Oxidative Stress and Preeclampsia-Associated Prothrombotic State


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Placental Development
3. Pathogenesis of PE
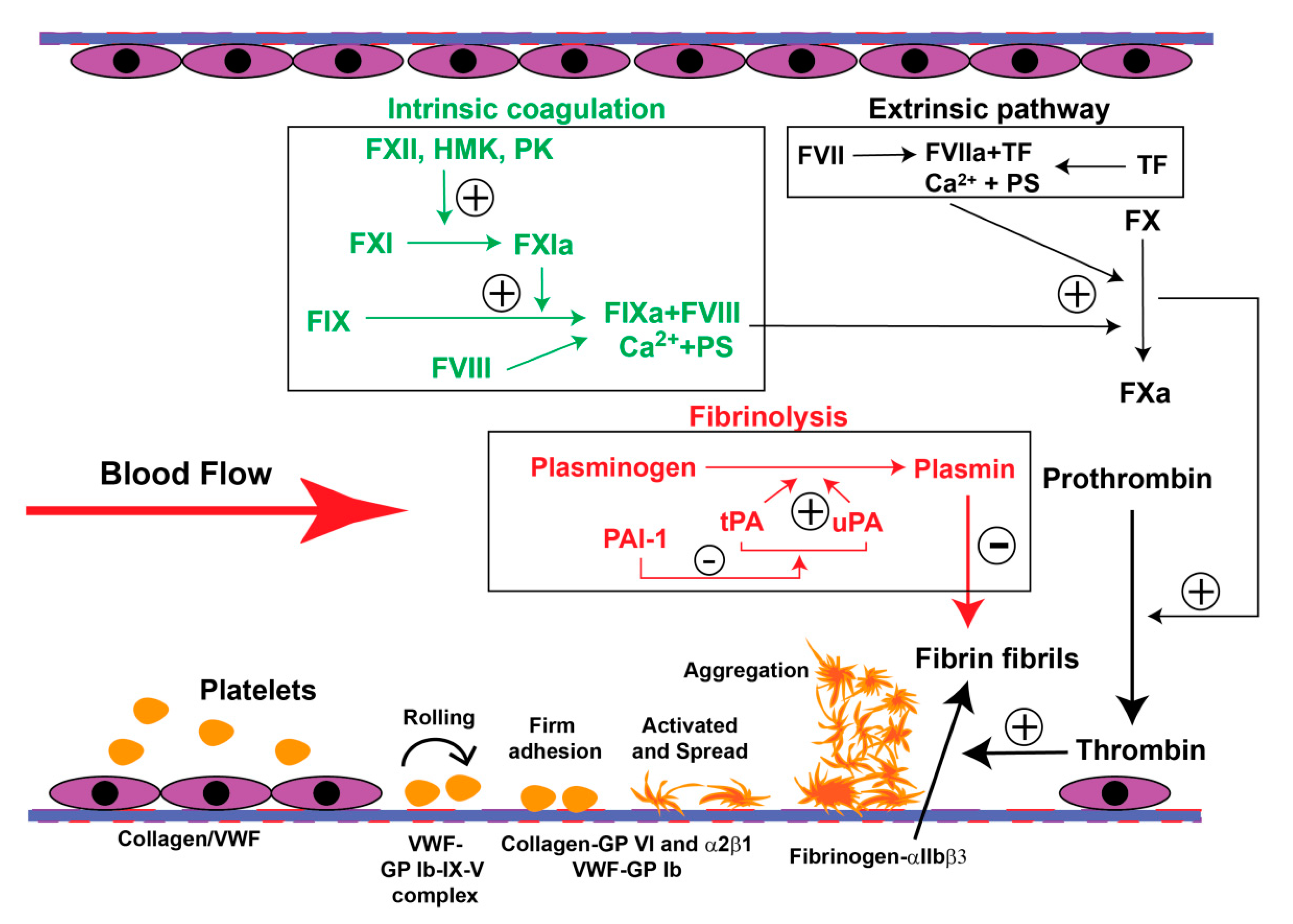
4. The PE-Associated Hypercoagulable State
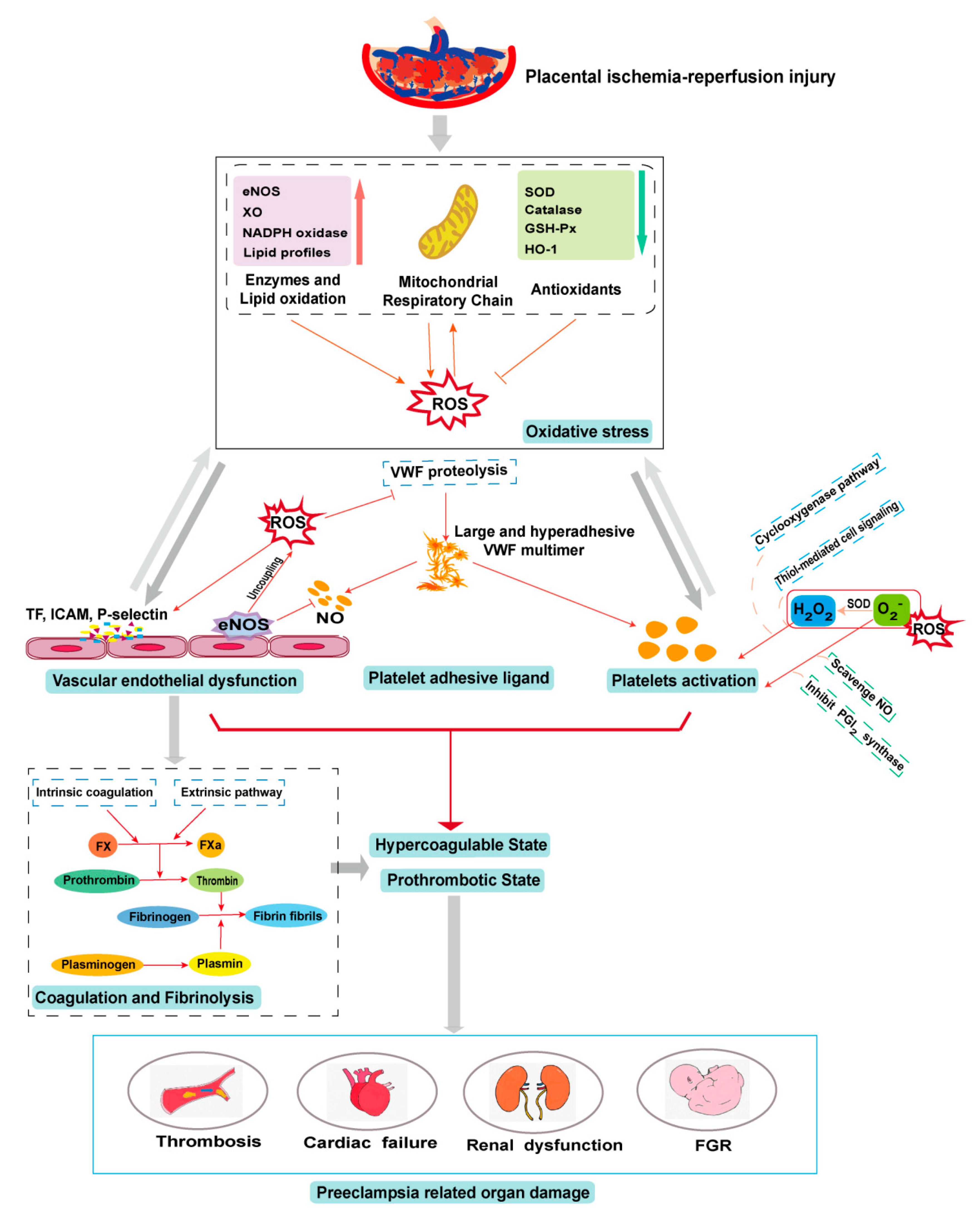
5. PE and Oxidative Stress
6. Oxidative Stress in the PE-Induced Prothrombotic State
6.1. Oxidative Stress on the Endothelium
6.2. Oxidative Stress on Platelets
6.3. Oxidative Stress on Platelet Adhesive Ligands
6.4. Oxidative Stress on Coagulation and Fibrinolysis
7. Antioxidant Therapies in PE
7.1. Endogenous Antioxidants
7.2. Therapeutic Antioxidants
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics. Gestational hypertension and preeclampsia: Acog practice bulletin, number 222. Obstet. Gynecol. 2020, 135, e237–e260. [Google Scholar] [CrossRef]
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Ananth, C.V.; Keyes, K.M.; Wapner, R.J. Pre-eclampsia rates in the united states, 1980–2010: Age-period-cohort analysis. BMJ (Clin. Res. Ed.) 2013, 347, f6564. [Google Scholar] [CrossRef]
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-eclampsia part 1: Current understanding of its pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466–480. [Google Scholar] [CrossRef]
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ (Clin. Res. Ed.) 2007, 335, 974. [Google Scholar] [CrossRef]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.L. Oxidative stress in placental pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E.; Watson, A.L. Maternal arterial connections to the placental intervillous space during the first trimester of human pregnancy: The boyd collection revisited. Am. J. Obstet. Gynecol. 1999, 181, 718–724. [Google Scholar] [CrossRef]
- Burton, G.J.; Hempstock, J.; Jauniaux, E. Oxygen, early embryonic metabolism and free radical-mediated embryopathies. Reprod. Biomed. Online 2003, 6, 84–96. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Ji, L.; Brkić, J.; Liu, M.; Fu, G.; Peng, C.; Wang, Y.L. Placental trophoblast cell differentiation: Physiological regulation and pathological relevance to preeclampsia. Mol. Asp. Med. 2013, 34, 981–1023. [Google Scholar] [CrossRef]
- Genbacev, O.; Zhou, Y.; Ludlow, J.W.; Fisher, S.J. Regulation of human placental development by oxygen tension. Science 1997, 277, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Crocker, I.P.; Wareing, M.; Ferris, G.R.; Jones, C.J.; Cartwright, J.E.; Baker, P.N.; Aplin, J.D. The effect of vascular origin, oxygen, and tumour necrosis factor alpha on trophoblast invasion of maternal arteries in vitro. J. Pathol. 2005, 206, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Caniggia, I.; Winter, J.; Lye, S.J.; Post, M. Oxygen and placental development during the first trimester: Implications for the pathophysiology of pre-eclampsia. Placenta 2000, 21 (Suppl. A), S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Falco, M.L.; Sivanathan, J.; Laoreti, A.; Thilaganathan, B.; Khalil, A. Placental histopathology associated with pre-eclampsia: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2017, 50, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134–135, 1–10. [Google Scholar] [CrossRef]
- Rajakumar, A.; Brandon, H.M.; Daftary, A.; Ness, R.; Conrad, K.P. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta 2004, 25, 763–769. [Google Scholar] [CrossRef]
- Tal, R.; Shaish, A.; Barshack, I.; Polak-Charcon, S.; Afek, A.; Volkov, A.; Feldman, B.; Avivi, C.; Harats, D. Effects of hypoxia-inducible factor-1alpha overexpression in pregnant mice: Possible implications for preeclampsia and intrauterine growth restriction. Am. J. Pathol. 2010, 177, 2950–2962. [Google Scholar] [CrossRef]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef]
- Pijnenborg, R.; Anthony, J.; Davey, D.A.; Rees, A.; Tiltman, A.; Vercruysse, L.; van Assche, A. Placental bed spiral arteries in the hypertensive disorders of pregnancy. Br. J. Obstet. Gynaecol. 1991, 98, 648–655. [Google Scholar] [CrossRef]
- Han, C.; Wang, C.; Chen, Y.; Wang, J.; Xu, X.; Hilton, T.; Cai, W.; Zhao, Z.; Wu, Y.; Li, K.; et al. Placenta-derived extracellular vesicles induce preeclampsia in mouse models. Haematologica 2020, 105, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, P.; Han, C.; Li, J.; Liu, L.; Zhao, Z.; Gao, Y.; Qin, Y.; Xu, Q.; Yan, Y.; et al. Association of placenta-derived extracellular vesicles with preeclampsia and associated hypercoagulability: A clinical observational study. BJOG Int. J. Obstet. Gynaecol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lisonkova, S.; Joseph, K.S. Incidence of preeclampsia: Risk factors and outcomes associated with early- versus late-onset disease. Am. J. Obstet. Gynecol. 2013, 209, 544.e1–544.e12. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M. Apoptotic changes occur in syncytiotrophoblast of human placental villi where fibrin type fibrinoid is deposited at discontinuities in the villous trophoblast. Placenta 1996, 17, 387–391. [Google Scholar] [CrossRef]
