Have We Looked in the Wrong Direction for More Than 100 Years? Delayed Onset Muscle Soreness Is, in Fact, Neural Microdamage Rather Than Muscle Damage


{kind=link}
Abstract
1. Introduction
2. Background of our Hypothesis
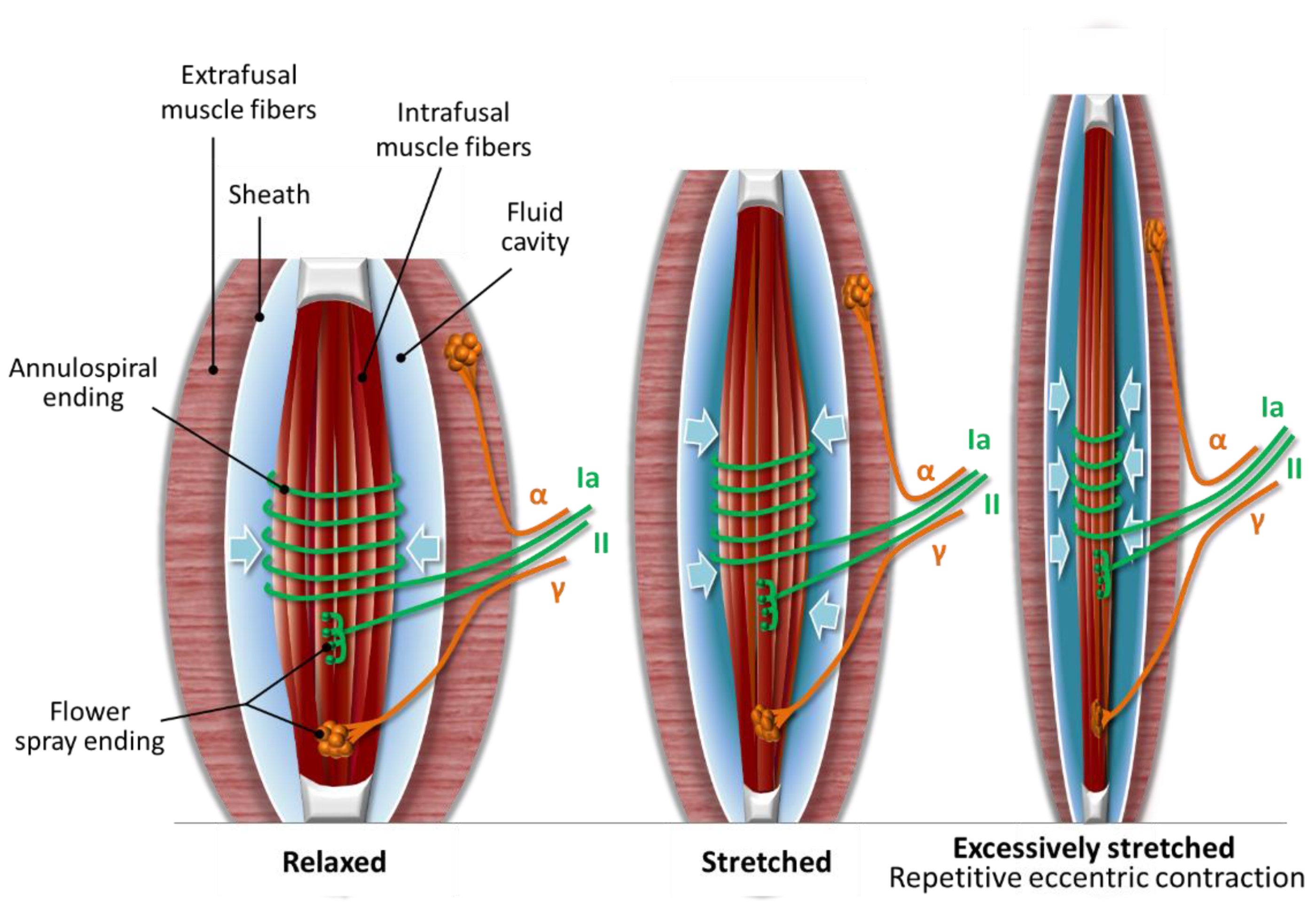
2.1. The Muscle Spindle
2.2. Compression Microinjury of the Nerve Endings in the Muscle Spindle
3. Hypothesis: DOMS is an Acute Compression Axonopathy with Possible Coinciding Tissue Microinjuries and Is Enhanced by Immune-Mediated Inflammation
3.1. Primary Damage Phase: Acute Compression Axonopathy and Possible Coinciding Tissue Microinjury during Eccentric Exercise
3.2. Secondary Phase: Microinjury-Induced Immune-Mediated Inflammation and Regeneration after Microdamaging Eccentric Exercise
3.2.1. Mechanical Hyperalgesia: Neuropathic Pain Enhanced by Inflammatory Pain
3.2.2. Immune-Mediated Inflammation
3.2.3. Decrease in Muscle Strength and Stiffness
4. Reactive Oxygen Species (ROS) and Nitric Oxide (NO)
5. DOMS and Ontogenesis
6. Conclusions
- DOMS could be an acute compression axonopathy of the nerve endings in the muscle spindle,
- The cause of DOMS could be the repetitive superposition of compression under cognitive demand and a resultant metabolic insult,
- DOMS could be initiated from the muscle spindle,
- The fluid cavity in the muscle spindle could play an important functional role in DOMS,
- Mitochondrial electron transport chain generated free radical involvement is suspected with a TAD-like lesion in the acute axonopathy of the sensory nerve endings in DOMS,
- Unaccustomed or strenuous eccentric exercise-induced SNS activity could be an essential underlying factor in DOMS initiation,
- DOMS could be initiated earlier than it is experienced, but at the beginning, it is suppressed by SNS activity,
- Delayed onset of soreness could be a result of the hypoalgesic state of the ‘closed gate’ caused by the enhanced firing of the microinjured Type Ia sensory fibers in addition to the initial SNS suppression,
- There could be a cross-talk on the PGE2 level between the pain pathways,
- Hyperexcited microinjured Type II sensory fibers in the muscle spindle could override, with the possible help of SNS, the conduction velocity reducing microinjured Type Ia sensory fibers’ inhibition with a delayed onset. The result will be an ‘open gate’ in the dorsal horn and the pathway to hyperalgesia in DOMS,
- Keeping a ‘closed gate’ with concentric exercise could have importance in non-pharmacological disease and neuropathic pain management by simultaneously alleviating pain and enjoying the positive anti-inflammatory characteristics of exercise; therefore, we call it a ‘closed gate exercise’,
- DOMS could cause a transient increase of the blood–spinal cord barrier and selective muscle spindle barrier permeability,
- DOMS could be a safety function in repetitive eccentric contractions as it resolves when the microinjury of the muscle spindle afferent sensory and motoneuron nerve endings are regenerated,
- Finally, we suspect that DOMS could play an important role in ontogenesis by triggering muscle growth and adapting the nervous system in the growth process.
Author Contributions
Funding
