Astaxanthin Treatment Induces Maturation and Functional Change of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Tumor Model
2.2. MDSC Isolation
2.3. Viability Assay
2.4. Phenotype Analysis of MDSCs
2.5. Real Time-quantitative Polymerase Chain Reaction (RT-qPCR)
2.6. ROS Detection
2.7. In Vivo CTL Assay
2.8. Adoptive Transfer of MDSCs
2.9. Statistical Analysis
3. Results
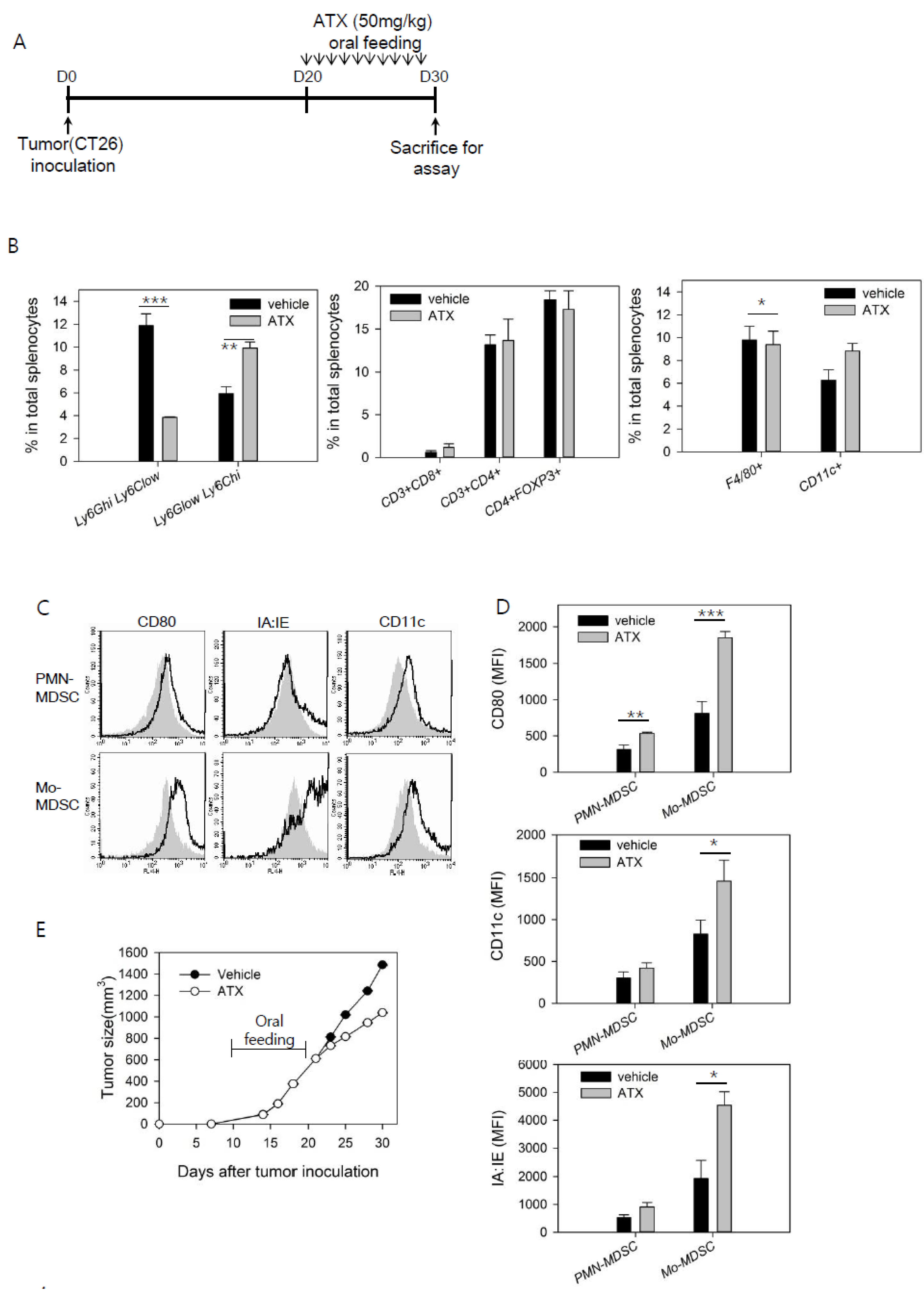
3.1. ATX Injection Induces Immunogenic Conversion of Mo-MDSCs with a Decreasing Percentage of PMN-MDSCs in Tumor-Bearing Mice
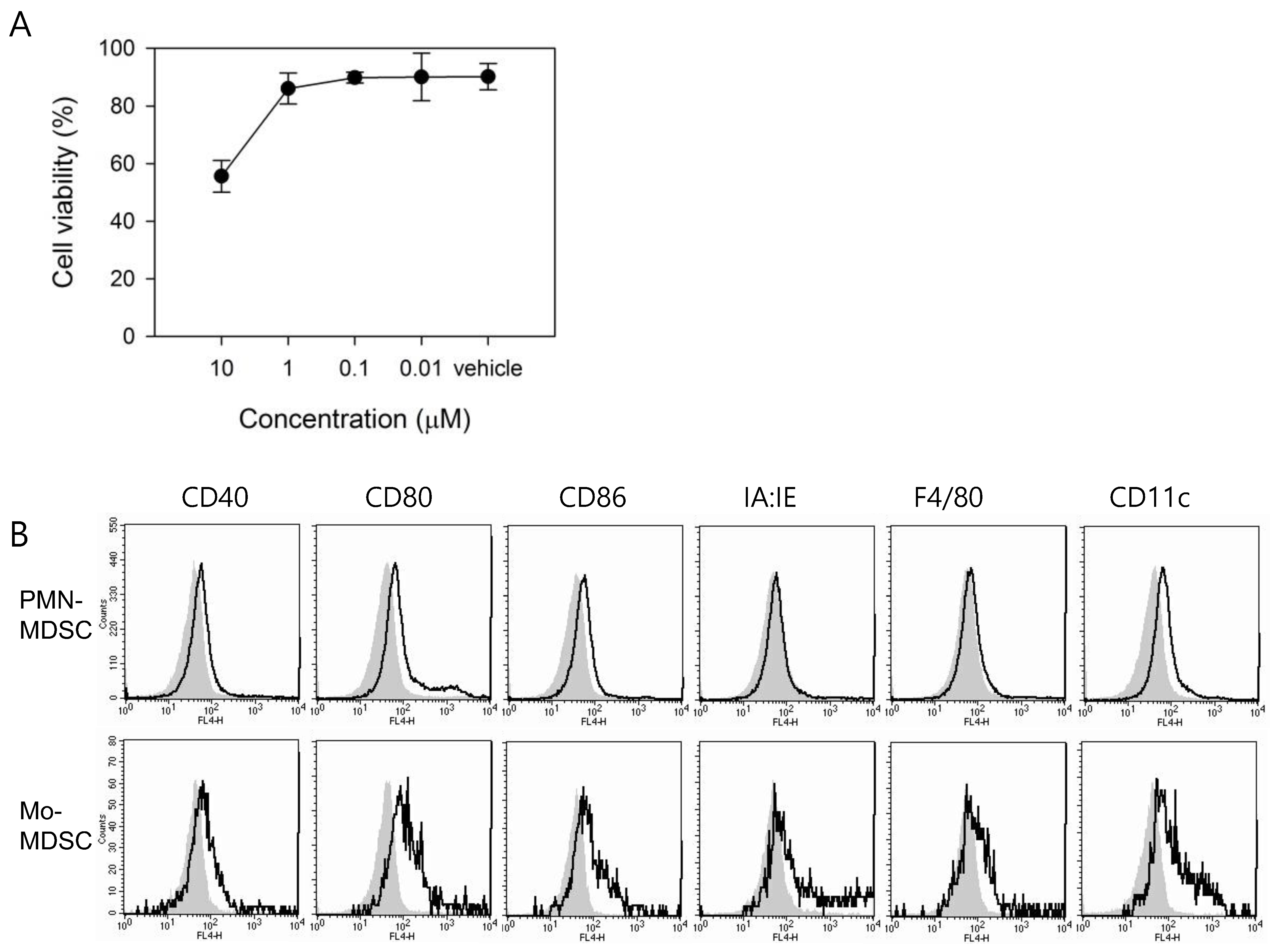
3.2. In Vitro Treatment with ATX Induces MDSC Differentiation
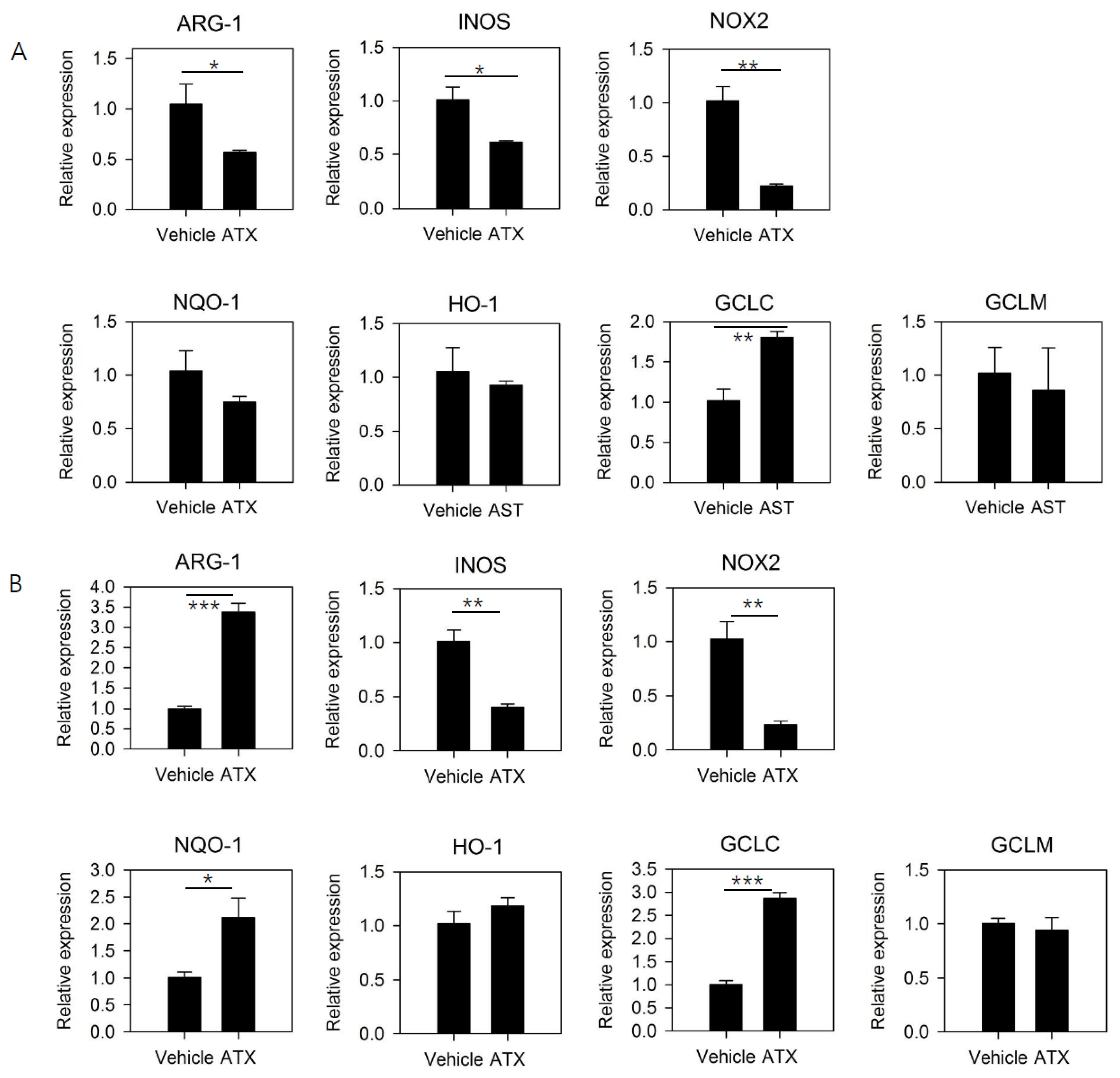
3.3. ATX Treatment Reduces the Expression of Functional Mediators of MDSCs and Induces the Expression of Genes Involved in GSH Synthesis
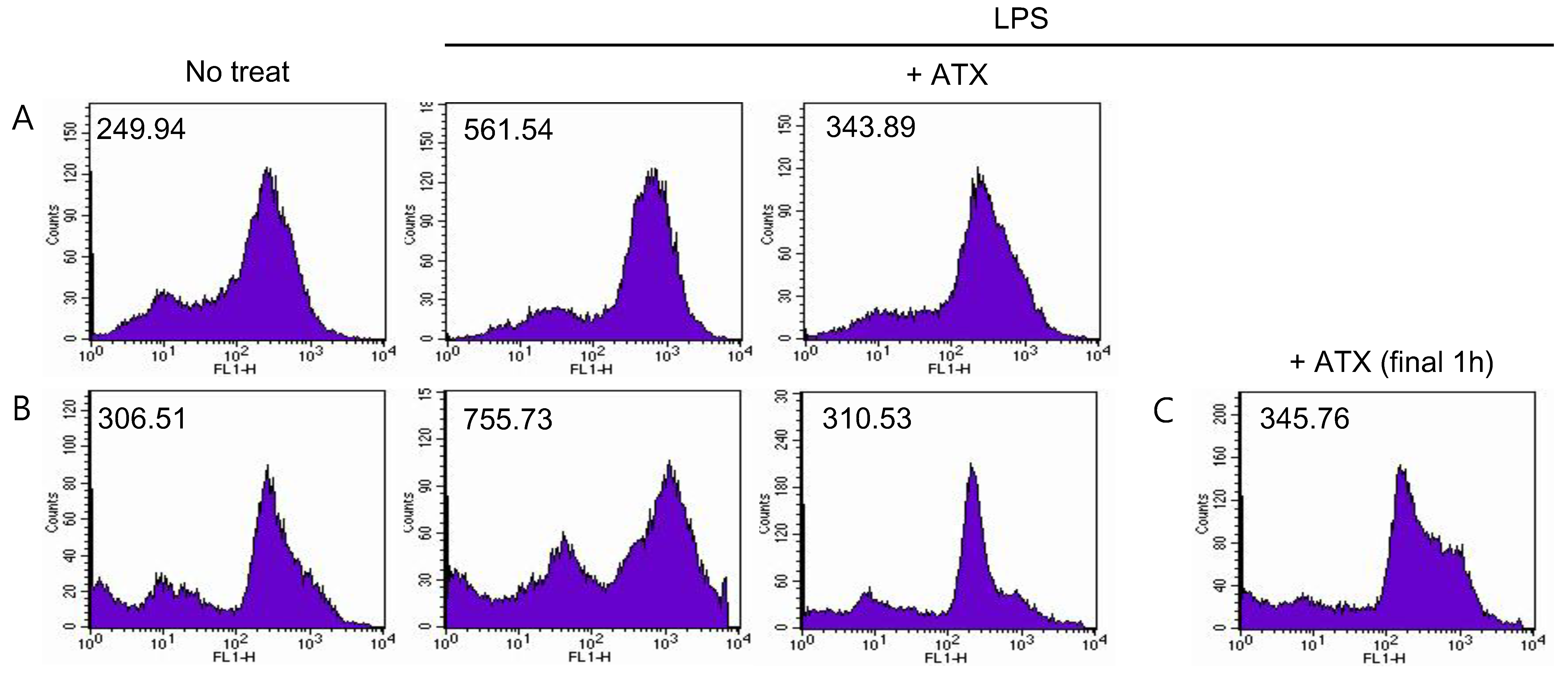
3.4. ATX Treatment Reduces ROS Level in MDSCs
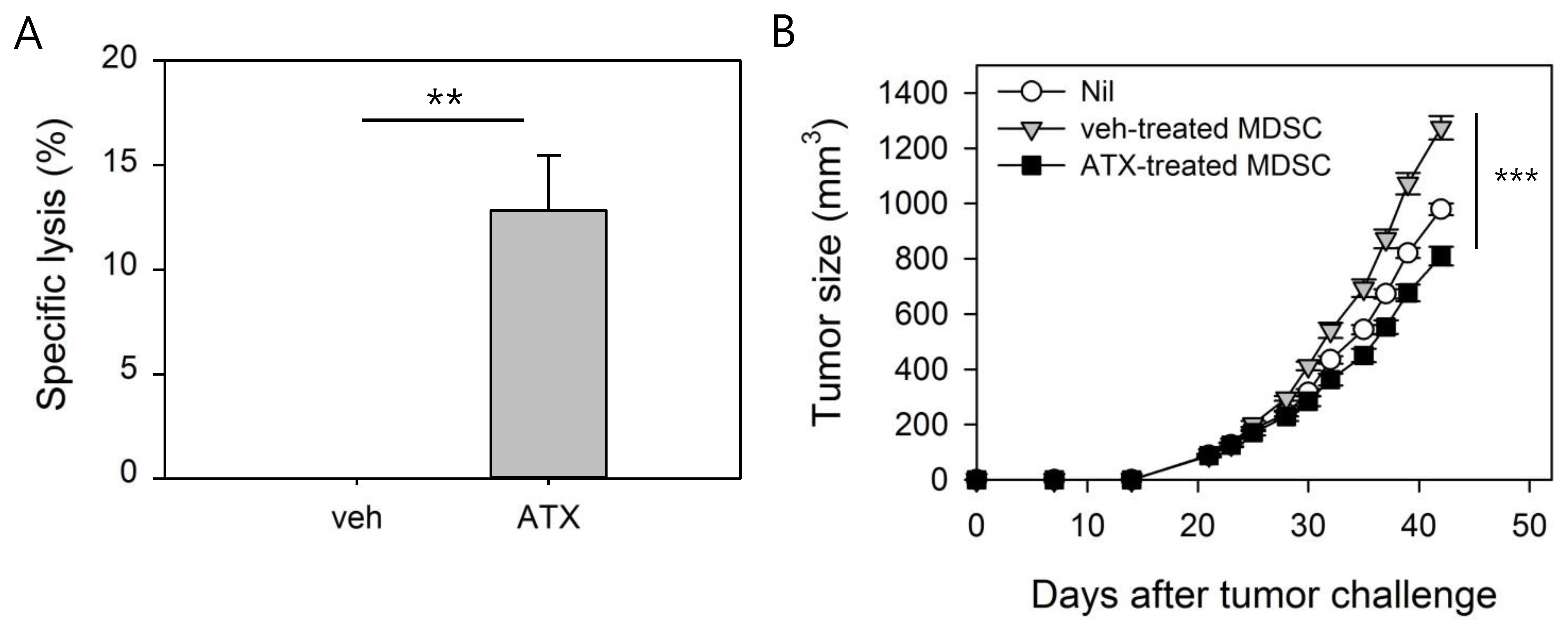
3.5. ATX-Treated MDSCs Act As Immunogenic APCs With Antitumor Activity
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Ghirelli, C.; Hagemann, T. Targeting immunosuppression for cancer therapy. J. Clin. Investig. 2013, 123, 2355–2357. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Coukos, G. Deciphering and reversing tumor immune suppression. Immunity 2013, 39, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, J.; Ren, J.P.; Wu, X.Y.; Morrison, Z.D.; Elgazzar, M.A.; Ning, S.; Moorman, J.P.; Yao, Z.Q. Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory T cells in HIV-1+ individuals. AIDS 2016, 30, 1521–1531. [Google Scholar] [CrossRef]
