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Zaroff, S.; Leone, P.; Markov, V.; Francis, J.S. Transcriptional regulation of N-acetylaspartate metabolism in the 5xFAD model of Alzheimer’s disease: Evidence for neuron-glia communication during energetic crisis. Mol. Cell. Neurosci. 2015, 65, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, P.; Singla, N.; Dhawan, D.K. Protective role of zinc during aluminum-induced hepatotoxicity. Environ. Toxicol. 2014, 29, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Kristal, B.S.; Effron, M.S.; Shestopalov, A.I.; Ullucci, P.A.; Sheu, K.F.; Blass, J.P.; Cooper, A.J. Zn2+ inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2000, 275, 13441–13447. [Google Scholar] [CrossRef] [PubMed]
- Arun, P.; Madhavarao, C.N.; Moffett, J.R.; Namboodiri, M.A. Regulation of N-acetylaspartate and N-acetylaspartylglutamate biosynthesis by protein kinase activators. J. Neurochem. 2006, 98, 2034–2042. [Google Scholar] [CrossRef]
- Walicke, P.A.; Patterson, P.H. On the role of Ca2+ in the transmitter choice made by cultured sympathetic neurons. J. Neurosci. 1981, 1, 343–350. [Google Scholar] [CrossRef]
- Paidi, R.K.; Nthenge-Ngumbau, D.N.; Singh, R.; Kankanala, T.; Mehta, H.; Mohanakumar, K.P. Mitochondrial deficits accompany cognitive decline following single bilateral intracerebroventricular streptozotocin. Curr. Alzheimer Res. 2015, 12, 785–795. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Sham Control | STZ | Theophylline |
|---|---|---|---|
| Body weight (g) | 290 (275–298) | 208 (194–222) *** | 310 (296–319) ** |
| Blood glucose (mg/dL) | 131 (120–133) | 518 (500–535) *** | 127 (108–141) |
| Urine acetoacetate (µmol/24 h) | 0.3 (0.2–0.4) | 1.8 (1.0–3.1) *** | 0.2 (0.1–0.6) |
| Brain hexokinase (µmol/min/mg protein) | 27.6 (27.5–38.4) | 62.1 (35.3–92.7) ** | 28.9 (23.3–96.4) |
| Brain β-hydroxybutyrate (nmol/mg protein) | 3.8 (1.6–13.8) | 17.1 (16.1–20.7) *** | 5.6 (1.5–10.4) |
| Gene Transcript | Primers | TaqMan Probe | Transcript of Reference Gene |
|---|---|---|---|
| Nat8l NM_001191681.1 | (F) tggctgacattgaacagtactaca (R) cacaacattgccgtccag | Universal ProbeLibrary Probe #83 (Roche, Cat #04689062001) | Universal ProbeLibrary Rat Actb Gene Assay (Roche, Cat #05046203001) |
| Parameters | Whole Cells | Cytoplasmic Fraction | Mitochondrial Fraction | Mitochondria % of Total Value |
|---|---|---|---|---|
| Enzymatic Markers | ||||
| Citrate synthase nmol/min/mg protein | 80.0 (66.3–91.7) | 4.7 (1.5–10.2) | 77.1 (70.0–81.9) | 96 |
| Glutamate dehydrogenase nmol/min/mg protein | 68.0 (60.1–76.5) | 6.3 (4.5–8.7) | 62.1 (55.3–66.6) | 91 |
| Western Blot Assay * | ||||
| Glyceraldehyde 3-phosphate dehydrogenase peak height value, Au | 8.1 (6.6–10.0) | 7.3 (5.8–9.4) | 0.8 (0.8–0.8) | 9 |
| α-ketoglutarate dehydrogenase peak height value, Au | 8.5 (8.2–9.5) | 0.3 (0.2–0.3) | 8.3 (7.9–9.3) | 97 |
| Parameter | Added | Whole Cells | Cytoplasmic Fraction | Mitochondrial Fraction | Mitochondria % of Total Value |
|---|---|---|---|---|---|
| Metabolic Parameters | |||||