- Huppertz, B.; Frank, H.G.; Reister, F.; Kingdom, J.; Korr, H.; Kaufmann, P. Apoptosis cascade progresses during turnover of human trophoblast: Analysis of villous cytotrophoblast and syncytial fragments in vitro. Lab. Investig. 1999, 79, 1687–1702. [Google Scholar]
- Smith, S.C.; Baker, P.N. Placental apoptosis is increased in post-term pregnancies. Br. J. Obstet. Gynaecol. 1999, 106, 861–862. [Google Scholar] [CrossRef]
- Devaux, P.F. Protein involvement in transmembrane lipid asymmetry. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 417–439. [Google Scholar] [CrossRef]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-dependent phospholipid scrambling by tmem16f. Nature 2010, 468, 834–838. [Google Scholar] [CrossRef]
- Zwaal, R.F.; Comfurius, P.; van Deenen, L.L. Membrane asymmetry and blood coagulation. Nature 1977, 268, 358–360. [Google Scholar] [CrossRef]
- Lentz, B.R. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog. Lipid Res. 2003, 42, 423–438. [Google Scholar] [CrossRef]
- Abumaree, M.H.; Stone, P.R.; Chamley, L.W. The effects of apoptotic, deported human placental trophoblast on macrophages: Possible consequences for pregnancy. J. Reprod. Immunol. 2006, 72, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Gris, J.C.; Bouvier, S.; Cochery-Nouvellon, É.; Mercier, É.; Mousty, È.; Pérez-Martin, A. The role of haemostasis in placenta-mediated complications. Thromb. Res. 2019, 181 (Suppl. 1), S10–S14. [Google Scholar] [CrossRef]
- Feldstein, O.; Kovo, M. The association between abnormal coagulation testing in preeclampsia, adverse pregnancy outcomes, and placental histopathology. Hypertens. Pregnancy 2019, 38, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, J.S.; Stanek, J.; Warshak, C.; Khoury, J.; Sibai, B. The frequency and severity of placental findings in women with preeclampsia are gestational age dependent. Am. J. Obstet. Gynecol. 2003, 189, 1173–1177. [Google Scholar] [CrossRef]
- Abildgaard, U.; Heimdal, K. Pathogenesis of the syndrome of hemolysis, elevated liver enzymes, and low platelet count (hellp): A review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 166, 117–123. [Google Scholar] [CrossRef]
- Sep, S.; Verbeek, J.; Koek, G.; Smits, L.; Spaanderman, M.; Peeters, L. Clinical differences between early-onset hellp syndrome and early-onset preeclampsia during pregnancy and at least 6 months postpartum. Am. J. Obstet. Gynecol. 2010, 202, 271.e1–271.e5. [Google Scholar] [CrossRef]
- van Runnard Heimel, P.J.; Kavelaars, A.; Heijnen, C.J.; Peters, W.H.; Huisjes, A.J.; Franx, A.; Bruinse, H.W. Hellp syndrome is associated with an increased inflammatory response, which may be inhibited by administration of prednisolone. Hypertens. Pregnancy 2008, 27, 253–265. [Google Scholar] [CrossRef]
- George, J.N.; Nester, C.M.; McIntosh, J.J. Syndromes of thrombotic microangiopathy associated with pregnancy. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 644–648. [Google Scholar] [CrossRef]
- Teng, Y.C.; Lin, Q.D.; Lin, J.H.; Ding, C.W.; Zuo, Y. Coagulation and fibrinolysis related cytokine imbalance in preeclampsia: The role of placental trophoblasts. J. Perinat. Med. 2009, 37, 343–348. [Google Scholar] [CrossRef][Green Version]
- Macey, M.G.; Bevan, S.; Alam, S.; Verghese, L.; Agrawal, S.; Beski, S.; Thuraisingham, R.; MacCallum, P.K. Platelet activation and endogenous thrombin potential in pre-eclampsia. Thromb. Res. 2010, 125, e76–e81. [Google Scholar] [CrossRef]
- Posma, J.J.; Posthuma, J.J.; Spronk, H.M. Coagulation and non-coagulation effects of thrombin. J. Thromb. Haemost. JTH 2016, 14, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Rabiet, M.J.; Plantier, J.L.; Dejana, E. Thrombin-induced endothelial cell dysfunction. Br. Med Bull. 1994, 50, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S.; Menton, D.N.; Laszlo, A.; Lum, H. Nature of thrombin-induced sustained increase in cytosolic calcium concentration in cultured endothelial cells. J. Biol. Chem. 1989, 264, 16771–16775. [Google Scholar] [PubMed]
- Hutt, R.; Ogunniyi, S.O.; Sullivan, M.H.; Elder, M.G. Increased platelet volume and aggregation precede the onset of preeclampsia. Obstet. Gynecol. 1994, 83, 146–149. [Google Scholar] [CrossRef]
- Janes, S.L.; Kyle, P.M.; Redman, C.; Goodall, A.H. Flow cytometric detection of activated platelets in pregnant women prior to the development of pre-eclampsia. Thromb. Haemost. 1995, 74, 1059–1063. [Google Scholar] [CrossRef]