- Monu, N.R.; Frey, A. Myeloid-derived suppressor cells and anti-tumor T cells: A complex relationship. Immunol. Investig. 2012, 41, 595–613. [Google Scholar] [CrossRef]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Youn, J.-I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of Myeloid-Derived Suppressor Cells in Tumor Bearing Mice1. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Kim, Y.-J.; Lee, J.-M.; Kim, E.K.; Park, Y.-J.; Choe, S.-K.; Ko, H.-J.; Kang, C.-Y. Functional Changes in Myeloid-Derived Suppressor Cells (MDSCs) during Tumor Growth: FKBP51 Contributes to the Regulation of the Immunosuppressive Function of MDSCs. J. Immunol. 2012, 188, 4226–4234. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Hoechst, B.; Duffy, A.; Gamrekelashvili, J.; Fioravanti, S.; Manns, M.P.; Greten, T.F.; Korangy, F. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology 2012, 136, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.-J.; Kim, Y.-J. Signal transducer and activator of transcription proteins: Regulators of myeloid-derived suppressor cell-mediated immunosuppression in cancer. Arch. Pharmacal Res. 2016, 39, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Le, H.K.; Graham, L.; Cha, E.; Morales, J.K.; Manjili, M.H.; Bear, H. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int. Immunopharmacol. 2009, 9, 900–909. [Google Scholar] [CrossRef]