Conflicts of Interest
References
- Clarkson, P.M.; Nosaka, K.; Braun, B. Muscle function after exercise-induced muscle damage and rapid adaptation. Med. Sci. Sports Exerc. 1992, 24, 512–520. [Google Scholar] [CrossRef]
- Hough, T. ERGOGRAPHIC STUDIES IN MUSCULAR SORENESS. Am. J. Physiol. Leg. Content 1902, 7, 76–92. [Google Scholar] [CrossRef]
- Newham, D.J. The consequences of eccentric contractions and their relationship to delayed onset muscle pain. Eur. J. Appl. Physiol. Occup. Physiol. 1988, 57, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Taguchi, T. Delayed onset muscle soreness: Involvement of neurotrophic factors. J. Physiol. Sci. 2016, 66, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.; Hume, P.; Maxwell, L. Delayed onset muscle soreness: Treatment strategies and performance factors. Sports Med. 2003, 33, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Colon, A.; Guo, X.; Akanda, N.; Cai, Y.; Hickman, J.J. Functional analysis of human intrafusal fiber innervation by human gamma-motoneurons. Sci. Rep. 2017, 7, 17202. [Google Scholar] [CrossRef] [PubMed]
- Stifani, N. Motor neurons and the generation of spinal motor neuron diversity. Front. Cell Neurosci. 2014, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Kanning, K.C.; Kaplan, A.; Henderson, C.E. Motor neuron diversity in development and disease. Annu. Rev. Neurosci. 2010, 33, 409–440. [Google Scholar] [CrossRef]
- Liu, J.X.; Thornell, L.E.; Pedrosa-Domellof, F. Muscle spindles in the deep muscles of the human neck: A morphological and immunocytochemical study. J. Histochem. Cytochem. 2003, 51, 175–186. [Google Scholar] [CrossRef]
- Radovanovic, D.; Peikert, K.; Lindstrom, M.; Domellof, F.P. Sympathetic innervation of human muscle spindles. J. Anat. 2015, 226, 542–548. [Google Scholar] [CrossRef]
- Hamill, O.P. A new stretch for muscle spindle research. J. Physiol. 2010, 588, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Ovalle, W.K.; Nahirney, P.C. Netter’s Essential Histology; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Bewick, G.S.; Banks, R.W. Mechanotransduction in the muscle spindle. Pflug. Arch. 2015, 467, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Boyd, I.A. The response of fast and slow nuclear bag fibres and nuclear chain fibres in isolated cat muscle spindles to fusimotor stimulation, and the effect of intrafusal contraction on the sensory endings. Q. J. Exp. Physiol. Cogn Med. Sci. 1976, 61, 203–254. [Google Scholar] [CrossRef]
- Poppele, R.E.; Quick, D.C. Effect of intrafusal muscle mechanics on mammalian muscle spindle sensitivity. J. Neurosci. 1985, 5, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Hody, S.; Croisier, J.L.; Bury, T.; Rogister, B.; Leprince, P. Eccentric Muscle Contractions: Risks and Benefits. Front. Physiol. 2019, 10, 536. [Google Scholar] [CrossRef]
- Morgan, D.L.; Allen, D.G. Early events in stretch-induced muscle damage. J. Appl. Physiol. 1999, 87, 2007–2015. [Google Scholar] [CrossRef]
- Michael, S.; Graham, K.S.; Davis, G.M.O. Cardiac Autonomic Responses during Exercise and Post-exercise Recovery Using Heart Rate Variability and Systolic Time Intervals-A Review. Front. Physiol. 2017, 8, 301. [Google Scholar] [CrossRef]