- Lukanov, T.H.; Bojinova, S.I.; Popova, V.S.; Emin, A.L.; Veleva, G.L.; Gecheva, S.P.; Konova, E.I. Flow cytometric investigation of cd40-cd40 ligand system in preeclampsia and normal pregnancy. Clin. Appl. Thromb./Hemost. 2010, 16, 306–312. [Google Scholar] [CrossRef]
- Major, H.D.; Campbell, R.A.; Silver, R.M.; Branch, D.W.; Weyrich, A.S. Synthesis of sflt-1 by platelet-monocyte aggregates contributes to the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol. 2014, 210, e541–e547. [Google Scholar] [CrossRef]
- Han, C.; Han, L.; Huang, P.; Chen, Y.; Wang, Y.; Xue, F. Syncytiotrophoblast-derived extracellular vesicles in pathophysiology of preeclampsia. Front. Physiol. 2019, 10, 1236. [Google Scholar] [CrossRef]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef]
- Raijmakers, M.T.; Peters, W.H.; Steegers, E.A.; Poston, L. Nad(p)h oxidase associated superoxide production in human placenta from normotensive and pre-eclamptic women. Placenta 2004, 25 (Suppl. A), S85–S89. [Google Scholar] [CrossRef]
- Matsubara, S.; Sato, I. Enzyme histochemically detectable nad(p)h oxidase in human placental trophoblasts: Normal, preeclamptic, and fetal growth restriction-complicated pregnancy. Histochem. Cell Biol. 2001, 116, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular nadph oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Lapaire, O.; Grill, S.; Lalevee, S.; Kolla, V.; Hösli, I.; Hahn, S. Microarray screening for novel preeclampsia biomarker candidates. Fetal Diagn. Ther. 2012, 31, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Karabulut, A.B.; Kafkasli, A.; Burak, F.; Gozukara, E.M. Maternal and fetal plasma adenosine deaminase, xanthine oxidase and malondialdehyde levels in pre-eclampsia. Cell Biochem. Funct. 2005, 23, 279–283. [Google Scholar] [CrossRef]
- Many, A.; Hubel, C.A.; Fisher, S.J.; Roberts, J.M.; Zhou, Y. Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am. J. Pathol. 2000, 156, 321–331. [Google Scholar] [CrossRef]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef]
- McCarthy, C.M.; Kenny, L.C. Mitochondrial [dys]function; culprit in pre-eclampsia? Clin. Sci. 2016, 130, 1179–1184. [Google Scholar] [CrossRef]
- Myatt, L. Review: Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta 2010, 31, S66–S69. [Google Scholar] [CrossRef]
- Vaka, V.R.; McMaster, K.M.; Cunningham, M.W., Jr.; Ibrahim, T.; Hazlewood, R.; Usry, N.; Cornelius, D.C.; Amaral, L.M.; LaMarca, B. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 2018, 72, 703–711. [Google Scholar] [CrossRef]
- Covarrubias, A.E.; Lecarpentier, E.; Lo, A.; Salahuddin, S.; Gray, K.J.; Karumanchi, S.A.; Zsengeller, Z.K. Ap39, a modulator of mitochondrial bioenergetics, reduces antiangiogenic response and oxidative stress in hypoxia-exposed trophoblasts: Relevance for preeclampsia pathogenesis. Am. J. Pathol. 2019, 189, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Cairns, C.B.; Moore, F.A.; Haenel, J.B.; Gallea, B.L.; Ortner, J.P.; Rose, S.J.; Moore, E.E. Evidence for early supply independent mitochondrial dysfunction in patients developing multiple organ failure after trauma. J. Trauma 1997, 42, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Smeding, L.; Plotz, F.B.; Groeneveld, A.B.; Kneyber, M.C. Structural changes of the heart during severe sepsis or septic shock. Shock 2012, 37, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Peterson, P.L.; Verweij, B.H.; Vinas, F.C.; Muizelaar, J.P.; Lee, C.P. Mitochondrial dysfunction after experimental traumatic brain injury: Combined efficacy of snx-111 and u-101033e. J. Neurotrauma 1998, 15, 531–544. [Google Scholar] [CrossRef]
- Azbill, R.D.; Mu, X.; Bruce-Keller, A.J.; Mattson, M.P.; Springer, J.E. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997, 765, 283–290. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, M.; Tian, Y.; Hilton, T.; Salsbery, B.; Zhou, E.Z.; Wu, X.; Thiagarajan, P.; Boilard, E.; Li, M.; et al. Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood 2016, 127, 2763–2772. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhou, Y.; Hilton, T.; Li, F.; Han, C.; Liu, L.; Yuan, H.; Li, Y.; Xu, X.; Wu, X.; et al. Extracellular mitochondria released from traumatized brains induced platelet procoagulant activity. Haematologica 2020, 105, 209–217. [Google Scholar] [CrossRef]
- Jedlicka, J.; Becker, B.F.; Chappell, D. Endothelial glycocalyx. Crit. Care Clin. 2020, 36, 217–232. [Google Scholar] [CrossRef]
- Kao, C.K.; Morton, J.S.; Quon, A.L.; Reyes, L.M.; Lopez-Jaramillo, P.; Davidge, S.T. Mechanism of vascular dysfunction due to circulating factors in women with pre-eclampsia. Clin. Sci. 2016, 130, 539–549. [Google Scholar] [CrossRef]
- Dong, J.F.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; Lopez, J.A. Adamts-13 rapidly cleaves newly secreted ultralarge von willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef]