- Huang, X.; Cui, S.; Shu, Y. Cisplatin selectively downregulated the frequency and immunoinhibitory function of myeloid-derived suppressor cells in a murine B16 melanoma model. Immunol. Res. 2015, 64, 160–170. [Google Scholar] [CrossRef]
- Kodumudi, K.N.; Woan, K.; Gilvary, D.L.; Sahakian, E.; Wei, S.; Djeu, J.Y. A novel chemoimmunomodulating property of docetaxel: Suppression of myeloid-derived suppressor cells in tumor bearers. Clin. Cancer Res. 2010, 16, 4583–4594. [Google Scholar] [CrossRef]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rebe, C.; Ghiringhelli, F. 5-Fluorouracil Selectively Kills Tumor-Associated Myeloid-Derived Suppressor Cells Resulting in Enhanced T Cell-Dependent Antitumor Immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef]
- Jayaraman, P.; Parikh, F.; Lopez-Rivera, E.; Hailemichael, Y.; Clark, A.; Ma, G.; Cannan, D.; Ramacher, M.; Kato, M.; Overwijk, W.W.; et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J. Immunol. 2012, 188, 5365–5376. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Zea, A.H.; DeSalvo, J.; Culotta, K.S.; Zabaleta, J.; Quiceno, D.G.; Ochoa, J.B.; Ochoa, A.C. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J. Immunol. 2003, 171, 1232–1239. [Google Scholar] [CrossRef]