- White, D.W.; Raven, P.B. Autonomic neural control of heart rate during dynamic exercise: Revisited. J. Physiol. 2014, 592, 2491–2500. [Google Scholar] [CrossRef]
- Thomas, G.D.; Segal, S.S. Neural control of muscle blood flow during exercise. J. Appl. Physiol. 2004, 97, 731–738. [Google Scholar] [CrossRef]
- Boulton, D.; Taylor, C.E.; Macefield, V.G.; Green, S. Effect of contraction intensity on sympathetic nerve activity to active human skeletal muscle. Front. Physiol. 2014, 5, 194. [Google Scholar] [CrossRef][Green Version]
- Illingworth, V. The Penguin Dictionary of Physics; Penguin Books: London, UK, 1991. [Google Scholar]
- Enoka, R.M.; Baudry, S.; Rudroff, T.; Farina, D.; Klass, M.; Duchateau, J. Unraveling the neurophysiology of muscle fatigue. J. Electromyogr. Kinesiol. 2011, 21, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.L.; Gandevia, S.C. A comparison of central aspects of fatigue in submaximal and maximal voluntary contractions. J. Appl. Physiol. 2008, 104, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Brownstone, R.M.; Lancelin, C. Escape from homeostasis: Spinal microcircuits and progression of amyotrophic lateral sclerosis. J. Neurophysiol. 2018, 119, 1782–1794. [Google Scholar] [CrossRef] [PubMed]
- Hospod, V.; Aimonetti, J.M.; Roll, J.P.; Ribot-Ciscar, E. Changes in human muscle spindle sensitivity during a proprioceptive attention task. J. Neurosci. 2007, 27, 5172–5178. [Google Scholar] [CrossRef]
- Macefield, G.; Hagbarth, K.E.; Gorman, R.; Gandevia, S.C.; Burke, D. Decline in spindle support to alpha-motoneurones during sustained voluntary contractions. J. Physiol. 1991, 440, 497–512. [Google Scholar] [CrossRef]
- Schwartzman, R.J.; Kerrigan, J. The movement disorder of reflex sympathetic dystrophy. Neurology 1990, 40, 57–61. [Google Scholar] [CrossRef]
- Hellstrom, F.; Roatta, S.; Thunberg, J.; Passatore, M.; Djupsjobacka, M. Responses of muscle spindles in feline dorsal neck muscles to electrical stimulation of the cervical sympathetic nerve. Exp. Brain Res. 2005, 165, 328–342. [Google Scholar] [CrossRef]
- Roatta, S.; Windhorst, U.; Ljubisavljevic, M.; Johansson, H.; Passatore, M. Sympathetic modulation of muscle spindle afferent sensitivity to stretch in rabbit jaw closing muscles. J. Physiol. 2002, 540, 237–248. [Google Scholar] [CrossRef]
- Schlereth, T.; Birklein, F. The sympathetic nervous system and pain. Neuromol. Med. 2008, 10, 141–147. [Google Scholar] [CrossRef]
- Rossi, A.; Mazzocchio, R.; Decchi, B. Effect of chemically activated fine muscle afferents on spinal recurrent inhibition in humans. Clin. Neurophysiol. 2003, 114, 279–287. [Google Scholar] [CrossRef]
- Melzack, R.; Wall, P.D. Pain mechanisms: A new theory. Science 1965, 150, 971–979. [Google Scholar] [CrossRef]
- Sufka, K.J.; Price, D.D. Gate control theory reconsidered. Brain Mind 2002, 3, 277–290. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef]
- Lund, J.P.; Sadeghi, S.; Athanassiadis, T.; Caram Salas, N.; Auclair, F.; Thivierge, B.; Arsenault, I.; Rompre, P.; Westberg, K.G.; Kolta, A. Assessment of the potential role of muscle spindle mechanoreceptor afferents in chronic muscle pain in the rat masseter muscle. PLoS ONE 2010, 5, e11131. [Google Scholar] [CrossRef] [PubMed]