- Jacobi, J.; Kristal, B.; Chezar, J.; Shaul, S.M.; Sela, S. Exogenous superoxide mediates pro-oxidative, proinflammatory, and procoagulatory changes in primary endothelial cell cultures. Free Radic. Biol. Med. 2005, 39, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Szotowski, B.; Antoniak, S.; Goldin-Lang, P.; Tran, Q.V.; Pels, K.; Rosenthal, P.; Bogdanov, V.Y.; Borchert, H.H.; Schultheiss, H.P.; Rauch, U. Antioxidative treatment inhibits the release of thrombogenic tissue factor from irradiation- and cytokine-induced endothelial cells. Cardiovasc. Res. 2007, 73, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.F.; Gardiner, E.E.; Kenny, D.; Andrews, R.K.; Berndt, M.C. Platelet receptor redox regulation. Platelets 2008, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Begonja, A.J.; Gambaryan, S.; Geiger, J.; Aktas, B.; Pozgajova, M.; Nieswandt, B.; Walter, U. Platelet nad(p)h-oxidase-generated ros production regulates alphaiibbeta3-integrin activation independent of the no/cgmp pathway. Blood 2005, 106, 2757–2760. [Google Scholar] [CrossRef] [PubMed]
- Krotz, F.; Sohn, H.Y.; Pohl, U. Reactive oxygen species: Players in the platelet game. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1988–1996. [Google Scholar] [CrossRef]
- Freedman, J.E. Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, s11–s16. [Google Scholar] [CrossRef]
- Arthur, J.F.; Qiao, J.; Shen, Y.; Davis, A.K.; Dunne, E.; Berndt, M.C.; Gardiner, E.E.; Andrews, R.K. Itam receptor-mediated generation of reactive oxygen species in human platelets occurs via syk-dependent and syk-independent pathways. J. Thromb. Haemost. JTH 2012, 10, 1133–1141. [Google Scholar] [CrossRef]
- Pietraforte, D.; Vona, R.; Marchesi, A.; de Jacobis, I.T.; Villani, A.; Del Principe, D.; Straface, E. Redox control of platelet functions in physiology and pathophysiology. Antioxid. Redox Signal. 2014, 21, 177–193. [Google Scholar] [CrossRef]
- Vara, D.; Campanella, M.; Pula, G. The novel nox inhibitor 2-acetylphenothiazine impairs collagen-dependent thrombus formation in a gpvi-dependent manner. Br. J. Pharmacol. 2013, 168, 212–224. [Google Scholar] [CrossRef]
- Fuentes, E.; Gibbins, J.M.; Holbrook, L.M.; Palomo, I. Nadph oxidase 2 (nox2): A key target of oxidative stress-mediated platelet activation and thrombosis. Trends Cardiovasc. Med. 2018, 28, 429–434. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. Ros in platelet biology: Functional aspects and methodological insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef] [PubMed]
- Leo, R.; Pratico, D.; Iuliano, L.; Pulcinelli, F.M.; Ghiselli, A.; Pignatelli, P.; Colavita, A.R.; FitzGerald, G.A.; Violi, F. Platelet activation by superoxide anion and hydroxyl radicals intrinsically generated by platelets that had undergone anoxia and then reoxygenated. Circulation 1997, 95, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Iuliano, L.; Ghiselli, A.; Alessandri, C.; Violi, F. Hydrogen peroxide as trigger of platelet aggregation. Haemostasis 1991, 21, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Wang, S.B.; Park, S.J.; Oh, G.T.; Lee, S.H.; Ho, Y.S.; Chang, T.S. Reactive oxygen species play a critical role in collagen-induced platelet activation via shp-2 oxidation. Antioxid. Redox Signal. 2014, 20, 2528–2540. [Google Scholar] [CrossRef]
- Jang, J.Y.; Wang, S.B.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Yu, D.Y.; Chang, T.S. Peroxiredoxin ii is an antioxidant enzyme that negatively regulates collagen-stimulated platelet function. J. Biol. Chem. 2015, 290, 11432–11442. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, R.K.; Bhardwaj, T.R. Therapeutic role of nitric oxide as emerging molecule. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 85, 182–201. [Google Scholar] [CrossRef]
- Essex, D.W.; Li, M.; Feinman, R.D.; Miller, A. Platelet surface glutathione reductase-like activity. Blood 2004, 104, 1383–1385. [Google Scholar] [CrossRef]
- Essex, D.W.; Chen, K.; Swiatkowska, M. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood 1995, 86, 2168–2173. [Google Scholar] [CrossRef]
- Manickam, N.; Ahmad, S.S.; Essex, D.W. Vicinal thiols are required for activation of the αiibβ3 platelet integrin. J. Thromb. Haemost. 2011, 9, 1207–1215. [Google Scholar] [CrossRef]
- Manickam, N.; Sun, X.; Li, M.; Gazitt, Y.; Essex, D.W. Protein disulphide isomerase in platelet function. Br. J. Haematol. 2008, 140, 223–229. [Google Scholar] [CrossRef]
- Lahav, J.; Wijnen, E.M.; Hess, O.; Hamaia, S.W.; Griffiths, D.; Makris, M.; Knight, C.G.; Essex, D.W.; Farndale, R.W. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood 2003, 102, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, Y.; Chen, F.; Wang, L.; Rauova, L.; Hayes, V.M.; Poncz, M.; Li, H.; Liu, T.; Liu, J.; et al. The disulfide isomerase erp72 supports arterial thrombosis in mice. Blood 2017, 130, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Essex, D.W. Redox control of platelet function. Antioxid. Redox Signal. 2009, 11, 1191–1225. [Google Scholar] [CrossRef] [PubMed]