- Veltman, J.D.; Lambers, M.E.; Van Nimwegen, M.; Hendriks, R.W.; Hoogsteden, H.C.; Aerts, J.; Hegmans, J.P. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 2010, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of All-TransRetinoic Acid Effect on Tumor-Associated Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef]
- Heim, C.E.; Vidlak, D.; Scherr, T.D.; Hartman, C.W.; Garvin, K.L.; Kielian, T. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J. Immunol. 2015, 194, 3861–3872. [Google Scholar] [CrossRef] [PubMed]
- Zoglmeier, C.; Bauer, H.; Wedekind, G.; Bittner, P.; Sandholzer, N.; Rapp, M.; Anz, D.; Endres, S.; Bourquin, C.; Nörenberg, D. CpG Blocks Immunosuppression by Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. Clin. Cancer Res. 2011, 17, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Zhao, Y.; Zhang, X.; Lin, X. Astaxanthin Inhibits Acetaldehyde-Induced Cytotoxicity in SH-SY5Y Cells by Modulating Akt/CREB and p38MAPK/ERK Signaling Pathways. Mar. Drugs 2016, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Liu, H.; Chen, X.; Shi, H.; Fan, Y.; Hou, D.; Zhang, X. Astaxanthin protects ARPE-19 cells from oxidative stress via upregulation of Nrf2-regulated phase II enzymes through activation of PI3K/Akt. Mol. Vis. 2013, 19, 1656–1666. [Google Scholar] [PubMed]