| Acetyl-CoA pmol/mg protein | Control | 27.0 (24.3–31.6) | 16.0 (12.2–18.5) | 12.3 (11.0–13.5) | 45 |
| 0.15-mM Zn2+ | 12.2 (10.8–17.8) aaa | 5.3 (4.7–10.1) aaa | 7.2 (6.1–8.1) aaa | 52 | |
| Oxaloacetate nmol/mg protein | Control | 12.4 (7.2–15.8) | 8.3 (3.1–12.6) | 4.5 (3.6–5.4) | 30 |
| 0.15-mM Zn2+ | 9.1 (5.2–10.1) a | 5.2 (2.1–7.3) a | 3.4 (2.5–4.0) a | 26 | |
| Aspartate nmol/mg protein | Control | 60.9 (46.7–73.1) | 37.1 (14.2–44.5) | 28.8 (21.5–31.6) | 47 |
| 0.15-mM Zn2+ | 43.1 (36.7–50.8) a | 32.9 (28.3–42.1) | 8.3 (6.5–11.2) aaa | 19 | |
| Malate nmol/mg protein | Control | 16.2 (11.5–20.1) | 8.9 (5.9–14.6) | 6.4 (5.0–7.1) | 40 |
| 0.15-mM Zn2+ | 14.0 (11.2–18.1) | 7.8 (4.2–11.4) | 6.5 (5.0–8.0) | 46 | |
| N-acetylaspartate nmol/mg protein | Control | 62.8 (52.5–73.7) | 6.7 (5.2–18.1) | 53.2 (47.7–58.4) | 85 |
| 0.15-mM Zn2+ | 30.2 (28.2–35.0) aaa | 2.2 (1.5–4.4) aa | 27.0 (26.3–30.7) aaa | 89 | |
| Enzymatic Parameters | |||||
| NAT8L pmol/min/ mg protein | Control | 82.4 (59.7–88.8) | 6.5 (5.2–13.3) | 69.6 (47.3–76.7) | 84 |
| 0.15-mM Zn2+ | 55.5 (46.1–73.0) a | 10.5 (7.2–16.9) | 38.6 (31.4–76.7) aa | 70 | |
| Malate dehydrogenase µmol/mig/mg protein | Control | 0.7 (0.6–0.8) | |||
| 0.15 mMZn2+ | 0.6 (0.6–0.7) | ||||
| Aspartate aminotransferase nmol/min/mg protein | Control | 56.0 (52.9–71.3) | |||
| 0.15-mM Zn2+ | 53.6 (50.2–59.0) | ||||
| Lactate dehydrogenase µmol/min/mg protein | 1.7 (1.2–2.1) | ||||
| 2.0 (1.6–2.3) | |||||
| Parameter | Added | Wistar rats’ Brain | |
|---|---|---|---|
| Brain Septum | Cerebellum | ||
| Enzymatic Parameters | |||
| Aspartate aminotransferase nmol/min/mg protein | Control STZ Theophylline | 1.0 (0.8–2.0) 0.9 (0.4–1.8) 1.4 (0.9–1.7) | 1.0 (0.9–1.4) 0.7 (0.4–1.3) 1.6 (0.8–2.2) |
| Lactate dehydrogenase µmol/min/mg protein | Control STZ Theophylline | 0.4 (0.3–0.4) 0.4 (0.3–0.5) 0.3 (0.2–0.5) | 0.4 (0.3–0.6) 0.4 (0.2–0.5) 0.3 (0.3–0.6) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zyśk, M.; Sakowicz-Burkiewicz, M.; Pikul, P.; Kowalski, R.; Michno, A.; Pawełczyk, T. The Impact of Acetyl-CoA and Aspartate Shortages on the N-Acetylaspartate Level in Different Models of Cholinergic Neurons. Antioxidants 2020, 9, 522. https://doi.org/10.3390/antiox9060522
Zyśk M, Sakowicz-Burkiewicz M, Pikul P, Kowalski R, Michno A, Pawełczyk T. The Impact of Acetyl-CoA and Aspartate Shortages on the N-Acetylaspartate Level in Different Models of Cholinergic Neurons. Antioxidants. 2020; 9(6):522. https://doi.org/10.3390/antiox9060522
Chicago/Turabian StyleZyśk, Marlena, Monika Sakowicz-Burkiewicz, Piotr Pikul, Robert Kowalski, Anna Michno, and Tadeusz Pawełczyk. 2020. "The Impact of Acetyl-CoA and Aspartate Shortages on the N-Acetylaspartate Level in Different Models of Cholinergic Neurons" Antioxidants 9, no. 6: 522. https://doi.org/10.3390/antiox9060522
APA StyleZyśk, M., Sakowicz-Burkiewicz, M., Pikul, P., Kowalski, R., Michno, A., & Pawełczyk, T. (2020). The Impact of Acetyl-CoA and Aspartate Shortages on the N-Acetylaspartate Level in Different Models of Cholinergic Neurons. Antioxidants, 9(6), 522. https://doi.org/10.3390/antiox9060522