- Baron, R. Neuropathic pain: A clinical perspective. Handb. Handb. Exp. Pharm. 2009, 3–30. [Google Scholar] [CrossRef]
- Sun, W.; Yang, F.; Wang, Y.; Fu, H.; Yang, Y.; Li, C.L.; Wang, X.L.; Lin, Q.; Chen, J. Contribution of large-sized primary sensory neuronal sensitization to mechanical allodynia by upregulation of hyperpolarization-activated cyclic nucleotide gated channels via cyclooxygenase 1 cascade. Neuropharmacology 2017, 113, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.F.; Wu, Q.; Henry, J.L. Changes in functional properties of A-type but not C-type sensory neurons in vivo in a rat model of peripheral neuropathy. J. Pain Res. 2012, 5, 175–192. [Google Scholar] [CrossRef][Green Version]
- Andersen, O.K.; Gracely, R.H.; Arendt-Nielsen, L. Facilitation of the human nociceptive reflex by stimulation of A beta-fibres in a secondary hyperalgesic area sustained by nociceptive input from the primary hyperalgesic area. Acta Physiol. Scand. 1995, 155, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Gracely, R.H.; Lynch, S.A.; Bennett, G.J. Painful neuropathy: Altered central processing maintained dynamically by peripheral input. Pain 1992, 51, 175–194. [Google Scholar] [CrossRef]
- Nosaka, K. Muscle Soreness and Damage and the Repetead-Bout Effect. In Skeletal Muscle Damage and Repair; Tiidus, P.M., Ed.; Human Kinetics: Champaign, IL, USA, 2008; pp. 59–76. [Google Scholar]
- Ferreira, S.H. Prostaglandins, aspirin-like drugs and analgesia. Nat. New Biol. 1972, 240, 200–203. [Google Scholar] [CrossRef]
- Turrini, P.; Gaetano, C.; Antonelli, A.; Capogrossi, M.C.; Aloe, L. Nerve growth factor induces angiogenic activity in a mouse model of hindlimb ischemia. Neurosci. Lett. 2002, 323, 109–112. [Google Scholar] [CrossRef]
- Amano, T.; Yamakuni, T.; Okabe, N.; Sakimura, K.; Takahashi, Y. Production of nerve growth factor in rat skeletal muscle. Neurosci. Lett. 1991, 132, 5–7. [Google Scholar] [CrossRef]
- Murase, S.; Terazawa, E.; Hirate, K.; Yamanaka, H.; Kanda, H.; Noguchi, K.; Ota, H.; Queme, F.; Taguchi, T.; Mizumura, K. Upregulated glial cell line-derived neurotrophic factor through cyclooxygenase-2 activation in the muscle is required for mechanical hyperalgesia after exercise in rats. J. Physiol. 2013, 591, 3035–3048. [Google Scholar] [CrossRef]
- Murase, S.; Terazawa, E.; Queme, F.; Ota, H.; Matsuda, T.; Hirate, K.; Kozaki, Y.; Katanosaka, K.; Taguchi, T.; Urai, H.; et al. Bradykinin and nerve growth factor play pivotal roles in muscular mechanical hyperalgesia after exercise (delayed-onset muscle soreness). J. Neurosci. 2010, 30, 3752–3761. [Google Scholar] [CrossRef]
- Lau, W.Y.; Blazevich, A.J.; Newton, M.J.; Wu, S.S.; Nosaka, K. Changes in electrical pain threshold of fascia and muscle after initial and secondary bouts of elbow flexor eccentric exercise. Eur. J. Appl. Physiol. 2015, 115, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Yasui, M.; Kubo, A.; Abe, M.; Kiyama, H.; Yamanaka, A.; Mizumura, K. Nociception originating from the crural fascia in rats. Pain 2013, 154, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Tesarz, J.; Hoheisel, U.; Wiedenhofer, B.; Mense, S. Sensory innervation of the thoracolumbar fascia in rats and humans. Neuroscience 2011, 194, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Francis, K.T.; Hoobler, T. Effects of aspirin on delayed muscle soreness. J. Sports Med. Phys. Fit. 1987, 27, 333–337. [Google Scholar]