- Raijmakers, M.T.; Zusterzeel, P.L.; Roes, E.M.; Steegers, E.A.; Mulder, T.P.; Peters, W.H. Oxidized and free whole blood thiols in preeclampsia. Obstet. Gynecol. 2001, 97, 272–276. [Google Scholar]
- Pimentel, A.M.; Pereira, N.R.; Costa, C.A.; Mann, G.E.; Cordeiro, V.S.; de Moura, R.S.; Brunini, T.M.; Mendes-Ribeiro, A.C.; Resende, A.C. L-arginine-nitric oxide pathway and oxidative stress in plasma and platelets of patients with pre-eclampsia. Hypertens. Res. 2013, 36, 783–788. [Google Scholar] [CrossRef]
- Larson, M.C.; Hillery, C.A.; Hogg, N. Circulating membrane-derived microvesicles in redox biology. Free Radic. Biol. Med. 2014, 73, 214–228. [Google Scholar] [CrossRef]
- Bhullar, J.; Bhopale, V.M.; Yang, M.; Sethuraman, K.; Thom, S.R. Microparticle formation by platelets exposed to high gas pressures—An oxidative stress response. Free Radic. Biol. Med. 2016, 101, 154–162. [Google Scholar] [CrossRef]
- Oe, Y.; Ko, M.; Fushima, T.; Sato, E.; Karumanchi, S.A.; Sato, H.; Sugawara, J.; Ito, S.; Takahashi, N. Hepatic dysfunction and thrombocytopenia induced by excess sflt1 in mice lacking endothelial nitric oxide synthase. Sci. Rep. 2018, 8, 102. [Google Scholar] [CrossRef]
- Koenig, M.; Roy, M.; Baccot, S.; Cuilleron, M.; de Filippis, J.P.; Cathebras, P. Thrombotic microangiopathy with liver, gut, and bone infarction (catastrophic antiphospholipid syndrome) associated with hellp syndrome. Clin. Rheumatol. 2005, 24, 166–168. [Google Scholar] [CrossRef]
- Abraham, K.A.; Kennelly, M.; Dorman, A.M.; Walshe, J.J. Pathogenesis of acute renal failure associated with the hellp syndrome: A case report and review of the literature. Eur. J. Obstet. Gynecol. Reprod. Biol. 2003, 108, 99–102. [Google Scholar] [CrossRef]
- Sadler, J.E. Biochemistry and genetics of von willebrand factor. Annu. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Wagner, D.D. In vitro multimerization of von willebrand factor is triggered by low ph. Importance of the propolypeptide and free sulfhydryls. J. Biol. Chem. 1989, 264, 13497–13503. [Google Scholar] [PubMed]
- Furlan, M. Von willebrand factor: Molecular size and functional activity. Ann. Hematol. 1996, 72, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Sporn, L.A.; Marder, V.J.; Wagner, D.D. Von willebrand factor released from weibel-palade bodies binds more avidly to extracellular matrix than that secreted constitutively. Blood 1987, 69, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Anvari, B.; Romo, G.M.; Cruz, M.A.; Dong, J.F.; McIntire, L.V.; Moake, J.L.; Lopez, J.A. Ultralarge multimers of von willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein ib-ix complex: Studies using optical tweezers. Blood 2002, 99, 3971–3977. [Google Scholar] [CrossRef]
- Furlan, M.; Robles, R.; Lammle, B. Partial purification and characterization of a protease from human plasma cleaving von willebrand factor to fragments produced by in vivo proteolysis. Blood 1996, 87, 4223–4234. [Google Scholar] [CrossRef]
- Tsai, H.M. Physiologic cleavage of von willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood 1996, 87, 4235–4244. [Google Scholar] [CrossRef]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the adamts gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–494. [Google Scholar] [CrossRef]
- Kroll, M.H.; Hellums, J.D.; McIntire, L.V.; Schafer, A.I.; Moake, J.L. Platelets and shear stress. Blood 1996, 88, 1525–1541. [Google Scholar] [CrossRef]
- Chen, J.; Fu, X.; Wang, Y.; Ling, M.; McMullen, B.; Kulman, J.; Chung, D.W.; López, J.A. Oxidative modification of von willebrand factor by neutrophil oxidants inhibits its cleavage by adamts13. Blood 2010, 115, 706–712. [Google Scholar] [CrossRef]
- Lancellotti, S.; De Filippis, V.; Pozzi, N.; Peyvandi, F.; Palla, R.; Rocca, B.; Rutella, S.; Pitocco, D.; Mannucci, P.M.; De Cristofaro, R. Formation of methionine sulfoxide by peroxynitrite at position 1606 of von willebrand factor inhibits its cleavage by adamts-13: A new prothrombotic mechanism in diseases associated with oxidative stress. Free Radic. Biol. Med. 2010, 48, 446–456. [Google Scholar] [PubMed]
- Choi, H.; Aboulfatova, K.; Pownall, H.J.; Cook, R.; Dong, J.F. Shear-induced disulfide bond formation regulates adhesion activity of von willebrand factor. J. Biol. Chem. 2007, 282, 35604–35611. [Google Scholar] [CrossRef]