- Li, J.; Xia, Y.; Liu, T.; Wang, J.; Dai, W.; Wang, F.; Zheng, Y.; Chen, K.; Li, S.; Abudumijiti, H.; et al. Protective Effects of Astaxanthin on ConA-Induced Autoimmune Hepatitis by the JNK/p-JNK Pathway-Mediated Inhibition of Autophagy and Apoptosis. PLoS ONE 2015, 10, e0120440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H. Multiple Mechanisms of Anti-Cancer Effects Exerted by Astaxanthin. Mar. Drugs 2015, 13, 4310–4330. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Y.; Wei, Y.; Liu, G.; Liu, Y.; Gao, Q.; Zou, L.; Zeng, W.; Zhang, N. Activation of AKT pathway by Nrf2/PDGFA feedback loop contributes to HCC progression. Oncotarget 2016, 7, 65389–65402. [Google Scholar] [CrossRef]
- Callegari, A.; Liu, Y.; White, C.C.; Chait, A.; Gough, P.; Raines, E.W.; Cox, D.; Kavanagh, T.J.; Rosenfeld, M.E. Gain and loss of function for glutathione synthesis: Impact on advanced atherosclerosis in apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2011, 31, 2473–2482. [Google Scholar] [CrossRef]
- Nagata, Y.; Furugen, R.; Hiasa, A.; Ikeda, H.; Ohta, N.; Furukawa, K.; Nakamura, H.; Furukawa, K.; Kanematsu, T.; Shiku, H. Peptides derived from a wild-type murine proto-oncogene c-erbB-2/HER2/neu can induce CTL and tumor suppression in syngeneic hosts. J. Immunol. 1997, 159, 1336–1343. [Google Scholar]
- Kim, Y.-J.; Ko, H.-J.; Kim, Y.-S.; Kim, D.-H.; Kang, S.; Kim, J.-M.; Chung, Y.; Kang, C.-Y. α-Galactosylceramide-loaded, antigen-expressing B cells prime a wide spectrum of antitumor immunity. Int. J. Cancer 2008, 122, 2774–2783. [Google Scholar] [CrossRef]