- Evans, W.J.; Meredith, C.N.; Cannon, J.G.; Dinarello, C.A.; Frontera, W.R.; Hughes, V.A.; Jones, B.H.; Knuttgen, H.G. Metabolic changes following eccentric exercise in trained and untrained men. J. Appl. Physiol. 1986, 61, 1864–1868. [Google Scholar] [CrossRef]
- Smith, L.L. Acute inflammation: The underlying mechanism in delayed onset muscle soreness? Med. Sci. Sports Exerc. 1991, 23, 542–551. [Google Scholar] [CrossRef]
- Hasson, S.M.; Wible, C.L.; Reich, M.; Barnes, W.S.; Williams, J.H. Dexamethasone iontophoresis: Effect on delayed muscle soreness and muscle function. Can. J. Sport Sci. 1992, 17, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Friden, J.; Sfakianos, P.N.; Hargens, A.R. Muscle soreness and intramuscular fluid pressure: Comparison between eccentric and concentric load. J. Appl. Physiol. 1986, 61, 2175–2179. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, J.T.; Char, D.; McDermott, J.; Goya, C. Immediate Postexercise Massage Does Not Attenuate Delayed Onset Muscle Soreness. J. Strength Cond. Res. 1997, 11, 119–124. [Google Scholar]
- Gulick, D.; Kimura, I. Delayed Onset Muscle Soreness: What Is It and How Do We Treat It? J. Sport Rehabil. 1996, 5, 234–243. [Google Scholar] [CrossRef]
- Armstrong, R.B. Mechanisms of exercise-induced delayed onset muscular soreness: A brief review. Med. Sci. Sports Exerc. 1984, 16, 529–538. [Google Scholar] [CrossRef]
- Armstrong, R.B. Initial events in exercise-induced muscular injury. Med. Sci. Sports Exerc. 1990, 22, 429–435. [Google Scholar]
- Pizza, F.X.; Koh, T.J.; McGregor, S.J.; Brooks, S.V. Muscle inflammatory cells after passive stretches, isometric contractions, and lengthening contractions. J. Appl. Physiol. 2002, 92, 1873–1878. [Google Scholar] [CrossRef]
- Best, T.M.; Fiebig, R.; Corr, D.T.; Brickson, S.; Ji, L. Free radical activity, antioxidant enzyme, and glutathione changes with muscle stretch injury in rabbits. J. Appl. Physiol. (1985) 1999, 87, 74–82. [Google Scholar] [CrossRef]
- Malm, C.; Sjodin, T.L.; Sjoberg, B.; Lenkei, R.; Renstrom, P.; Lundberg, I.E.; Ekblom, B. Leukocytes, cytokines, growth factors and hormones in human skeletal muscle and blood after uphill or downhill running. J. Physiol. 2004, 556, 983–1000. [Google Scholar] [CrossRef]
- Hayashi, K.; Abe, M.; Yamanaka, A.; Mizumura, K.; Taguchi, T. Degenerative histological alteration is not required for the induction of muscular mechanical hyperalgesia after lengthening contraction in rats. J. Physiol. Sci. 2015, 65, S277. [Google Scholar]
- Kuphal, K.E.; Fibuch, E.E.; Taylor, B.K. Extended swimming exercise reduces inflammatory and peripheral neuropathic pain in rodents. J. Pain 2007, 8, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Echeverry, S.; Shi, X.Q.; Rivest, S.; Zhang, J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J. Neurosci. 2011, 31, 10819–10828. [Google Scholar] [CrossRef] [PubMed]