- Li, Y.; Choi, H.; Zhou, Z.; Nolasco, L.; Pownall, H.J.; Voorberg, J.; Moake, J.L.; Dong, J.F. Covalent regulation of ulvwf string formation and elongation on endothelial cells under flow conditions. J. Thromb. Haemost. 2008, 6, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Butera, D.; Passam, F.; Ju, L.; Cook, K.M.; Woon, H.; Aponte-Santamaria, C.; Gardiner, E.; Davis, A.K.; Murphy, D.A.; Bronowska, A.; et al. Autoregulation of von willebrand factor function by a disulfide bond switch. Sci. Adv. 2018, 4, eaaq1477. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Ling, M.; Lopez, J.A.; Chung, D.W.; Fu, X. Hypochlorous acid generated by neutrophils inactivates adamts13: An oxidative mechanism for regulating adamts13 proteolytic activity during inflammation. J. Biol. Chem. 2015, 290, 1422–1431. [Google Scholar] [PubMed]
- Yeh, H.C.; Zhou, Z.; Choi, H.; Tekeoglu, S.; May, W., III; Wang, C.; Turner, N.; Scheiflinger, F.; Moake, J.L.; Dong, J.F. Disulfide bond reduction of von willebrand factor by adamts-13. J. Thromb. Haemost. 2010, 8, 2778–2788. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aref, S.; Goda, H. Increased vwf antigen levels and decreased adamts13 activity in preeclampsia. Hematology 2013, 18, 237–241. [Google Scholar] [CrossRef]
- Szpera-Gozdziewicz, A.; Gozdziewicz, T.; Boruczkowski, M.; Dworacki, G.; Breborowicz, G.H. Relationship between the von willebrand factor plasma concentration and ultrasonographic doppler findings in pregnancies complicated by hypertensive disorders: A pilot study. Gynecol. Obstet. Investig. 2018, 83, 252–258. [Google Scholar] [CrossRef]
- Hulstein, J.J.; van Runnard Heimel, P.J.; Franx, A.; Lenting, P.J.; Bruinse, H.W.; Silence, K.; de Groot, P.G.; Fijnheer, R. Acute activation of the endothelium results in increased levels of active von willebrand factor in hemolysis, elevated liver enzymes and low platelets (hellp) syndrome. J. Thromb. Haemost. JTH 2006, 4, 2569–2575. [Google Scholar] [CrossRef]
- Lattuada, A.; Rossi, E.; Calzarossa, C.; Candolfi, R.; Mannucci, P.M. Mild to moderate reduction of a von willebrand factor cleaving protease (adamts-13) in pregnant women with hellp microangiopathic syndrome. Haematologica 2003, 88, 1029–1034. [Google Scholar]
- Palos, L.A. Oxidation of the coagulation factors. Nature 1949, 164, 926. [Google Scholar] [CrossRef] [PubMed]
- Bayele, H.K.; Murdock, P.J.; Perry, D.J.; Pasi, K.J. Simple shifts in redox/thiol balance that perturb blood coagulation. FEBS Lett. 2002, 510, 67–70. [Google Scholar] [CrossRef]
- Chen, V.M.; Ahamed, J.; Versteeg, H.H.; Berndt, M.C.; Ruf, W.; Hogg, P.J. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry 2006, 45, 12020–12028. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Ruf, W. Thiol pathways in the regulation of tissue factor prothrombotic activity. Curr. Opin. Hematol. 2011, 18, 343–348. [Google Scholar] [CrossRef]
- Chen, V.M. Tissue factor de-encryption, thrombus formation, and thiol-disulfide exchange. Semin. Thromb. Hemost. 2013, 39, 40–47. [Google Scholar] [CrossRef]
- Harutyunyan, H.A. Prothrombin and fibrinogen carbonylation: How that can affect the blood clotting. Redox Rep. Commun. Free Radic. Res. 2017, 22, 160–165. [Google Scholar] [CrossRef][Green Version]
- Roitman, E.V.; Azizova, O.A.; Morozov, Y.A.; Aseichev, A.V. Effect of oxidized fibrinogens on blood coagulation. Bull. Exp. Biol. Med. 2004, 138, 245–247. [Google Scholar] [CrossRef]
- Weigandt, K.M.; White, N.; Chung, D.; Ellingson, E.; Wang, Y.; Fu, X.; Pozzo, D.C. Fibrin clot structure and mechanics associated with specific oxidation of methionine residues in fibrinogen. Biophys. J. 2012, 103, 2399–2407. [Google Scholar] [CrossRef]
- White, N.J.; Wang, Y.; Fu, X.; Cardenas, J.C.; Martin, E.J.; Brophy, D.F.; Wade, C.E.; Wang, X.; St John, A.E.; Lim, E.B.; et al. Post-translational oxidative modification of fibrinogen is associated with coagulopathy after traumatic injury. Free Radic. Biol. Med. 2016, 96, 181–189. [Google Scholar] [CrossRef]
- Mello, G.; Parretti, E.; Marozio, L.; Pizzi, C.; Lojacono, A.; Frusca, T.; Facchinetti, F.; Benedetto, C. Thrombophilia is significantly associated with severe preeclampsia: Results of a large-scale, case-controlled study. Hypertension 2005, 46, 1270–1274. [Google Scholar] [CrossRef]