- Mei, S.; Xin, J.; Liu, Y.; Zhang, Y.; Liang, X.; Su, X.; Yan, H.; Huang, Y.; Yang, R. MicroRNA-200c Promotes Suppressive Potential of Myeloid-Derived Suppressor Cells by Modulating PTEN and FOG2 Expression. PLoS ONE 2015, 10, e0135867. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, G.; Cheng, W.A.; Dong, Z.; Sun, H.; Lee, V.Y.; Cha, S.-C.; Smith, L.; Kwak, L.W.; Qin, H. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Hiramoto, K.; Satoh, H.; Suzuki, T.; Moriguchi, T.; Pi, J.; Shimosegawa, T.; Yamamoto, M. Myeloid Lineage-Specific Deletion of Antioxidant System Enhances Tumor Metastasis. Cancer Prev. Res. 2014, 7, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Fragoulis, A.; Klemm, P.; Baumeister, J.; Klock, W.; Verjans, E.; Böll, S.; Möllmann, J.; Lehrke, M.; Costa, I.; et al. Nrf2 Is a Central Regulator of Metabolic Reprogramming of Myeloid-Derived Suppressor Cells in Steady State and Sepsis. Front. Immunol. 2018, 9, 1552. [Google Scholar] [CrossRef]
- Weldy, C.S.; Luttrell, I.P.; White, C.C.; Morgan-Stevenson, V.; Bammler, T.K.; Beyer, R.P.; Afsharinejad, Z.; Kim, F.; Chitaley, K.; Kavanagh, T.J. Glutathione (GSH) and the GSH synthesis gene Gclm modulate vascular reactivity in mice. Free Radic. Biol. Med. 2012, 53, 1264–1278. [Google Scholar] [CrossRef]