- Beggs, S.; Liu, X.J.; Kwan, C.; Salter, M.W. Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Mol. Pain 2010, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Costigan, M.; Moss, A.; Latremoliere, A.; Johnston, C.; Verma-Gandhu, M.; Herbert, T.A.; Barrett, L.; Brenner, G.J.; Vardeh, D.; Woolf, C.J.; et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J. Neurosci. 2009, 29, 14415–14422. [Google Scholar] [CrossRef] [PubMed]
- Radu, B.M.; Bramanti, P.; Osculati, F.; Flonta, M.L.; Radu, M.; Bertini, G.; Fabene, P.F. Neurovascular unit in chronic pain. Mediat. Inflamm. 2013, 2013, 648268. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Nahrendorf, M.; Swirski, F.K. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum. J. Am. Coll Cardiol. 2016, 67, 1091–1103. [Google Scholar] [CrossRef]
- Chapman, D.; Newton, M.; Sacco, P.; Nosaka, K. Greater muscle damage induced by fast versus slow velocity eccentric exercise. Int. J. Sports Med. 2006, 27, 591–598. [Google Scholar] [CrossRef]
- Mori, T.; Agata, N.; Itoh, Y.; Miyazu-Inoue, M.; Sokabe, M.; Taguchi, T.; Kawakami, K. Stretch speed-dependent myofiber damage and functional deficits in rat skeletal muscle induced by lengthening contraction. Physiol. Rep. 2014, 2. [Google Scholar] [CrossRef]
- Friden, J.; Lieber, R.L. Structural and mechanical basis of exercise-induced muscle injury. Med. Sci. Sports Exerc. 1992, 24, 521–530. [Google Scholar] [CrossRef]
- Crameri, R.M.; Aagaard, P.; Qvortrup, K.; Langberg, H.; Olesen, J.; Kjaer, M. Myofibre damage in human skeletal muscle: Effects of electrical stimulation versus voluntary contraction. J. Physiol. 2007, 583, 365–380. [Google Scholar] [CrossRef]
- Connolly, D.A.; Sayers, S.P.; McHugh, M.P. Treatment and prevention of delayed onset muscle soreness. J. Strength Cond. Res. 2003, 17, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Close, G.L.; Ashton, T.; Cable, T.; Doran, D.; MacLaren, D.P. Eccentric exercise, isokinetic muscle torque and delayed onset muscle soreness: The role of reactive oxygen species. Eur. J. Appl. Physiol. 2004, 91, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Camus, G.; Deby-Dupont, G.; Duchateau, J.; Deby, C.; Pincemail, J.; Lamy, M. Are similar inflammatory factors involved in strenuous exercise and sepsis? Intensive Care Med. 1994, 20, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Borras, C.; Gomez-Cabrera, M.C.; Orr, W.C. Part of the series: From dietary antioxidants to regulators in cellular signalling and gene expression. Role of reactive oxygen species and (phyto)oestrogens in the modulation of adaptive response to stress. Free Radic Res. 2006, 40, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Naito, H.; Taylor, A.W.; Goto, S. Nitric oxide: Is it the cause of muscle soreness? Nitric Oxide 2012, 26, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Pucsok, J.; Mecseki, S.; Csont, T.; Ferdinandy, P. Muscle soreness-induced reduction in force generation is accompanied by increased nitric oxide content and DNA damage in human skeletal muscle. Free Radic Biol. Med. 1999, 26, 1059–1063. [Google Scholar] [CrossRef]