- Dudding, T.; Heron, J.; Thakkinstian, A.; Nurk, E.; Golding, J.; Pembrey, M.; Ring, S.M.; Attia, J.; Scott, R.J. Factor v leiden is associated with pre-eclampsia but not with fetal growth restriction: A genetic association study and meta-analysis. J. Thromb. Haemost. JTH 2008, 6, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Schlembach, D.; Beinder, E.; Zingsem, J.; Wunsiedler, U.; Beckmann, M.W.; Fischer, T. Association of maternal and/or fetal factor v leiden and g20210a prothrombin mutation with hellp syndrome and intrauterine growth restriction. Clin. Sci. 2003, 105, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Tranquilli, A.L.; Giannubilo, S.R.; Dell’Uomo, B.; Grandone, E. Adverse pregnancy outcomes are associated with multiple maternal thrombophilic factors. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 117, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.R.; Platt, R.; McNamara, H.; Rozen, R.; Chen, M.F.; Genest, J., Jr.; Goulet, L.; Lydon, J.; Seguin, L.; Dassa, C.; et al. Inherited thrombophilia and preeclampsia within a multicenter cohort: The montreal preeclampsia study. Am. J. Obstet. Gynecol. 2009, 200, 151.e1–151.e9. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.M.; Zhao, Y.; Spong, C.Y.; Sibai, B.; Wendel, G., Jr.; Wenstrom, K.; Samuels, P.; Caritis, S.N.; Sorokin, Y.; Miodovnik, M.; et al. Prothrombin gene g20210a mutation and obstetric complications. Obstet. Gynecol. 2010, 115, 14–20. [Google Scholar] [CrossRef]
- Hiltunen, L.M.; Laivuori, H.; Rautanen, A.; Kaaja, R.; Kere, J.; Krusius, T.; Paunio, M.; Rasi, V. Blood group ab and factor v leiden as risk factors for pre-eclampsia: A population-based nested case-control study. Thromb. Res. 2009, 124, 167–173. [Google Scholar] [CrossRef]
- Stief, T.W. Oxidized fibrin stimulates the activation of pro-urokinase and is the preferential substrate of human plasmin. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 1993, 4, 117–121. [Google Scholar] [CrossRef]
- Pretorius, E.; Bester, J.; Vermeulen, N.; Lipinski, B. Oxidation inhibits iron-induced blood coagulation. Curr. Drug Targets 2013, 14, 13–19. [Google Scholar] [CrossRef][Green Version]
- Stief, T.W.; Martin, E.; Jimenez, J.; Digon, J.; Rodriguez, J.M. Effect of oxidants on proteases of the fibrinolytic system: Possible role for methionine residues in the interaction between tissue type plasminogen activator and fibrin. Thromb. Res. 1991, 61, 191–200. [Google Scholar] [CrossRef]
- Stief, T.W.; Kurz, J.; Doss, M.O.; Fareed, J. Singlet oxygen inactivates fibrinogen, factor v, factor viii, factor x, and platelet aggregation of human blood. Thromb. Res. 2000, 97, 473–480. [Google Scholar] [CrossRef]
- Wang, Y.; Walsh, S.W. Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta 1998, 19, 581–586. [Google Scholar] [CrossRef]
- Afonso, C.B.; Spickett, C.M. Lipoproteins as targets and markers of lipoxidation. Redox Biol. 2019, 23, 101066. [Google Scholar] [CrossRef] [PubMed]
- Slatter, D.A.; Percy, C.L.; Allen-Redpath, K.; Gajsiewicz, J.M.; Brooks, N.J.; Clayton, A.; Tyrrell, V.J.; Rosas, M.; Lauder, S.N.; Watson, A.; et al. Enzymatically oxidized phospholipids restore thrombin generation in coagulation factor deficiencies. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Belo, L.; Caslake, M.; Gaffney, D.; Santos-Silva, A.; Pereira-Leite, L.; Quintanilha, A.; Rebelo, I. Changes in ldl size and hdl concentration in normal and preeclamptic pregnancies. Atherosclerosis 2002, 162, 425–432. [Google Scholar] [CrossRef]
- Witztum, J.L. Susceptibility of low-density lipoprotein to oxidative modification. Am. J. Med. 1993, 94, 347–349. [Google Scholar] [CrossRef]
- Qiu, C.; Phung, T.T.; Vadachkoria, S.; Muy-Rivera, M.; Sanchez, S.E.; Williams, M.A. Oxidized low-density lipoprotein (oxidized ldl) and the risk of preeclampsia. Physiol. Res. 2006, 55, 491–500. [Google Scholar]
- Ali Akbar, S.; Nicolaides, K.H.; Brown, P.R. Measurement of cu/zn sod in placenta, cultured cells, various fetal tissues, decidua and semen by elisa. J. Obstet. Gynaecol. J. Inst. Obstet. Gynaecol. 1998, 18, 331–335. [Google Scholar] [CrossRef]
- Wang, Y.; Walsh, S.W. Antioxidant activities and mrna expression of superoxide dismutase, catalase, and glutathione peroxidase in normal and preeclamptic placentas. J. Soc. Gynecol. Investig. 1996, 3, 179–184. [Google Scholar] [CrossRef]
- Mistry, H.D.; Kurlak, L.O.; Williams, P.J.; Ramsay, M.M.; Symonds, M.E.; Broughton Pipkin, F. Differential expression and distribution of placental glutathione peroxidases 1, 3 and 4 in normal and preeclamptic pregnancy. Placenta 2010, 31, 401–408. [Google Scholar] [CrossRef]
- Sahay, A.S.; Sundrani, D.P.; Wagh, G.N.; Mehendale, S.S.; Joshi, S.R. Regional differences in the placental levels of oxidative stress markers in pre-eclampsia. Int. J. Gynaecol. Obstet. 2015, 129, 213–218. [Google Scholar] [CrossRef]