- Nkabyo, Y.S.; Ziegler, T.R.; Gu, L.H.; Watson, W.H.; Jones, D.P. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am. J. Physiol. Liver Physiol. 2002, 283, G1352–G1359. [Google Scholar] [CrossRef]
- Ko, H.-J.; Lee, J.-M.; Kim, Y.-J.; Kim, Y.-S.; Lee, K.-A.; Kang, C.-Y. Immunosuppressive Myeloid-Derived Suppressor Cells Can Be Converted into Immunogenic APCs with the Help of Activated NKT Cells: An Alternative Cell-Based Antitumor Vaccine. J. Immunol. 2009, 182, 1818–1828. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J. Leukoc. Biol. 2003, 74, 186–196. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.M.; Kim, Y.-J. Astaxanthin Treatment Induces Maturation and Functional Change of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. Antioxidants 2020, 9, 350. https://doi.org/10.3390/antiox9040350
Jeong SM, Kim Y-J. Astaxanthin Treatment Induces Maturation and Functional Change of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. Antioxidants. 2020; 9(4):350. https://doi.org/10.3390/antiox9040350
Chicago/Turabian StyleJeong, Seong Mun, and Yeon-Jeong Kim. 2020. "Astaxanthin Treatment Induces Maturation and Functional Change of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice" Antioxidants 9, no. 4: 350. https://doi.org/10.3390/antiox9040350
APA StyleJeong, S. M., & Kim, Y.-J. (2020). Astaxanthin Treatment Induces Maturation and Functional Change of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. Antioxidants, 9(4), 350. https://doi.org/10.3390/antiox9040350