- Reid, M.B.; Kobzik, L.; Bredt, D.S.; Stamler, J.S. Nitric oxide modulates excitation-contraction coupling in the diaphragm. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 1998, 119, 211–218. [Google Scholar] [CrossRef]
- Pouvreau, S.; Jacquemond, V. Nitric oxide synthase inhibition affects sarcoplasmic reticulum Ca2+ release in skeletal muscle fibres from mouse. J. Physiol. 2005, 567, 815–828. [Google Scholar] [CrossRef]
- Lee, J.S.; Zhang, Y.; Ro, J.Y. Involvement of neuronal, inducible and endothelial nitric oxide synthases in capsaicin-induced muscle hypersensitivity. Eur. J. Pain 2009, 13, 924–928. [Google Scholar] [CrossRef]
- Leiter, J.R.; Anderson, J.E. Satellite cells are increasingly refractory to activation by nitric oxide and stretch in aged mouse-muscle cultures. Int. J. Biochem. Cell Biol. 2010, 42, 132–136. [Google Scholar] [CrossRef]
- Cashman, C.R.; Hoke, A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 2015, 596, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Metodiewa, D.; Koska, C. Reactive oxygen species and reactive nitrogen species: Relevance to cyto(neuro)toxic events and neurologic disorders. An overview. Neurotox Res. 2000, 1, 197–233. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.M.; Russell, J.W.; Low, P.; Feldman, E.L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 2004, 25, 612–628. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.; Doyle, T.; Bryant, L.; Esposito, E.; Cuzzocrea, S.; Ryerse, J.; Bennett, G.J.; Salvemini, D. Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy-induced neuropathic pain resulting from peroxynitrite-mediated post-translational nitration of mitochondrial superoxide dismutase. Pain 2013, 154, 2432–2440. [Google Scholar] [CrossRef]
- Jiang, Y.; Guo, C.; Vasko, M.R.; Kelley, M.R. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008, 68, 6425–6434. [Google Scholar] [CrossRef]
- Scuteri, A.; Galimberti, A.; Maggioni, D.; Ravasi, M.; Pasini, S.; Nicolini, G.; Bossi, M.; Miloso, M.; Cavaletti, G.; Tredici, G. Role of MAPKs in platinum-induced neuronal apoptosis. Neurotoxicology 2009, 30, 312–319. [Google Scholar] [CrossRef]
- Bennett, G.J.; Liu, G.K.; Xiao, W.H.; Jin, H.W.; Siau, C. Terminal arbor degeneration—A novel lesion produced by the antineoplastic agent paclitaxel. Eur. J. Neurosci. 2011, 33, 1667–1676. [Google Scholar] [CrossRef]
- Holland, N.R.; Crawford, T.O.; Hauer, P.; Cornblath, D.R.; Griffin, J.W.; McArthur, J.C. Small-fiber sensory neuropathies: Clinical course and neuropathology of idiopathic cases. Ann. Neurol. 1998, 44, 47–59. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell Biol. 2006, 18, 453–459. [Google Scholar] [CrossRef]
- Mironov, S.L. ADP regulates movements of mitochondria in neurons. Biophys. J. 2007, 92, 2944–2952. [Google Scholar] [CrossRef]
- Elefteriou, F.; Campbell, P.; Ma, Y. Control of bone remodeling by the peripheral sympathetic nervous system. Calcif. Tissue Int. 2014, 94, 140–151. [Google Scholar] [CrossRef]
- Gray, C.; Hukkanen, M.; Konttinen, Y.T.; Terenghi, G.; Arnett, T.R.; Jones, S.J.; Burnstock, G.; Polak, J.M. Rapid neural growth: Calcitonin gene-related peptide and substance P-containing nerves attain exceptional growth rates in regenerating deer antler. Neuroscience 1992, 50, 953–963. [Google Scholar] [CrossRef]