- Nakamura, M.; Sekizawa, A.; Purwosunu, Y.; Okazaki, S.; Farina, A.; Wibowo, N.; Shimizu, H.; Okai, T. Cellular mrna expressions of anti-oxidant factors in the blood of preeclamptic women. Prenat. Diagn. 2009, 29, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Levytska, K.; Kingdom, J.; Baczyk, D.; Drewlo, S. Heme oxygenase-1 in placental development and pathology. Placenta 2013, 34, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Ehsanipoor, R.M.; Fortson, W.; Fitzmaurice, L.E.; Liao, W.X.; Wing, D.A.; Chen, D.B.; Chan, K. Nitric oxide and carbon monoxide production and metabolism in preeclampsia. Reprod. Sci. 2013, 20, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Wruck, C.J.; Huppertz, B.; Bose, P.; Brandenburg, L.O.; Pufe, T.; Kadyrov, M. Role of a fetal defence mechanism against oxidative stress in the aetiology of preeclampsia. Histopathology 2009, 55, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Guo, D.; Gu, H.; Zhang, L.; Lv, S. Selenium and preeclampsia: A systematic review and meta-analysis. Biol. Trace Elem. Res. 2016, 171, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liu, L.; Zhou, K.; Ding, L.; Zeng, J.; Zhang, W. Anti-oxidant and anti-endothelial dysfunctional properties of nano-selenium in vitro and in vivo of hyperhomocysteinemic rats. Int. J. Nanomed. 2020, 15, 4501–4521. [Google Scholar] [CrossRef]
- Khera, A.; Vanderlelie, J.J.; Perkins, A.V. Selenium supplementation protects trophoblast cells from mitochondrial oxidative stress. Placenta 2013, 34, 594–598. [Google Scholar] [CrossRef]
- Khera, A.; Vanderlelie, J.J.; Holland, O.; Perkins, A.V. Overexpression of endogenous anti-oxidants with selenium supplementation protects trophoblast cells from reactive oxygen species-induced apoptosis in a bcl-2-dependent manner. Biol. Trace Elem. Res. 2017, 177, 394–403. [Google Scholar] [CrossRef]
- Teran, E.; Hernandez, I.; Nieto, B.; Tavara, R.; Ocampo, J.E.; Calle, A. Coenzyme q10 supplementation during pregnancy reduces the risk of pre-eclampsia. Int. J. Gynaecol. Obstet. 2009, 105, 43–45. [Google Scholar] [CrossRef]
- Gao, L.; Mao, Q.; Cao, J.; Wang, Y.; Zhou, X.; Fan, L. Effects of coenzyme q10 on vascular endothelial function in humans: A meta-analysis of randomized controlled trials. Atherosclerosis 2012, 221, 311–316. [Google Scholar] [CrossRef]
- Kerley, R.N.; McCarthy, C.; Kell, D.B.; Kenny, L.C. The potential therapeutic effects of ergothioneine in pre-eclampsia. Free Radic. Biol. Med. 2018, 117, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.D.; McCarthy, F.P.; Manna, S.; Groarke, E.; Kell, D.B.; Kenny, L.C.; McCarthy, C.M. L-(+)-ergothioneine significantly improves the clinical characteristics of preeclampsia in the reduced uterine perfusion pressure rat model. Hypertension 2020, 75, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Roberge, S.; Demers, S.; Nicolaides, K.H.; Bureau, M.; Côté, S.; Bujold, E. Prevention of pre-eclampsia by low-molecular-weight heparin in addition to aspirin: A meta-analysis. Ultrasound Obstet. Gynecol. 2016, 47, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Taubert, D.; Berkels, R.; Grosser, N.; Schröder, H.; Gründemann, D.; Schömig, E. Aspirin induces nitric oxide release from vascular endothelium: A novel mechanism of action. Br. J. Pharmacol. 2004, 143, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Nascimento-Silva, V.; Arruda, M.A.; Barja-Fidalgo, C.; Fierro, I.M. Aspirin-triggered lipoxin a4 blocks reactive oxygen species generation in endothelial cells: A novel antioxidative mechanism. Thromb. Haemost. 2007, 97, 88–98. [Google Scholar]
- Gil-Villa, A.M.; Alvarez, A.M. Role of aspirin-triggered lipoxin a4, aspirin, and salicylic acid in the modulation of the oxidative and inflammatory responses induced by plasma from women with pre-eclampsia. Am. J. Reprod. Immunol. 2020, 83, e13207. [Google Scholar] [CrossRef]
- Dzieciuchowicz, Ł.; Checiński, P.; Krauss, H. Heparin reduces oxidative stress in the postoperative period. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2002, 8, Cr657–Cr660. [Google Scholar]
- Manduteanu, I.; Dragomir, E.; Voinea, M.; Capraru, M.; Simionescu, M. Enoxaparin reduces h2o2-induced activation of human endothelial cells by a mechanism involving cell adhesion molecules and nuclear transcription factors. Pharmacology 2007, 79, 154–162. [Google Scholar] [CrossRef]
- Chlopicki, S.; Olszanecki, R.; Janiszewski, M.; Laurindo, F.R.; Panz, T.; Miedzobrodzki, J. Functional role of nadph oxidase in activation of platelets. Antioxid. Redox Signal. 2004, 6, 691–698. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, C.; Huang, P.; Lyu, M.; Dong, J. Oxidative Stress and Preeclampsia-Associated Prothrombotic State. Antioxidants 2020, 9, 1139. https://doi.org/10.3390/antiox9111139
Han C, Huang P, Lyu M, Dong J. Oxidative Stress and Preeclampsia-Associated Prothrombotic State. Antioxidants. 2020; 9(11):1139. https://doi.org/10.3390/antiox9111139
Chicago/Turabian StyleHan, Cha, Pengzhu Huang, Meilu Lyu, and Jingfei Dong. 2020. "Oxidative Stress and Preeclampsia-Associated Prothrombotic State" Antioxidants 9, no. 11: 1139. https://doi.org/10.3390/antiox9111139
APA StyleHan, C., Huang, P., Lyu, M., & Dong, J. (2020). Oxidative Stress and Preeclampsia-Associated Prothrombotic State. Antioxidants, 9(11), 1139. https://doi.org/10.3390/antiox9111139