- Singh, I.J.; Herskovits, M.S.; Chiego, D.J., Jr.; Klein, R.M. Modulation of osteoblastic activity by sensory and autonomic innervation of bone. Prog. Clin. Biol Res. 1982, 101, 535–551. [Google Scholar]
- Suttie, J.M.; Fennessy, P.F. Regrowth of amputated velvet antlers with and without innervation. J. Exp. Zool. 1985, 234, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, M.; Konttinen, Y.T.; Santavirta, S.; Paavolainen, P.; Gu, X.H.; Terenghi, G.; Polak, J.M. Rapid proliferation of calcitonin gene-related peptide-immunoreactive nerves during healing of rat tibial fracture suggests neural involvement in bone growth and remodelling. Neuroscience 1993, 54, 969–979. [Google Scholar] [CrossRef]
- Li, J.; Ahmad, T.; Spetea, M.; Ahmed, M.; Kreicbergs, A. Bone reinnervation after fracture: A study in the rat. J. Bone Min. Res. 2001, 16, 1505–1510. [Google Scholar] [CrossRef]
- Strange-Vognsen, H.H.; Laursen, H. Nerves in human epiphyseal uncalcified cartilage. J. Pediatr. Orthop. B 1997, 6, 56–58. [Google Scholar] [CrossRef]
- Mahns, D.A.; Ivanusic, J.J.; Sahai, V.; Rowe, M.J. An intact peripheral nerve preparation for monitoring the activity of single, periosteal afferent nerve fibres. J. Neurosci. Methods 2006, 156, 140–144. [Google Scholar] [CrossRef]
- Mach, D.B.; Rogers, S.D.; Sabino, M.C.; Luger, N.M.; Schwei, M.J.; Pomonis, J.D.; Keyser, C.P.; Clohisy, D.R.; Adams, D.J.; O’Leary, P.; et al. Origins of skeletal pain: Sensory and sympathetic innervation of the mouse femur. Neuroscience 2002, 113, 155–166. [Google Scholar] [CrossRef]
- Fan, W.; Bouwense, S.A.; Crawford, R.; Xiao, Y. Structural and cellular features in metaphyseal and diaphyseal periosteum of osteoporotic rats. J. Mol. Histol. 2010, 41, 51–60. [Google Scholar] [CrossRef]
- Berger, J.M.; Singh, P.; Khrimian, L.; Morgan, D.A.; Chowdhury, S.; Arteaga-Solis, E.; Horvath, T.L.; Domingos, A.I.; Marsland, A.L.; Yadav, V.K.; et al. Mediation of the Acute Stress Response by the Skeleton. Cell Metab. 2019, 30, 890–902.e898. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonkodi, B.; Berkes, I.; Koltai, E. Have We Looked in the Wrong Direction for More Than 100 Years? Delayed Onset Muscle Soreness Is, in Fact, Neural Microdamage Rather Than Muscle Damage. Antioxidants 2020, 9, 212. https://doi.org/10.3390/antiox9030212
Sonkodi B, Berkes I, Koltai E. Have We Looked in the Wrong Direction for More Than 100 Years? Delayed Onset Muscle Soreness Is, in Fact, Neural Microdamage Rather Than Muscle Damage. Antioxidants. 2020; 9(3):212. https://doi.org/10.3390/antiox9030212
Chicago/Turabian StyleSonkodi, Balazs, Istvan Berkes, and Erika Koltai. 2020. "Have We Looked in the Wrong Direction for More Than 100 Years? Delayed Onset Muscle Soreness Is, in Fact, Neural Microdamage Rather Than Muscle Damage" Antioxidants 9, no. 3: 212. https://doi.org/10.3390/antiox9030212
APA StyleSonkodi, B., Berkes, I., & Koltai, E. (2020). Have We Looked in the Wrong Direction for More Than 100 Years? Delayed Onset Muscle Soreness Is, in Fact, Neural Microdamage Rather Than Muscle Damage. Antioxidants, 9(3), 212. https://doi.org/10.3390/antiox9030212